Antitumor Mechanisms of Amino Acid Hydroxyurea Derivatives in the Metastatic Colon Cancer Model



Abstract
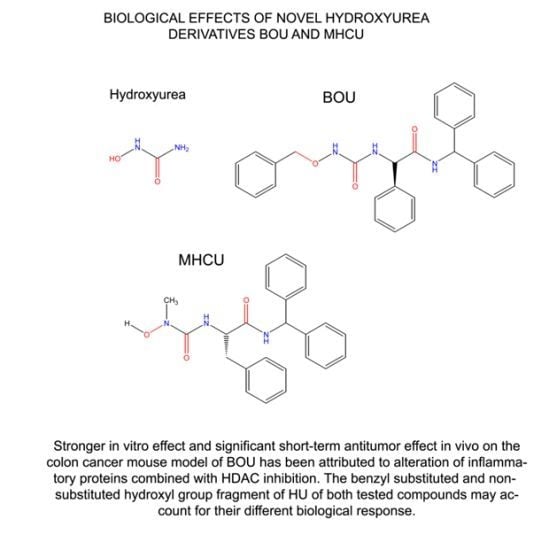
:
1. Introduction
2. Results and Discussion
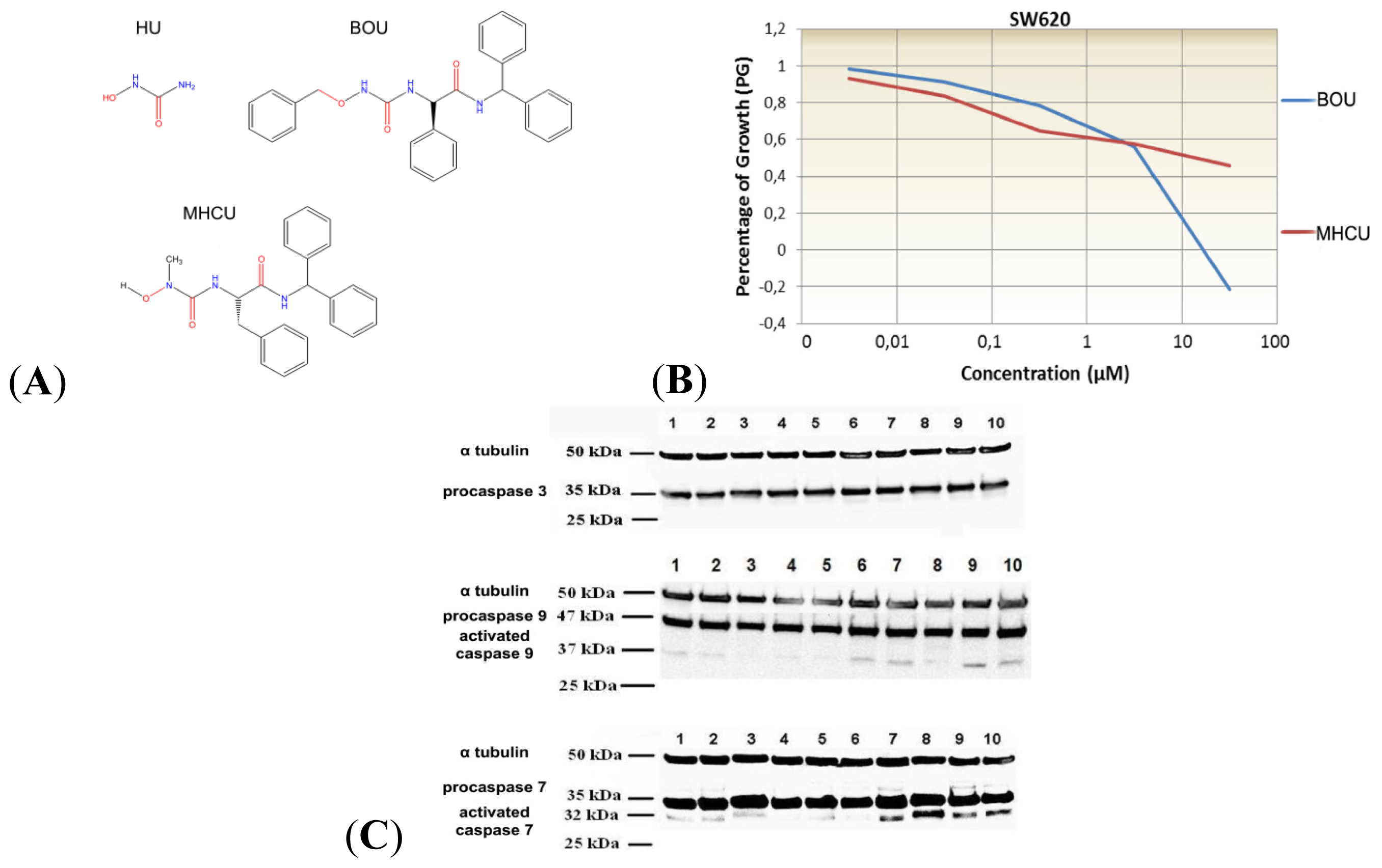
2.1. Amino Acid Hydroxyurea Derivatives BOU and MHCU Inhibit Proliferation of the Colon Cancer Cell Line SW620
2.2. Effects of BOU and MHCU on the Cell Cycle and Induction of Apoptosis
2.3. Protein Alterations in SW620 Cells Treated with BOU and MHCU
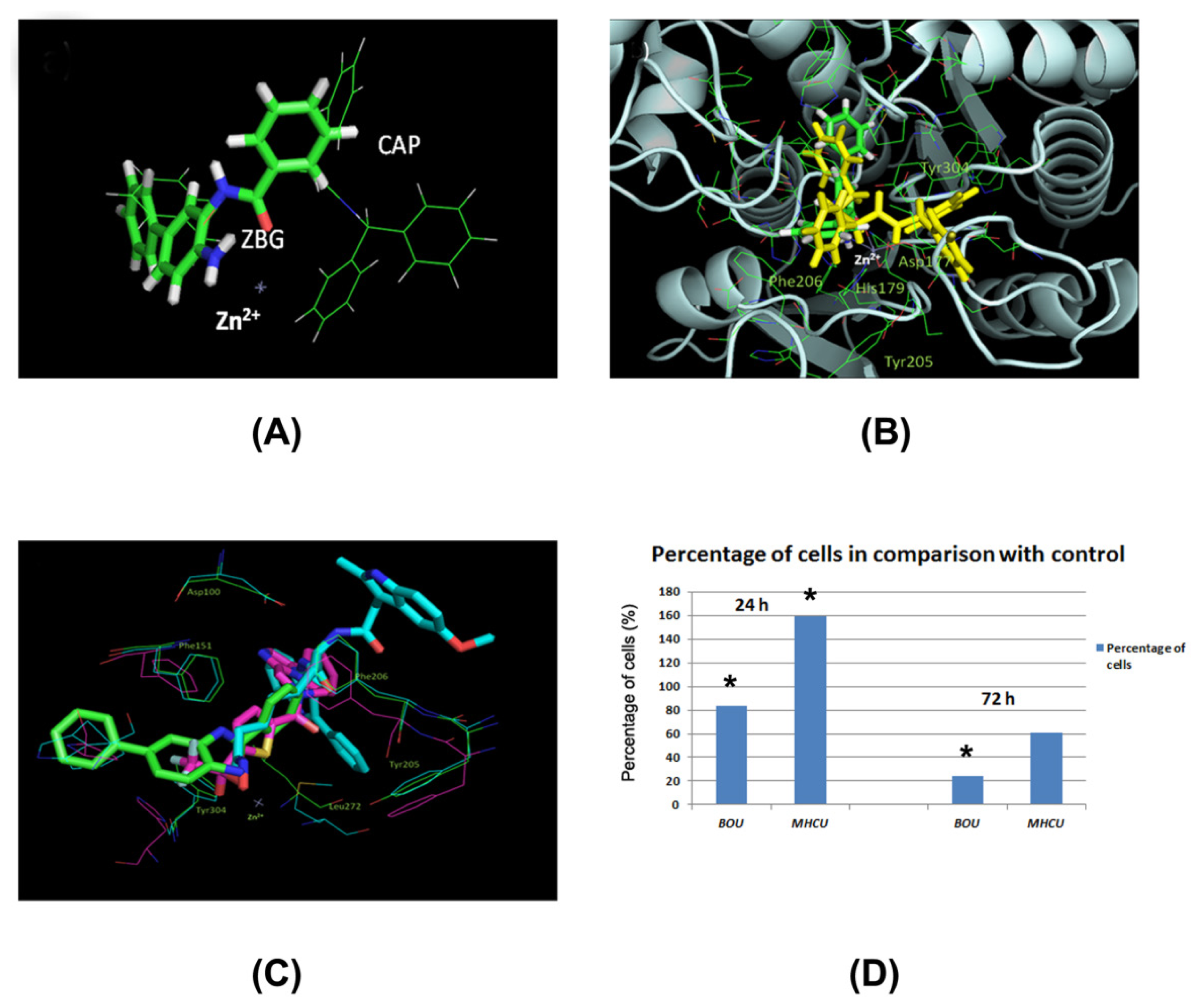
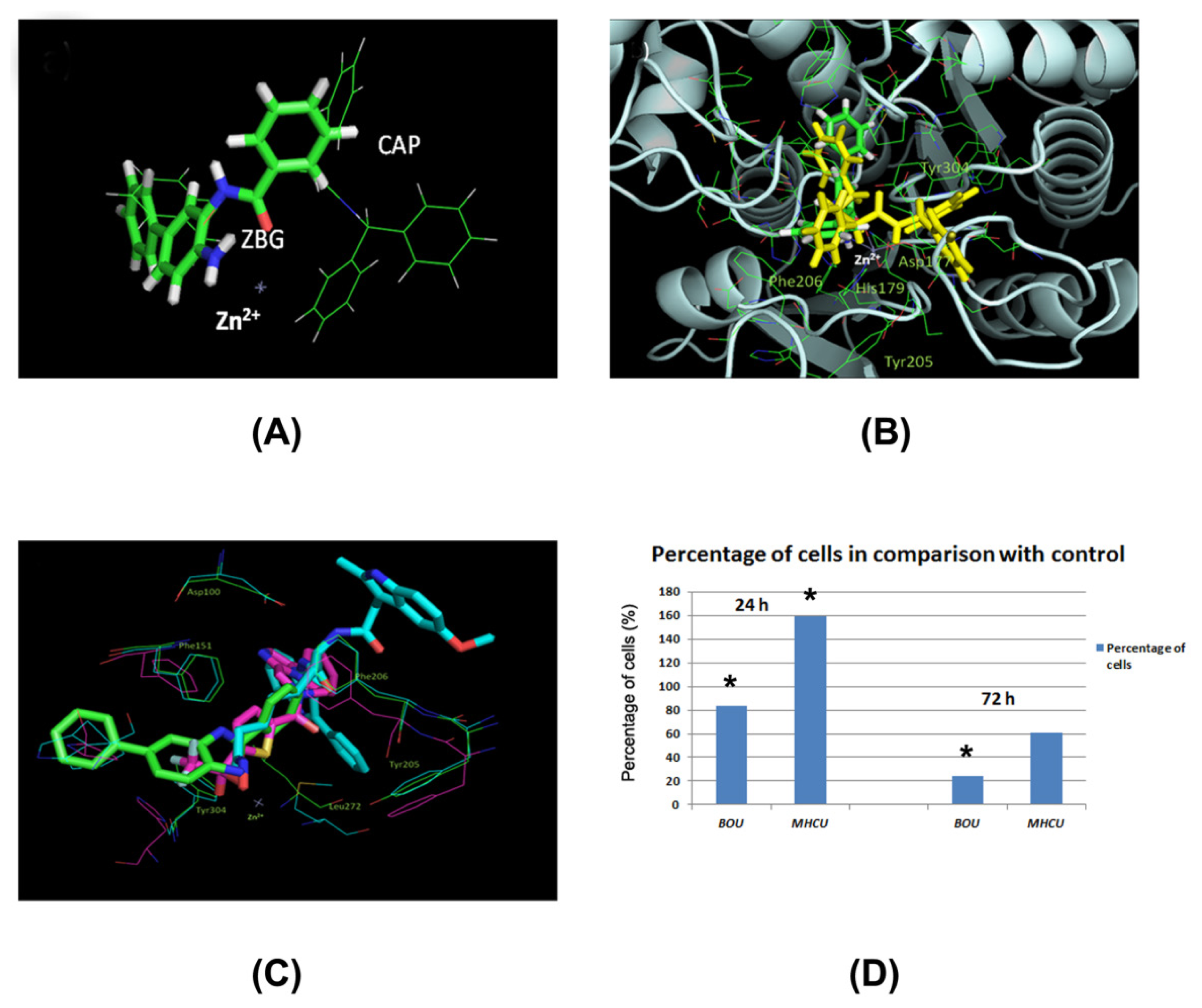
2.4. Docking of BOU and MHCU within HDAC Enzymes and the HDAC Inhibition Assay
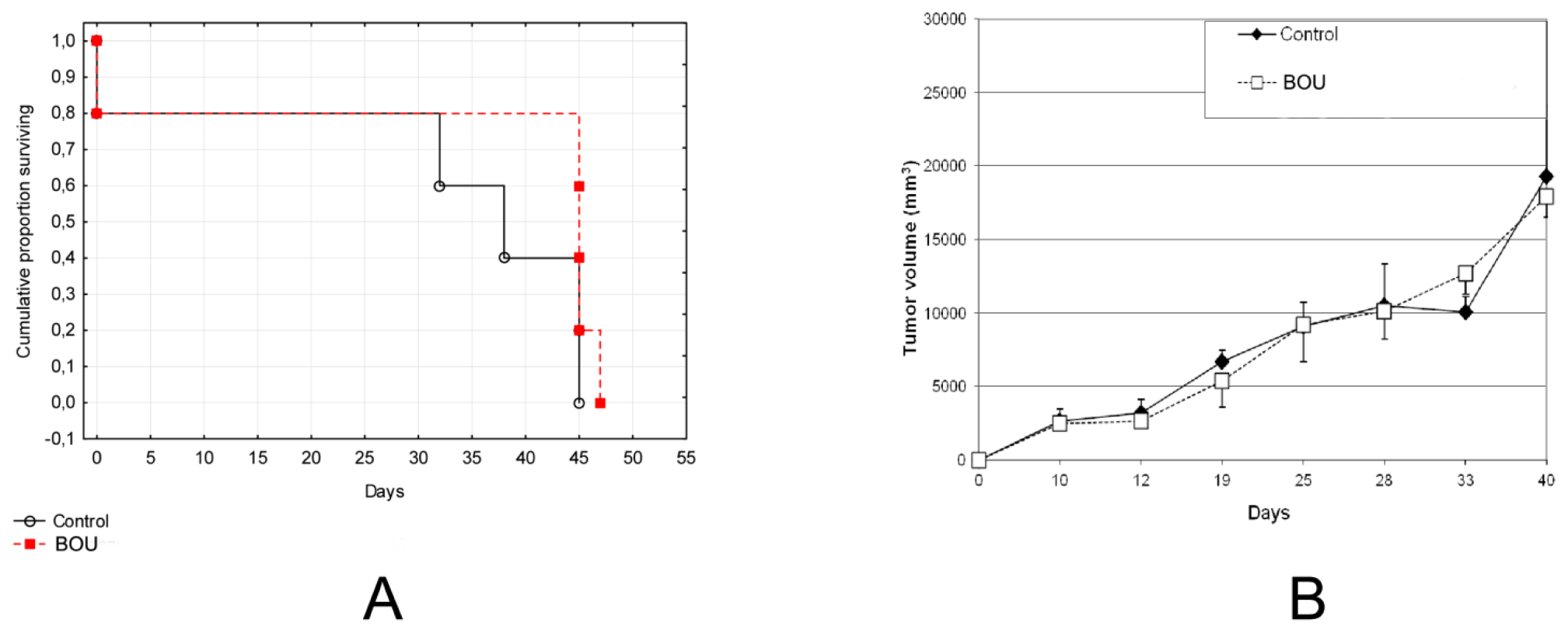
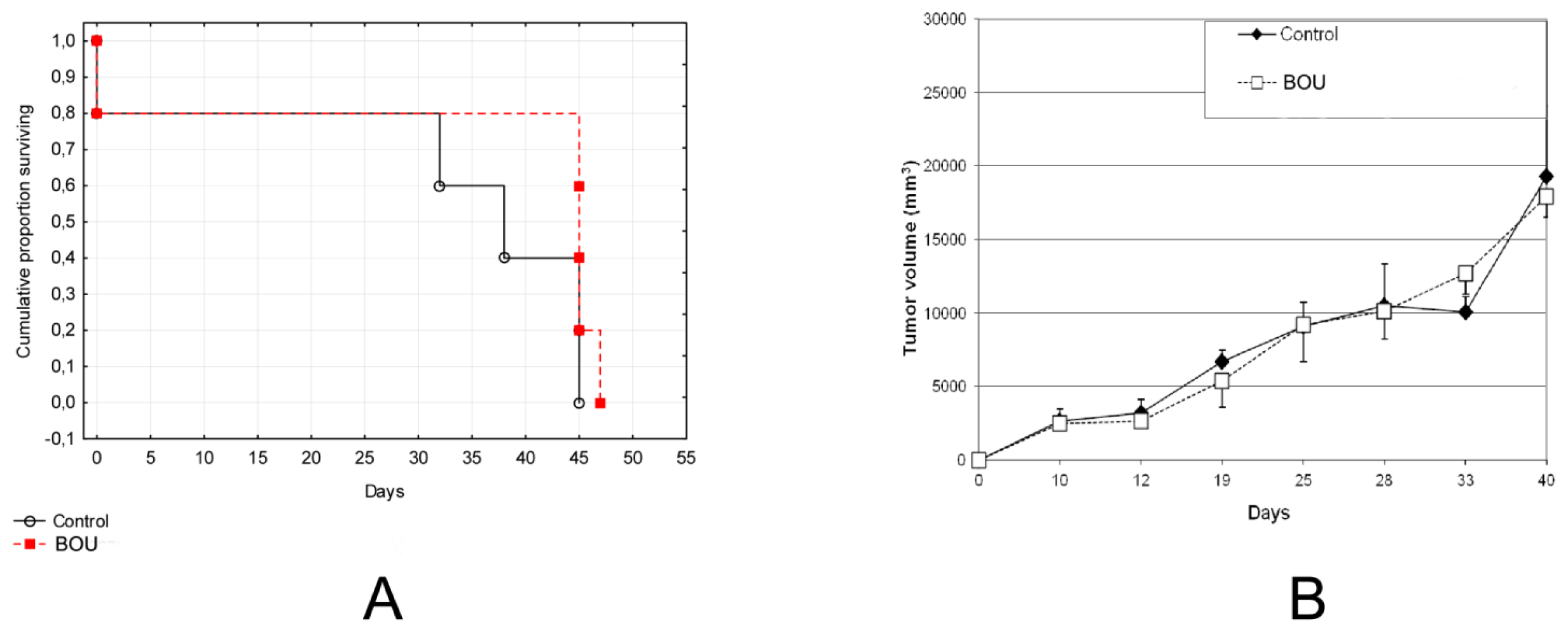
2.5. In Vivo Activity of BOU
3. Experimental Section
3.1. Tested Compounds
3.2. In Vitro Analyses
3.2.1. Cell Culturing
3.2.2. Cell Viability Assay
3.2.3. Cell Cycle Analyses
3.2.4. Annexin Test for Detection and Quantification of Apoptosis
3.2.5. Western Blot Analysis
3.2.6. Global Proteomic Profiling by 2D-Gel Electrophoresis and Mass Spectrometry
3.2.7. HDAC Colorimetric Activity Assay Kit for Screening HDAC Inhibitory Compounds
3.3. In Vivo Analyses
3.3.1. Animals
3.3.2. Tumor Cell Line and Culture Conditions
3.3.3. Production of a Tumor in the Muscle Tissue of the Right Hind Leg
3.3.4. Survival Analysis
3.3.5. Statistics
3.4. In Silico Analyses
4. Conclusions
Supplementary Information
ijms-14-23654-s001.pdfAcknowledgments
Conflicts of Interest
References
- Masi, G.; Vasile, E.; Loupakis, F.; Bursi, S.; Ricci, S.; Petrini, I.; Fontana, A.; Allegrini, G.; Falcone, A. Triplet combination of fluoropyrimidines, oxaliplatin, and irinotecan in the first-line treatment of metastatic colorectal cancer. Clin. Colorectal Cancer 2008, 7, 7–14. [Google Scholar]
- Banerjee, S.; Flores-Rozas, H. Monoclonal antibodies for targeted therapy in colorectal cancer. Cancer Biol. Ther 2010, 9, 563–571. [Google Scholar]
- Howlader, N.; Noone, A.M.; Krapcho, M.; Neyman, N.; Aminou, R.; Altekruse, S.F.; Kosary, C.L.; Ruhl, J.; Tatalovich, Z.; Cho, H.; et al. SEER Cancer Statistics Review, 1975–2009 (Vintage 2009 Populations). Available online: http://seer.cancer.gov/csr/1975_2009_pops09/ (accessed on 20 August 2012).
- Pavelić Kraljević, S.; Sedić, M.; Bosnjak, H.; Spaventi, S.; Pavelić, K. Metastasis: New perspectives on an old problem. Mol. Cancer 2011, 10, 10–22. [Google Scholar]
- Šaban, N.; Bujak, M. Hydroxyurea and hydroxamic acid derivatives as antitumor drugs. Cancer Chemoth. Pharm 2009, 64, 213–221. [Google Scholar]
- Perković, I.; Butula, I.; Zorc, B.; Hock, K.; Kraljević Pavelić, S.; Pavelić, K.; Clercq, E.D.; Balzarini, J.; Mintas, M. Novel lipophilic hydroxyurea derivatives: Synthesis, cytostatic and antiviral activity evaluations. Chem. Biol. Drug Des 2008, 71, 546–553. [Google Scholar]
- Opačić, N.; Barbarić, M.; Zorc, B.; Cetina, M.; Nagl, A.; Frković, D.; Kralj, M.; Pavelić, K.; Balzarini, J.; Andrei, G.; et al. The novel l- and d-amino acid derivatives of hydroxyurea and hydantoins: Synthesis, X-ray crystal structure study, and cytostatic and antiviral activity evaluations. J. Med. Chem 2005, 48, 475–482. [Google Scholar]
- Kajstura, M.; Halicka, H.D.; Pryjma, J.; Darzynkiewicz, Z. Discontinuous fragmentation of nuclear DNA during apoptosis revealed by discrete “sub-G1” peaks on DNA content histograms. Cytometry A 2007, 71, 125–131. [Google Scholar]
- Walczak, H.; Krammer, P.H. The CD95 (APO-1/Fas) and the TRAIL (APO-2L) apoptosis systems. Exp. Cell Res 2000, 256, 58–66. [Google Scholar]
- Fulda, S.; Debatin, K.M. Extrinsic versus intrinsic apoptosis pathways in anticancer chemotherapy. Oncogene 2006, 25, 4798–4811. [Google Scholar]
- Lamkanfi, M.; Kanneganti, T.D.; van Damme, P.; vanden Berghe, T.; Vanoverberghe, I.; Vandekerckhove, J.; Vandenabeele, P.; Gevaert, K.; Núñez, G. Targeted peptidecentric proteomics reveals caspase-7 as a substrate of the caspase-1 inflammasomes. Mol. Cell Proteomics 2008, 7, 2350–2363. [Google Scholar]
- Henriquez, M.; Armisén, R.; Stutzin, A.; Quest, A.F.G. Cell death by necrosis, a regulated way to go. Curr. Mol. Med 2008, 8, 187–206. [Google Scholar]
- Vakifahmetoglu, H.; Olsson, M.; Zhivotovsky, B. Death through a tragedy: Mitotic catastrophe. Cell Death Differ 2008, 15, 1153–1162. [Google Scholar]
- Masud, A.; Mohapatra, A.; Lakhani, S.A.; Ferrandino, A.; Hakem, R.; Flavell, R.A. Endoplasmic reticulum stress-induced death of mouse embryonic fibroblasts requires the intrinsic pathway of apoptosis. J. Biol. Chem 2007, 282, 14132–14139. [Google Scholar]
- Gaymes, T.J.; Padua, R.A.; Pla, M.; Orr, S.; Omidvar, N.; Chomienne, C.; Mufti, G.J.; Rassool, F.V. Histone deacetylase inhibitors (HDI) cause DNA damage in leukemia cells: A mechanism for leukemia-specific HDI-dependent apoptosis? Mol. Cancer Res 2006, 4, 563–573. [Google Scholar]
- Rosato, R.R.; Almenara, J.A.; Maggio, S.C.; Coe, S.; Atadja, P.; Dent, P.; Grant, S. Role of histone deacetylase inhibitor-induced reactive oxygen species and DNA damage in LAQ-824/fludarabine antileukemic interactions. Mol. Cancer Ther 2008, 7, 3285–3297. [Google Scholar]
- Sedić, M.; Poznić, M.; Gehrig, P.; Scott, M.; Schlapbach, R.; Hranjec, M.; Karminski-Zamola, G.; Pavelic, K.; Kraljevic Pavelic, S. Differential antiproliferative mechanisms of novel derivative of benzimidazo[1,2-alpha]quinoline in colon cancer cells depending on their p53 status. Mol. Cancer Ther 2008, 7, 2121–2132. [Google Scholar]
- Ushigome, M.; Ubagai, T.; Fukuda, H.; Tsuchiya, N.; Sugimura, T.; Takatsuka, J.; Nakagama, H. Up-regulation of hnRNP A1 gene in sporadic human colorectal cancers. Int. J. Oncol 2005, 26, 635–640. [Google Scholar]
- Jang, M.; Park, B.C.; Kang, S.; Chi, S.W.; Cho, S.; Chung, S.J.; Lee, S.C.; Bae, K.H.; Park, S.G. Far upstream element-binding protein-1, a novel caspase substrate, acts as a cross-talker between apoptosis and the c-myc oncogene. Oncogene 2009, 28, 1529–1536. [Google Scholar]
- Lindstrom, M.S.; Wallin, K.L. Prognostic Role of Proliferating Cell Nuclear Antigen (PCNA) in Cancer and other Diseases. In Proliferating Cell Nuclear Antigen (PCNA); Lee, H., Ed.; Research Signpost: Kerala, India, 2006; pp. 181–204. [Google Scholar]
- Zhang, Y.; Ye, Y.; Shen, D.; Jiang, K.; Zhang, H.; Sun, W.; Zhang, J.; Xu, F.; Cui, Z.; Wang, S. Identification of transgelin-2 as a biomarker of colorectal cancer by laser capture microdissection and quantitative proteome analysis. Cancer Sci 2010, 101, 523–529. [Google Scholar]
- Takenaka, Y.; Fukumori, T.; Raz, A. Galectin-3 and metastasis. Glycoconjugate J 2004, 19, 543–549. [Google Scholar]
- Sun, W.; Guo, C.; Meng, X.; Yu, Y.; Jin, Y.; Tong, D. Differential expression of PAI-RBP1, C1orf142, and COTL1 in non-small cell lung cancer cell lines with different tumor metastatic potential. J. Investig. Med 2012, 60, 689–694. [Google Scholar]
- Zhang, Z.; Huang, L.; Zhao, W.; Rigas, B. Annexin 1 induced by anti-inflammatory drugs binds to NF-κB and inhibits iIts activation: Anticancer effects in vitro and in vivo. Cancer Res. 2010, 70, 2379–2388. [Google Scholar]
- Lanaro, C.; Franco-Penteado, C.F.; Albuqueque, D.M.; Saad, S.T.; Conran, N.; Costa, F.F. Altered levels of cytokines and inflammatory mediators in plasma and leukocytes of sickle cell anemia patients and effects of hydroxyurea therapy. J. Leukoc. Biol 2009, 85, 235–242. [Google Scholar]
- Weichert, W.; Röske, A.; Niesporek, S.; Noske, A.; Buckendahl, A.C.; Dietel, M.; Gekeler, V.; Boehm, M.; Beckers, T.; Denkert, C. Class I histone deacetylase expression has independent prognostic impact in human colorectal cancer: Specific role of class I histone deacetylases in vitro and in vivo. Clin. Cancer Res. 2008, 14, 1669–1677. [Google Scholar]
- Parrish, D.A.; Zou, Z.; Allen, C.L.; Day, C.S.; King, S.B. A convenient method for the synthesis of N-hydroxyureas. Tetrahedron Lett 2005, 46, 8841–8843. [Google Scholar]
- Kouzarides, T. Histone acetylases and deacetylases in cell proliferation. Curr. Opin. Genet. Dev 1999, 9, 40–48. [Google Scholar]
- Finnin, M.S.; Donigian, J.R.; Cohen, A.; Richon, V.M.; Rifkind, R.A.; Marks, P.A.; Breslow, R.; Pavletich, N.P. Structures of a histone deacetylase homologue bound to the TSA and SAHA inhibitors. Nature 1999, 401, 188–193. [Google Scholar]
- Vanommeslaeghe, K.; de Proft, F.; Loverix, S.; Tourwe, D.; Geerlings, P. Theoretical study revealing the functioning of a novel combination of catalytic motifs in histone deacetylase. Bioorg. Med. Chem 2005, 13, 3987–3992. [Google Scholar]
- Bertrand, P. Inside HDAC with HDAC inhibitors. Eur. J. Med. Chem 2010, 45, 2095–2116. [Google Scholar]
- Hanessian, S.; Vinci, V.; Auzzas, L.; Marzi, M.; Giannini, G. Exploring alternative Zn-binding groups in the design of HDAC inhibitors: Squaric acid, N-hydroxyurea, and oxazoline analogues of SAHA. Bioorg. Med. Chem. Lett 2006, 16, 4784–4787. [Google Scholar]
- Bressi, J.C.; Jennings, A.J.; Skene, R.; Wu, Y.; Melkus, R.; de Jong, R.; O’Connell, S.; Grimshaw, C.E.; Navre, M.; Gangloff, A.R. Exploration of the HDAC2 foot pocket: Synthesis and SAR of substituted N-(2-aminophenyl)benzamides. Bioorg. Med. Chem. Lett 2010, 20, 3142–3145. [Google Scholar]
- Kang, M.R.; Kang, J.S.; Han, S.B.; Kim, J.H.; Kim, D.M.; Lee, K.; Lee, C.W.; Lee, K.H.; Lee, C.H.; Han, G.; et al. A novel delta-lactam-based histone deacetylase inhibitor, KBH-A42, induces cell cycle arrest and apoptosis in colon cancer cells. Biochem. Pharmacol 2009, 78, 486–494. [Google Scholar]
- Adcock, I.M. HDAC inhibitors as anti-inflammatory agents. Br. J. Pharmacol 2007, 150, 829–831. [Google Scholar]
- Iwata, K.; Tomita, K.; Sano, H.; Fujii, Y.; Yamasaki, A.; Shimizu, E. Trichostatin A, a histone deacetylase inhibitor, down-regulates interleukin-12 transcription in SV-40-transformed lung epithelial cells. Cell Immunol 2002, 218, 26–33. [Google Scholar]
- Leoni, F.; Fossati, G.; Lewis, E.C.; Lee, J.K.; Porro, G.; Pagani, P.; Modena, D.; Moras, M.L.; Pozzi, P.; Reznikov, L.L.; et al. The histone deacetylase inhibitor ITF2357 reduces production of pro-inflammatory cytokines in vitro and systemic inflammation in vivo. Mol. Med. 2005, 11, 1–15. [Google Scholar]
- Lin, H.Y.; Chen, C.S.; Lin, S.P.; Weng, J.R.; Chen, C.S. Targeting histone deacetylase in cancer therapy. Med. Res. Rev 2006, 26, 397–413. [Google Scholar]
- Ito, K.; Lim, S.; Caramori, G.; Borja, C.; Chung, K.F.; Adcock, I.M.; Barnes, P.J. A molecular mechanism of action of theophylline: Induction of histone deacetylase activity to decrease inflammatory gene expression. Proc. Natl. Acad. Sci. USA 2002, 99, 8921–8926. [Google Scholar]
- Ito, K.; Barnes, P.J.; Adcock, I.M. Glucocorticoid receptor recruitment of histone deacetylase 2 inhibits interleukin-1β-induced histone h4 acetylation on lysines 8 and 12. Mol. Cell Biol 2000, 20, 6891–6903. [Google Scholar]
- Boucher, P.D.; Ostruszka, L.J.; Murphy, P.J.; Shewach, D.S. Hydroxyurea significantly enhances tumor growth delay in vivo with herpes simplex virus thymidine kinase/ganciclovir gene therapy. Gene Ther 2002, 9, 1023–1030. [Google Scholar]
- Dethlefsen, L.A.; Sorensen, S.P.; Riley, R.M. Effects of double and multiple doses of hydroxyurea on mouse duodenum and mammary tumors. Cancer Res 1975, 35, 694–699. [Google Scholar]
- Lee, M.J.; Kim, Y.S.; Kummar, S.; Giaccone, G.; Trepel, J.B. Histone deacetylase inhibitors in cancer therapy. Curr. Opin. Oncol 2008, 20, 639–649. [Google Scholar]
- Joosten, L.A.; Leoni, F.; Meghji, S.; Mascagni, P. Inhibition of HDAC activity by ITF2357 ameliorates joint inflammation and prevents cartilage and bone destruction in experimental arthritis. Mol. Med 2011, 17, 391–396. [Google Scholar]
- Gazivoda, T.; Raić-Malić, S.; Krištafor, V.; Makuc, D.; Plavec, J.; Bratulić, S.; Kraljević-Pavelić, S.; Pavelić, K.; Naesens, L.; Andrei, G.; et al. Synthesis, cytostatic and anti-hiv evaluations of the new unsaturated acyclic c-5 pyrimidine nucleoside analogues. Bioorg. Med. Chem 2008, 16, 5624–5634. [Google Scholar]
- Drug Information Online. Available online: http://www.drugs.com/pro/hydroxyurea.html (accessed on 9 September 2013).
- Koedam, J.A.; Hoogerbrugge, C.M.; van Buul-Offers, S.C. Insulin-like growth factor binding proteins-3 and −5 form sodium dodecyl sulfate-stable multimers. Biochem. Biophys. Res. Commun 1997, 240, 707–714. [Google Scholar]
- Kollmar, O.; Rupertus, K.; Scheuer, C.; Nickels, R.M.; Haberl, G.C.; Tilton, B.; Menger, M.D.; Schilling, M.K. CXCR4 and CXCR7 regulate angiogenesis and CT26.WT tumor growth independent from SDF-1. Int. J. Cancer 2010, 126, 1302–1315. [Google Scholar]
- Yoshikawa, T.; Kokura, S.; Tainaka, K.; Naito, Y.; Kondo, M.A. Novel cancer therapy based on oxygen radicals. Cancer Res 1995, 55, 1617–1620. [Google Scholar]
- Oršolić, N.; Bašić, I. Immunomodulation by water-soluble derivative of propolis: A factor of antitumor reactivity. J. Ethnopharm 2003, 84, 265–273. [Google Scholar]
- Plowman, J.; Dykes, D.J.; Hollingshead, M.; Simpson-Herren, L.; Alley, M.C. Human Tumor Xenograft Models in NCI Drug Development. In Anticancer Drug Development Guide: Preclinical Screening, Clinical Trials, and Approval; Teicher, B., Ed.; Humana Press: Totowa, NJ, USA, 1995; p. 101. [Google Scholar]
- Kaplan, E.L.; Meier, P. Nonparametric estimation from incomplete observations. J. Am. Stat. Assoc 1958, 53, 457–465. [Google Scholar]
- Mantel, N.; Haenszel, W. Statistical aspects of the analysis of data from retrospective studies of disease. J. Natl. Cancer Inst 1959, 22, 719–748. [Google Scholar]
- The PubChem Project. Available online: http://pubchem.ncbi.nlm.nih.gov/ (accessed on 28 November 2013).
- ChEMBL Database. Available online: https://www.ebi.ac.uk/chembldb/ (accessed on 28 November 2013).
- Bernstein, F.C.; Koetzle, T.F.; Williams, G.J.B.; Meyer, E.F.; Brice, M.D.; Rodgers, J.R.; Kennard, O.; Shimanouchi, T.; Tasumi, M. The protein data bank: A computer-based archival file for macromolecular structures. J. Mol. Biol 1977, 112, 535–542. [Google Scholar]
- ArgusLab molecular modeling, graphics, and drug design program. Available online: http://www.arguslab.com/arguslab.com/ArgusLab.html (accessed on 28 November 2013).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Treatment | Cell percentage (%) ± standard deviation | ||||
|---|---|---|---|---|---|
| subG1 | G1 | S | G2/M | ||
| 24 h | Control | 5.4 ± 0.7 | 37.0 ± 0.5 | 45.6 ± 1.2 | 17.4 ± 0.9 |
| BOU, 1 μM | 5.3 ± 0.6 | 34.6 ± 0.7 * | 48.7 ± 1.4 | 16.7 ± 1.0 | |
| BOU, 50 μM | 36.6 ± 8.8 * | 36.5 ± 0.7 | 47.3 ±2.4 | 16.2 ± 2.0 | |
| MHCU, 1 μM | 5.0 ± 0.7 | 35.9 ± 0.7 | 48.5 ± 0.9 | 15.5 ± 0.6 | |
| MHCU, 50 μM | 7.5 ± 0.3 * | 35.6 ± 1.7 | 47.6 ± 1.5 | 16.8 ± 0.5 | |
| 72 h | Control | 5.5 ± 2.0 | 56.4 ± 1.7 | 28.5 ± 1.1 | 15.1 ± 1.5 |
| BOU, 1 μM | 5.5 ± 2.0 | 57.2 ± 2.0 | 25.9 ± 1.3 | 15.4 ± 2.0 | |
| BOU, 50 μM | 13.3 ± 1.5 * | 53.7 ± 2.3 * | 34.2 ± 2.2 * | 12.1 ± 0.8 | |
| MHCU, 1 μM | 4.3 ± 0.5 | 57.1 ± 2.7 | 29.7 ± 2.2 | 13.2 ± 1.7 | |
| MHCU, 50 μM | 9.8 ± 0.8 * | 58.6 ± 1.6 | 24.5 ± 1.8 | 16.9 ± 1.1 | |
© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Šaban, N.; Stepanić, V.; Vučinić, S.; Horvatić, A.; Cindrić, M.; Perković, I.; Zorc, B.; Oršolić, N.; Mintas, M.; Pavelić, K.; et al. Antitumor Mechanisms of Amino Acid Hydroxyurea Derivatives in the Metastatic Colon Cancer Model. Int. J. Mol. Sci. 2013, 14, 23654-23671. https://doi.org/10.3390/ijms141223654
Šaban N, Stepanić V, Vučinić S, Horvatić A, Cindrić M, Perković I, Zorc B, Oršolić N, Mintas M, Pavelić K, et al. Antitumor Mechanisms of Amino Acid Hydroxyurea Derivatives in the Metastatic Colon Cancer Model. International Journal of Molecular Sciences. 2013; 14(12):23654-23671. https://doi.org/10.3390/ijms141223654
Chicago/Turabian StyleŠaban, Nina, Višnja Stepanić, Srđan Vučinić, Anita Horvatić, Mario Cindrić, Ivana Perković, Branka Zorc, Nada Oršolić, Mladen Mintas, Krešimir Pavelić, and et al. 2013. "Antitumor Mechanisms of Amino Acid Hydroxyurea Derivatives in the Metastatic Colon Cancer Model" International Journal of Molecular Sciences 14, no. 12: 23654-23671. https://doi.org/10.3390/ijms141223654