Tackling Structures of Long Noncoding RNAs

Abstract
:1. Introduction
2. RNA Structure
3. Approaches to Determine 2-D Structures
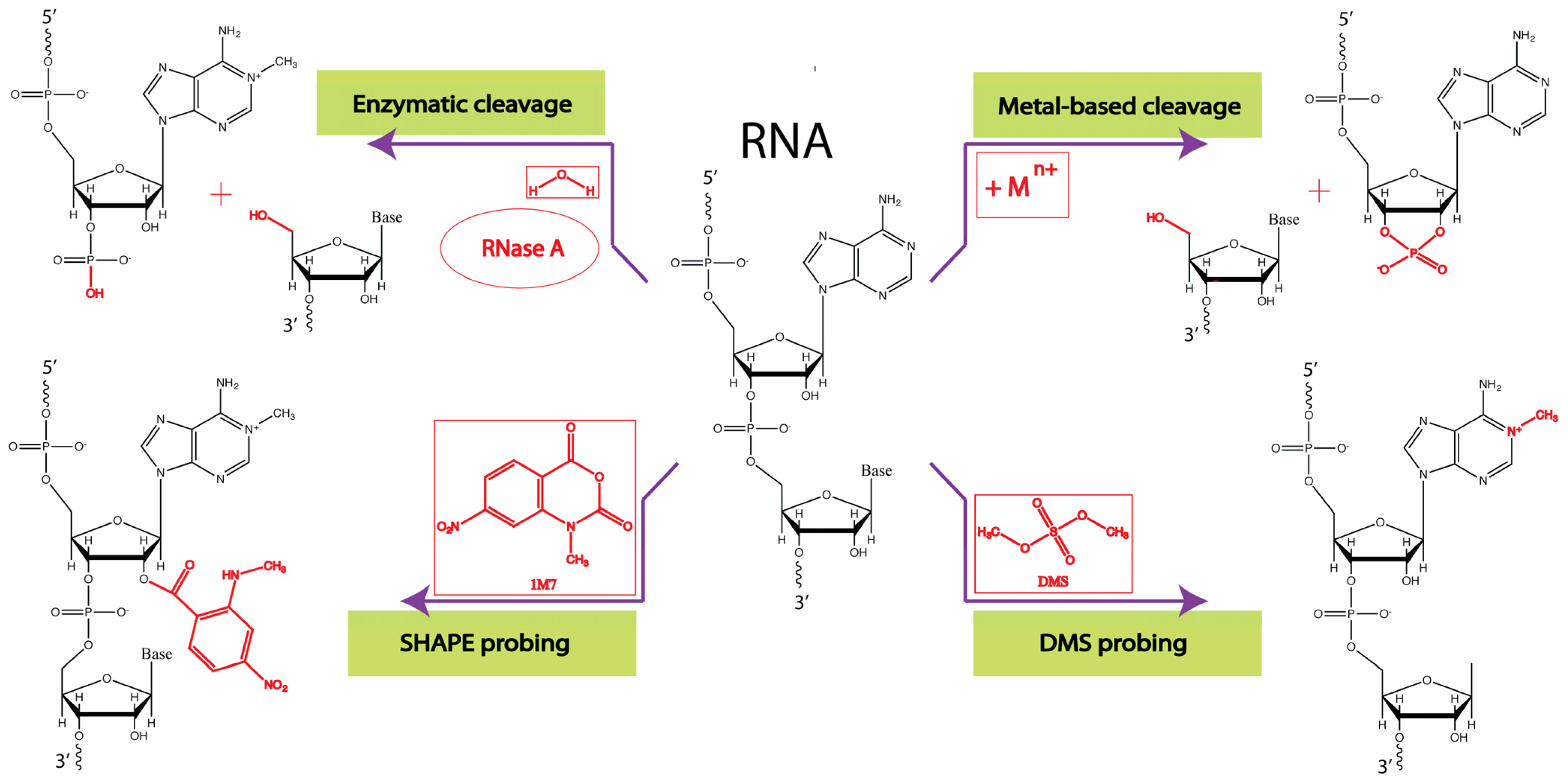
3.1. Enzymatic Probes
3.2. Chemical Probes
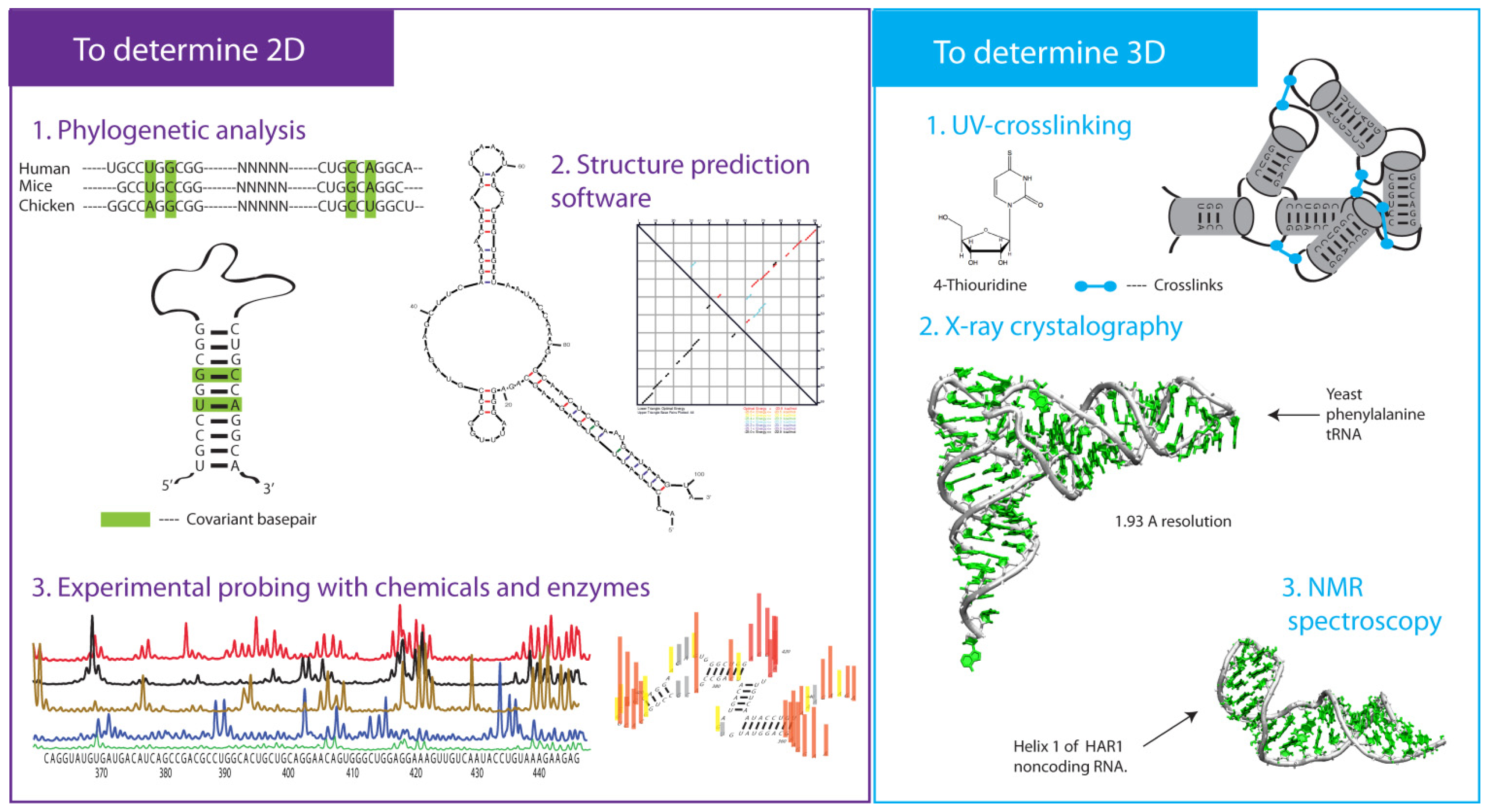
3.3. Phylogenetic Analysis
4. 2-D RNA Structure Predictions
5. Approaches to Determine 3-D Structures
6. Genome-Wide RNA Structural Studies and Sequencing Platforms
7. The Next Step: In Vivo RNA Structure Determination
8. Conclusions
Acknowledgments
Conflicts of Interest
References
- Rinn, J.L.; Chang, H.Y. Genome regulation by long noncoding RNAs. Annu. Rev. Biochem 2012, 81, 145–166. [Google Scholar]
- Will, C.L.; Luhrmann, R. Spliceosome structure and function. Csh. Perspect. Biol 2011. [Google Scholar] [CrossRef]
- Steitz, T.A. A structural understanding of the dynamic ribosome machine. Nat. Rev. Mol. Cell Biol 2008, 9, 242–253. [Google Scholar]
- Gong, C.; Maquat, L.E. lncRNAs transactivate STAU1-mediated mRNA decay by duplexing with 3′ UTRs via Alu elements. Nature 2011, 470, 284–288. [Google Scholar]
- Swiezewski, S.; Liu, F.; Magusin, A.; Dean, C. Cold-induced silencing by long antisense transcripts of an Arabidopsis Polycomb target. Nature 2009, 462, 799–802. [Google Scholar]
- Gutschner, T.; Hammerle, M.; Eissmann, M.; Hsu, J.; Kim, Y.; Hung, G.; Revenko, A.; Arun, G.; Stentrup, M.; Gross, M.; et al. The noncoding RNA MALAT1 is a critical regulator of the metastasis phenotype of lung cancer cells. Cancer Res 2013, 73, 1180–1189. [Google Scholar]
- Tani, H.; Torimura, M.; Akimitsu, N. The RNA degradation pathway regulates the function of GAS5 a non-coding RNA in Mammalian cells. PLoS One 2013, 8, e55684. [Google Scholar]
- Klattenhoff, C.A.; Scheuermann, J.C.; Surface, L.E.; Bradley, R.K.; Fields, P.A.; Steinhauser, M.L.; Ding, H.; Butty, V.L.; Torrey, L.; Haas, S.; et al. Braveheart, a long noncoding RNA required for cardiovascular lineage commitment. Cell 2013, 152, 570–583. [Google Scholar]
- Zhao, J.; Ohsumi, T.K.; Kung, J.T.; Ogawa, Y.; Grau, D.J.; Sarma, K.; Song, J.J.; Kingston, R.E.; Borowsky, M.; Lee, J.T. Genome-wide identification of polycomb-associated RNAs by rip-seq. Mol. Cell 2010, 40, 939–953. [Google Scholar]
- Martianov, I.; Ramadass, A.; Barros, A.S.; Chow, N.; Akoulitchev, A. Repression of the human dihydrofolate reductase gene by a non-coding interfering transcript. Nature 2007, 445, 666–670. [Google Scholar]
- Cesana, M.; Cacchiarelli, D.; Legnini, I.; Santini, T.; Sthandier, O.; Chinappi, M.; Tramontano, A.; Bozzoni, I. A long noncoding RNA controls muscle differentiation by functioning as a competing endogenous RNA. Cell 2011, 147, 947–947. [Google Scholar]
- Lanz, R.B.; McKenna, N.J.; Onate, S.A.; Albrecht, U.; Wong, J.M.; Tsai, S.Y.; Tsai, M.J.; O’Malley, B.W. A steroid receptor coactivator, SRA, functions as an RNA and is present in an src-1 complex. Cell 1999, 97, 17–27. [Google Scholar]
- Kino, T.; Hurt, D.E.; Ichijo, T.; Nader, N.; Chrousos, G.P. Noncoding RNA gas5 is a growth arrest- and starvation-associated repressor of the glucocorticoid receptor. Sci. Signal 2010. [Google Scholar] [CrossRef]
- Beltran, M.; Puig, I.; Pena, C.; Garcia, J.M.; Alvarez, A.B.; Pena, R.; Bonilla, F.; de Herreros, A.G. A natural antisense transcript regulates zeb2/sip1 gene expression during snail1-induced epithelial-mesenchymal transition. Gene Dev 2008, 22, 756–769. [Google Scholar]
- Carthew, R.W.; Sontheimer, E.J. Origins and mechanisms of miRNAs and siRNAs. Cell 2009, 136, 642–655. [Google Scholar]
- Leontis, N.B.; Westhof, E. Analysis of RNA motifs. Curr. Opin. Struct. Biol 2003, 13, 300–308. [Google Scholar]
- Butcher, S.E.; Pyle, A.M. The molecular interactions that stabilize RNA tertiary structure: RNA motifs, patterns, and networks. Accounts Chem. Res 2011, 44, 1302–1311. [Google Scholar]
- Novikova, I.V.; Hassan, B.H.; Mirzoyan, M.G.; Leontis, N.B. Engineering cooperative tecto-RNA complexes having programmable stoichiometries. Nucleic Acids Res 2011, 39, 2903–2917. [Google Scholar]
- Cech, T.R. Ribozymes, the first 20 years. Biochem. Soc. T 2002, 30, 1162–1166. [Google Scholar]
- Winkler, W.; Nahvi, A.; Breaker, R.R. Thiamine derivatives bind messenger RNAs directly to regulate bacterial gene expression. Nature 2002, 419, 952–956. [Google Scholar]
- Tung, C.S.; Sanbonmatsu, K.Y. Atomic model of the Thermus thermophilus 70S ribosome developed in silico. Biophys. J. 2004, 87, 2714–2722. [Google Scholar]
- Sanbonmatsu, K.Y. Alignment/misalignment hypothesis for tRNA selection by the ribosome. Biochimie 2006, 88, 1075–1089. [Google Scholar]
- Brion, P.; Westhof, E. Hierarchy and dynamics of RNA folding. Annu. Rev. Biophys. Biomol. Struct 1997, 26, 113–137. [Google Scholar]
- Whitford, P.C.; Schug, A.; Saunders, J.; Hennelly, S.P.; Onuchic, J.N.; Sanbonmatsu, K.Y. Nonlocal helix formation is key to understanding S-adenosylmethionine-1 riboswitch function. Biophys. J 2009, 96, L7–L9. [Google Scholar]
- Duncan, C.D.; Weeks, K.M. Nonhierarchical ribonucleoprotein assembly suggests a strain-propagation model for protein-facilitated RNA folding. Biochemistry 2010, 49, 5418–5425. [Google Scholar]
- Ziegeler, M.; Cevec, M.; Richter, C.; Schwalbe, H. NMR studies of HAR1 RNA secondary structures reveal conformational dynamics in the human RNA. Chembiochem 2012, 13, 2100–2112. [Google Scholar]
- Winkler, W.C.; Nahvi, A.; Sudarsan, N.; Barrick, J.E.; Breaker, R.R. An mRNA structure that controls gene expression by binding S-adenosylmethionine. Nat. Struct. Biol 2003, 10, 701–707. [Google Scholar]
- Stoddard, C.D.; Montange, R.K.; Hennelly, S.P.; Rambo, R.P.; Sanbonmatsu, K.Y.; Batey, R.T. Free state conformational sampling of the SAM-I riboswitch aptamer domain. Structure 2010, 18, 787–797. [Google Scholar]
- Schroeder, K.T.; Daldrop, P.; Lilley, D.M. RNA tertiary interactions in a riboswitch stabilize the structure of a kink turn. Structure 2011, 19, 1233–1240. [Google Scholar]
- Knapp, G. Enzymatic approaches to probing of RNA secondary and tertiary structure. Method. Enzymol 1989, 180, 192–212. [Google Scholar]
- Nichols, N.M.; Yue, D. Ribonucleases. In Current Protocols in Molecular Biology; John Wiley & Sons Inc: New York, NY, USA, 2008; Volume Chapter 3, Unit 3, p. 13. [Google Scholar]
- Ziehler, W.A.; Engelke, D.R. Probing RNA Structure with Chemical Reagents and Enzymes. In Current Protocols in Nucleic Acid Chemistry; John Wiley & Sons Inc: New York, NY, USA, 2001; Volume Chapter 6, Unit 6, p. 1. [Google Scholar]
- Brunel, C.; Romby, P. Probing RNA structure and RNA-ligand complexes with chemical probes. Method. Enzymol 2000, 318, 3–21. [Google Scholar]
- Forconi, M.; Herschlag, D. Metal ion-based RNA cleavage as a structural probe. Method. Enzymol 2009, 468, 91–106. [Google Scholar]
- Regulski, E.E.; Breaker, R.R. In-line probing analysis of riboswitches. Method. Mol. Biol 2008, 419, 53–67. [Google Scholar]
- Wilkinson, K.A.; Merino, E.J.; Weeks, K.M. Selective 2′-hydroxyl acylation analyzed by primer extension (SHAPE): Quantitative RNA structure analysis at single nucleotide resolution. Nat. Prot 2006, 1, 1610–1616. [Google Scholar]
- Mortimer, S.A.; Weeks, K.M. A fast-acting reagent for accurate analysis of RNA secondary and tertiary structure by SHAPE chemistry. J. Am. Chem. Soc 2007, 129, 4144–4145. [Google Scholar]
- McGinnis, J.L.; Dunkle, J.A.; Cate, J.H.; Weeks, K.M. The mechanisms of RNA SHAPE chemistry. J. Am. Chem. Soc 2012, 134, 6617–6624. [Google Scholar]
- Weeks, K.M. Advances in RNA structure analysis by chemical probing. Curr. Opin. Struct. Biol 2010, 20, 295–304. [Google Scholar]
- Fox, G.W.; Woese, C.R. 5s RNA secondary structure. Nature 1975, 256, 505–507. [Google Scholar]
- Noller, H.F.; Woese, C.R. Secondary structure of 16s ribosomal RNA. Science 1981, 212, 403–411. [Google Scholar]
- Woese, C.R.; Magrum, L.J.; Gupta, R.; Siegel, R.B.; Stahl, D.A.; Kop, J.; Crawford, N.; Brosius, J.; Gutell, R.; Hogan, J.J.; et al. Secondary structure model for bacterial 16s ribosomal RNA: Phylogenetic, enzymatic and chemical evidence. Nucleic Acids Res 1980, 8, 2275–2293. [Google Scholar]
- Noller, H.F.; Kop, J.; Wheaton, V.; Brosius, J.; Gutell, R.R.; Kopylov, A.M.; Dohme, F.; Herr, W.; Stahl, D.A.; Gupta, R.; et al. Secondary structure model for 23s ribosomal RNA. Nucleic Acids Res 1981, 9, 6167–6189. [Google Scholar]
- Shi, H.; Moore, P.B. The crystal structure of yeast phenylalanine tRNA at 1.93 a resolution: A classic structure revisited. RNA 2000, 6, 1091–1105. [Google Scholar]
- Derrien, T.; Johnson, R.; Bussotti, G.; Tanzer, A.; Djebali, S.; Tilgner, H.; Guernec, G.; Martin, D.; Merkel, A.; Knowles, D.G.; et al. The gencode v7 catalog of human long noncoding RNAs: Analysis of their gene structure, evolution, and expression. Genome Res 2012, 22, 1775–1789. [Google Scholar]
- Pang, K.C.; Frith, M.C.; Mattick, J.S. Rapid evolution of noncoding RNAs: Lack of conservation does not mean lack of function. Trends Genet 2006, 22, 1–5. [Google Scholar]
- Watts, J.M.; Dang, K.K.; Gorelick, R.J.; Leonard, C.W.; Bess, J.W., Jr.; Swanstrom, R.; Burch, C.L.; Weeks, K.M. Architecture and secondary structure of an entire HIV-1 RNA genome. Nature 2009, 460, 711–716. [Google Scholar]
- Hennelly, S.P.; Novikova, I.V.; Sanbonmatsu, K.Y. The expression platform and the aptamer: Cooperativity between Mg2+ and ligand in the SAM-I riboswitch. Nucleic Acids Res 2013, 41, 1922–1935. [Google Scholar]
- Kenyon, J.C.; Tanner, S.J.; Legiewicz, M.; Phillip, P.S.; Rizvi, T.A.; Le Grice, S.F.; Lever, A.M. SHAPE analysis of the FIV leader RNA reveals a structural switch potentially controlling viral packaging and genome dimerization. Nucleic Acids Res 2011, 39, 6692–6704. [Google Scholar]
- Legiewicz, M.; Zolotukhin, A.S.; Pilkington, G.R.; Purzycka, K.J.; Mitchell, M.; Uranishi, H.; Bear, J.; Pavlakis, G.N.; Le Grice, S.F.; Felber, B.K. The RNA transport element of the murine MUSD retrotransposon requires long-range intramolecular interactions for function. J. Biol. Chem 2010, 285, 42097–42104. [Google Scholar]
- Novikova, I.V.; Hennelly, S.P.; Sanbonmatsu, K.Y. Structural architecture of the human long non-coding RNA, steroid receptor RNA activator. Nucleic Acids Res 2012, 40, 5034–5051. [Google Scholar]
- Ghosh, S.K.; Patton, J.R.; Spanjaard, R.A. A small RNA derived from RNA coactivator SRA blocks steroid receptor signaling via inhibition of PUS1p-mediated pseudouridylation of SRA: Evidence of a novel RNA binding domain in the N-terminus of steroid receptors. Biochemistry 2012, 51, 8163–8172. [Google Scholar]
- Colley, S.M.; Leedman, P.J. Sra and its binding partners: An expanding role for RNA-binding coregulators in nuclear receptor-mediated gene regulation. Crit. Rev. Biochem. Mol. Biol 2009, 44, 25–33. [Google Scholar]
- Smit, S.; Yarus, M.; Knight, R. Natural selection is not required to explain universal compositional patterns in rRNA secondary structure categories. RNA 2006, 12, 1–14. [Google Scholar]
- Zuker, M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res 2003, 31, 3406–3415. [Google Scholar]
- Reuter, J.S.; Mathews, D.H. RNAstructure: Software for RNA secondary structure prediction and analysis. BMC Bioinforma 2010, 11, 129. [Google Scholar]
- Hofacker, I.L. Vienna RNA secondary structure server. Nucleic Acids Res 2003, 31, 3429–3431. [Google Scholar]
- Schroeder, S.J. Advances in RNA structure prediction from sequence: New tools for generating hypotheses about viral RNA structure-function relationships. J. Virol 2009, 83, 6326–6334. [Google Scholar]
- Zuker, M.; Jacobson, A.B. “Well-determined” regions in RNA secondary structure prediction: Analysis of small subunit ribosomal RNA. Nucleic Acids Res 1995, 23, 2791–2798. [Google Scholar]
- Bernhart, S.H.; Hofacker, I.L.; Will, S.; Gruber, A.R.; Stadler, P.F. RNAalifold: Improved consensus structure prediction for RNA alignments. BMC Bioinforma 2008, 9, 474. [Google Scholar]
- Wilm, A.; Linnenbrink, K.; Steger, G. Construct: Improved construction of RNA consensus structures. BMC Bioinforma 2008, 9, 219. [Google Scholar]
- Yao, Z.; Weinberg, Z.; Ruzzo, W.L. CMfinder—A covariance model based RNA motif finding algorithm. Bioinformatics 2006, 22, 445–452. [Google Scholar]
- Deigan, K.E.; Li, T.W.; Mathews, D.H.; Weeks, K.M. Accurate SHAPE-directed RNA structure determination. Proc. Natl. Acad. Sci. USA 2009, 106, 97–102. [Google Scholar]
- Ouyang, Z.; Snyder, M.P.; Chang, H.Y. Seqfold: Genome-scale reconstruction of RNA secondary structure integrating high-throughput sequencing data. Genome Res 2013, 23, 377–387. [Google Scholar]
- Duan, S.; Mathews, D.H.; Turner, D.H. Interpreting oligonucleotide microarray data to determine RNA secondary structure: Application to the 3′-end of Bombyx mori R2 RNA. Biochemistry 2006, 45, 9819–9832. [Google Scholar]
- Mathews, D.H.; Moss, W.H.; Turner, D.H. Folding and finding RNA secondary structure. Cold Spring Harbor Perspect. Biol 2010, 12, a003665. [Google Scholar]
- Puton, T.; Kozlowski, L.P.; Rother, K.M.; Bujnicki, J.M. CompaRNA: A server for continuous benchmarking of automated methods for RNA secondary structure prediction. Nucleic Acids Res 2013, 41, 4307–4323. [Google Scholar]
- Harris, M.E.; Christian, E.L. RNA crosslinking methods. Method. Enzymol 2009, 468, 127–146. [Google Scholar]
- Das, R.; Kudaravalli, M.; Jonikas, M.; Laederach, A.; Fong, R.; Schwans, J.P.; Baker, D.; Piccirilli, J.A.; Altman, R.B.; Herschlag, D. Structural inference of native and partially folded RNA by high-throughput contact mapping. Proc. Natl. Acad. Sci. USA 2008, 105, 4144–4149. [Google Scholar]
- Tullius, T.D.; Greenbaum, J.A. Mapping nucleic acid structure by hydroxyl radical cleavage. Curr. Opin. Chem. Biol 2005, 9, 127–134. [Google Scholar]
- Ban, N.; Nissen, P.; Hansen, J.; Moore, P.B.; Steitz, T.A. The complete atomic structure of the large ribosomal subunit at 2.4 a resolution. Science 2000, 289, 905–920. [Google Scholar]
- Ben-Shem, A.; Garreau de Loubresse, N.; Melnikov, S.; Jenner, L.; Yusupova, G.; Yusupov, M. The structure of the eukaryotic ribosome at 3.0 a resolution. Science 2011, 334, 1524–1529. [Google Scholar]
- Scott, L.G.; Hennig, M. RNA structure determination by NMR. Method. Mol. Biol 2008, 452, 29–61. [Google Scholar]
- Westhof, E.; Romby, P. The RNA structurome: High-throughput probing. Nat. Method 2010, 7, 965–967. [Google Scholar]
- Kertesz, M.; Wan, Y.; Mazor, E.; Rinn, J.L.; Nutter, R.C.; Chang, H.Y.; Segal, E. Genome-wide measurement of RNA secondary structure in yeast. Nature 2010, 467, 103–107. [Google Scholar]
- Wan, Y.; Qu, K.; Ouyang, Z.; Kertesz, M.; Li, J.; Tibshirani, R.; Makino, D.L.; Nutter, R.C.; Segal, E.; Chang, H.Y. Genome-wide measurement of RNA folding energies. Mol. Cell 2012, 48, 169–181. [Google Scholar]
- Underwood, J.G.; Uzilov, A.V.; Katzman, S.; Onodera, C.S.; Mainzer, J.E.; Mathews, D.H.; Lowe, T.M.; Salama, S.R.; Haussler, D. Fragseq: Transcriptome-wide RNA structure probing using high-throughput sequencing. Nat. Method 2010, 7, 995–1001. [Google Scholar]
- Lucks, J.B.; Mortimer, S.A.; Trapnell, C.; Luo, S.; Aviran, S.; Schroth, G.P.; Pachter, L.; Doudna, J.A.; Arkin, A.P. Multiplexed RNA structure characterization with selective 2′-hydroxyl acylation analyzed by primer extension sequencing (SHAPE-seq). Proc. Natl. Acad. Sci. USA 2011, 108, 11063–11068. [Google Scholar]
- Pan, T.; Sosnick, T. RNA folding during transcription. Annu. Rev. Biophys. Biomol. Struct 2006, 35, 161–175. [Google Scholar]
- Kilburn, D.; Roh, J.H.; Guo, L.; Briber, R.M.; Woodson, S.A. Molecular crowding stabilizes folded RNA structure by the excluded volume effect. J. Am. Chem. Soc 2010, 132, 8690–8696. [Google Scholar]
- Rajkowitsch, L.; Chen, D.; Stampfl, S.; Semrad, K.; Waldsich, C.; Mayer, O.; Jantsch, M.F.; Konrat, R.; Blasi, U.; Schroeder, R. RNA chaperones, RNA annealers and RNA helicases. RNA Biol 2007, 4, 118–130. [Google Scholar]
- Kortmann, J.; Narberhaus, F. Bacterial RNA thermometers: Molecular zippers and switches. Nat. Rev. Microbiol 2012, 10, 255–265. [Google Scholar]
- Nechooshtan, G.; Elgrably-Weiss, M.; Sheaffer, A.; Westhof, E.; Altuvia, S. A pH-responsive riboregulator. Genes Dev 2009, 23, 2650–2662. [Google Scholar]
- Charpentier, B.; Stutz, F.; Rosbash, M. A dynamic in vivo view of the HIV-I rev-RRE interaction. J. Mol. Biol 1997, 266, 950–962. [Google Scholar]
- Wells, S.E.; Hughes, J.M.; Igel, A.H.; Ares, M., Jr. Use of dimethyl sulfate to probe RNA structure in vivo. Method. Enzymol. 2000, 318, 479–493. [Google Scholar]
- Spitale, R.C.; Crisalli, P.; Flynn, R.A.; Torre, E.A.; Kool, E.T.; Chang, H.Y. RNA shape analysis in living cells. Nat. Chem. Biol 2013, 9, 18–20. [Google Scholar]



{kind=link}
{kind=link}
{kind=link}
© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Novikova, I.V.; Hennelly, S.P.; Sanbonmatsu, K.Y. Tackling Structures of Long Noncoding RNAs. Int. J. Mol. Sci. 2013, 14, 23672-23684. https://doi.org/10.3390/ijms141223672
Novikova IV, Hennelly SP, Sanbonmatsu KY. Tackling Structures of Long Noncoding RNAs. International Journal of Molecular Sciences. 2013; 14(12):23672-23684. https://doi.org/10.3390/ijms141223672
Chicago/Turabian StyleNovikova, Irina V., Scott P. Hennelly, and Karissa Y. Sanbonmatsu. 2013. "Tackling Structures of Long Noncoding RNAs" International Journal of Molecular Sciences 14, no. 12: 23672-23684. https://doi.org/10.3390/ijms141223672