Mevalonate Kinase Deficiency and Neuroinflammation: Balance between Apoptosis and Pyroptosis

 , ,
, ,  ,
,
Abstract
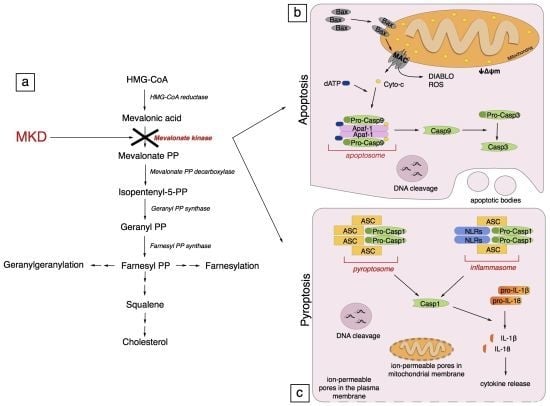
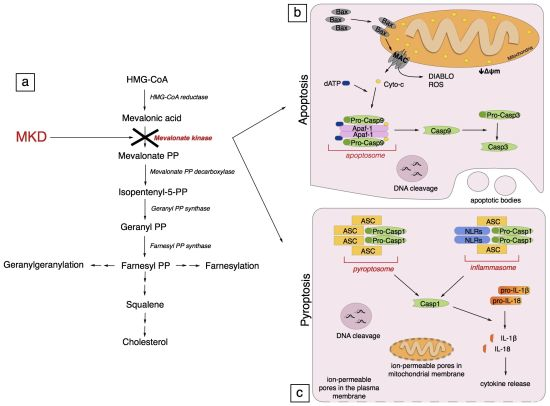
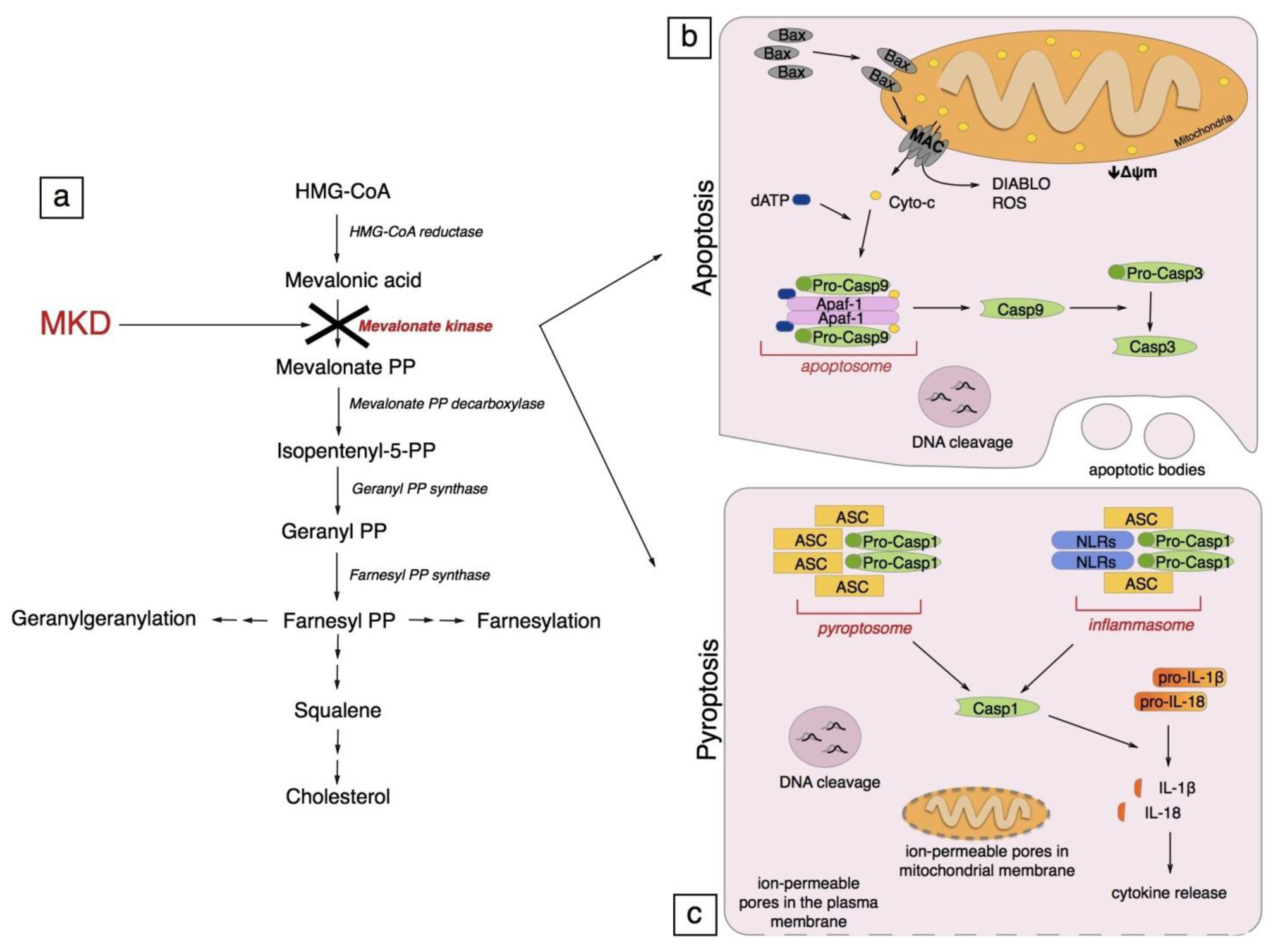
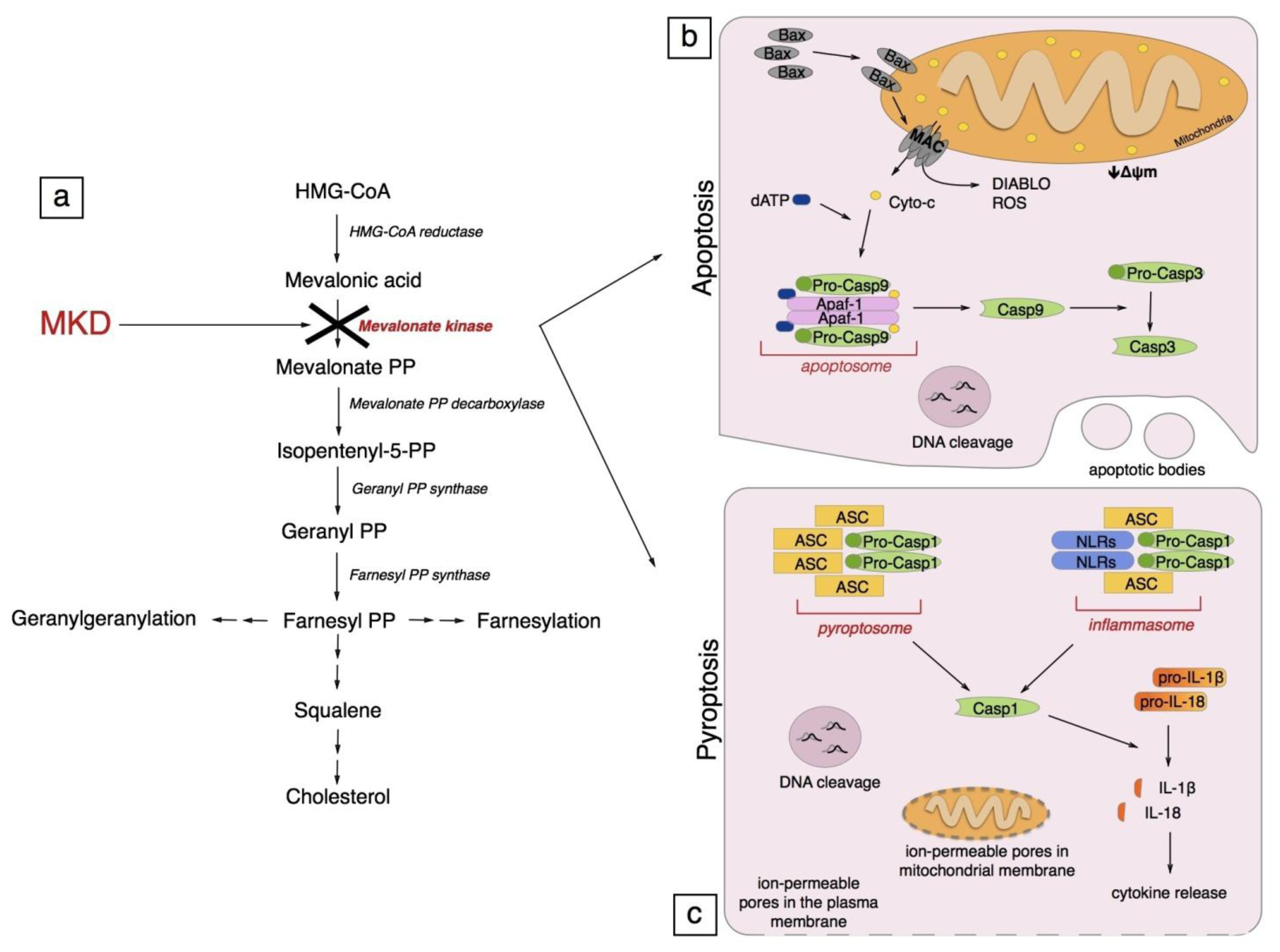
:
{kind=link}
{kind=link}
1. Introduction
2. Generalities on Mevalonate Pathway in the Central Nervous System
2.1. Neuroinflammation and the Mevalonate Pathway
2.2. Cholesterol Metabolism in the Central Nervous System
3. Models of MKD: In Vitro and in Vivo
4. Pathogenesis of Mevalonate Kinase Deficiency
5. Programmed Cell Death
5.1. Apoptosis
5.1.1. Generalities on Apoptosis
5.1.2. Apoptosis in Mevalonate Kinase Deficiency
5.2. Pyroptosis
5.2.1. Generalities on Pyroptosis
5.2.2. Pyroptosis in Mevalonate Kinase Deficiency
6. Oxidative Stress and Mitochondrial Damage through the Mevalonate Pathway
7. Concluding Remarks
Acknowledgments
Conflicts of Interest
References
- Goldstein, J.L.; Brown, M.S. Regulation of the mevalonate pathway. Nature 1990, 343, 425–430. [Google Scholar]
- Haas, D.; Hoffmann, G.F. Mevalonate kinase deficiency and autoinflammatory disorders. N. Engl. J. Med 2007, 356, 2671–2673. [Google Scholar]
- Drenth, J.P.; Haagsma, C.J.; van der Meer, J.W. Hyperimmunoglobulinemia D and periodic fever syndrome. The clinical spectrum in a series of 50 patients. International Hyper-IgD Study Group. Medicine 1994, 73, 133–144. [Google Scholar]
- Mandey, S.H.; Kuijk, L.M.; Frenkel, J.; Waterham, H.R. A role for geranylgeranylation in interleukin-1beta secretion. Arthritis Rheum 2006, 54, 3690–3695. [Google Scholar]
- Marcuzzi, A.; Pontillo, A.; de Leo, L.; Tommasini, A.; Decorti, G.; Not, T.; Ventura, A. Natural isoprenoids are able to reduce inflammation in a mouse model of mevalonate kinase deficiency. Pediatr. Res 2008, 64, 177–182. [Google Scholar]
- Frenkel, J.; Rijkers, G.T.; Mandey, S.H.; Buurman, S.W.; Houten, S.M.; Wanders, R.J.; Waterham, H.R.; Kuis, W. Lack of isoprenoid products raises ex vivo interleukin-1beta secretion in hyperimmunoglobulinemia D and periodic fever syndrome. Arthritis Rheum 2002, 46, 2794–2803. [Google Scholar]
- Marcuzzi, A.; de Leo, L.; Decorti, G.; Crovella, S.; Tommasini, A.; Pontillo, A. The farnesyltransferase inhibitors tipifarnib and lonafarnib inhibit cytokines secretion in a cellular model of mevalonate kinase deficiency. Pediatr. Res 2011, 70, 78–82. [Google Scholar]
- Marcuzzi, A.; Piscianz, E.; Girardelli, M.; Crovella, S.; Pontillo, A. Defect in mevalonate pathway induces pyroptosis in Raw 264.7 murine monocytes. Apoptosis 2011, 16, 882–888. [Google Scholar]
- Pontillo, A.; Paoluzzi, E.; Crovella, S. The inhibition of mevalonate pathway induces upregulation of NALP3 expression: New insight in the pathogenesis of mevalonate kinase deficiency. Eur. J. Hum. Genet 2010, 18, 844–847. [Google Scholar]
- Zhou, R.; Yazdi, A.S.; Menu, P.; Tschopp, J. A role for mitochondria in NLRP3 inflammasome activation. Nature 2011, 469, 221–225. [Google Scholar]
- Rubartelli, A.; Gattorno, M.; Netea, M.G.; Dinarello, C.A. Interplay between redox status and inflammasome activation. Trends Immunol 2011, 32, 559–566. [Google Scholar]
- Shimada, K.; Crother, T.R.; Karlin, J.; Dagvadorj, J.; Chiba, N.; Chen, S.; Ramanujan, V.K.; Wolf, A.J.; Vergnes, L.; Ojcius, D.M.; et al. Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity 2012, 36, 401–414. [Google Scholar]
- Cappellano, G.; Carecchio, M.; Fleetwood, T.; Magistrelli, L.; Cantello, R.; Dianzani, U.; Comi, C. Immunity and inflammation in neurodegenerative diseases. Am. J. Neurodegener. Dis 2013, 2, 89–107. [Google Scholar]
- Amor, S.; Puentes, F.; Baker, D.; van der Valk, P. Inflammation in neurodegenerative diseases. Immunology 2010, 129, 154–169. [Google Scholar]
- Abbott, N.J.; Patabendige, A.A.; Dolman, D.E.; Yusof, S.R.; Begley, D.J. Structure and function of the blood-brain barrier. Neurobiol. Dis 2010, 37, 13–25. [Google Scholar]
- Ransohoff, R.M.; Brown, M.A. Innate immunity in the central nervous system. J. Clin. Investig 2012, 122, 1164–1171. [Google Scholar]
- Erickson, M.A.; Banks, W.A. Blood-brain barrier dysfunction as a cause and consequence of Alzheimer’s disease. J. Cereb. Blood Flow Metab 2013, 33, 1500–1513. [Google Scholar]
- Galea, I.; Bechmann, I.; Perry, V.H. What is immune privilege (not)? Trends Immunol 2007, 28, 12–18. [Google Scholar]
- Dobolyi, A.; Vincze, C.; Pál, G.; Lovas, G. The neuroprotective functions of transforming growth factor beta proteins. Int. J. Mol. Sci 2012, 13, 8219–8258. [Google Scholar]
- Strle, K.; Zhou, J.H.; Shen, W.H.; Broussard, S.R.; Johnson, R.W.; Freund, G.G.; Dantzer, R.; Kelley, K.W. Interleukin-10 in the brain. Crit. Rev. Immunol 2001, 21, 427–449. [Google Scholar]
- Rubio-Perez, J.M.; Morillas-Ruiz, J.M. A review: Inflammatory process in Alzheimer’s disease, role of cytokines. Sci. World J 2012, 2012, 756357. [Google Scholar]
- Owens, T.; Bechmann, I.; Engelhardt, B. Perivascular spaces and the two steps to neuroinflammation. J. Neuropathol. Exp. Neurol 2008, 67, 1113–1121. [Google Scholar]
- Olah, M.; Amor, S.; Brouwer, N.; Vinet, J.; Eggen, B.; Biber, K.; Boddeke, H.W. Identification of a microglia phenotype supportive of remyelination. Glia 2012, 60, 306–321. [Google Scholar]
- Sierra, A.; Encinas, J.M.; Deudero, J.J.; Chancey, J.H.; Enikolopov, G.; Overstreet-Wadiche, L.S.; Tsirka, S.E.; Maletic-Savatic, M. Microglia shape adult hippocampal neurogenesis through apoptosis-coupled phagocytosis. Cell Stem Cell 2010, 7, 483–495. [Google Scholar]
- Vinet, J.; Weering, H.R.; Heinrich, A.; Kälin, R.E.; Wegner, A.; Brouwer, N.; Heppner, F.L.; Rooijen, N.; Boddeke, H.W.; Biber, K. Neuroprotective function for ramified microglia in hippocampal excitotoxicity. J. Neuroinflamm 2012, 9, 27. [Google Scholar] [Green Version]
- Shastri, A.; Bonifati, D.M.; Kishore, U. Innate immunity and neuroinflammation. Mediat. Inflamm 2013. [Google Scholar] [CrossRef]
- Kreutzberg, G.W. Microglia: A sensor for pathological events in the CNS. Trends Neurosci 1996, 19, 312–318. [Google Scholar]
- Olson, J.K.; Miller, S.D. Microglia initiate central nervous system innate and adaptive immune responses through multiple TLRs. J. Immunol 2004, 173, 3916–3924. [Google Scholar]
- Sofroniew, M.V.; Vinters, H.V. Astrocytes: Biology and pathology. Acta Neuropathol 2010, 119, 7–35. [Google Scholar]
- Pfrieger, F.W.; Ungerer, N. Cholesterol metabolism in neurons and astrocytes. Prog. Lipid Res 2011, 50, 357–371. [Google Scholar]
- Saito, M.; Benson, E.P.; Saito, M.; Rosenberg, A. Metabolism of cholesterol and triacylglycerol in cultured chick neuronal cells, glial cells, and fibroblasts: Accumulation of esterified cholesterol in serum-free culture. J. Neurosci. Res 1987, 18, 319–325. [Google Scholar]
- Pitas, R.E.; Boyles, J.K.; Lee, S.H.; Hui, D.; Weisgraber, K.H. Lipoproteins and their receptors in the central nervous system. Characterization of the lipoproteins in cerebrospinal fluid and identification of apolipoprotein B,E (LDL) receptors in the brain. J. Biol. Chem 1987, 262, 14352–14360. [Google Scholar]
- Mailman, T.; Hariharan, M.; Karten, B. Inhibition of neuronal cholesterol biosynthesis with lovastatin leads to impaired synaptic vesicle release even in the presence of lipoproteins or. geranylgeraniol. J. Neurochem 2011, 119, 1002–10015. [Google Scholar]
- Marcuzzi, A.; Tommasini, A.; Crovella, S.; Pontillo, A. Natural isoprenoids inhibit LPS-induced-production of cytokines and nitric oxide in aminobisphosphonate-treated monocytes. Int. Immunopharmacol 2010, 10, 639–642. [Google Scholar]
- Kuijk, L.M.; Mandey, S.H.; Schellens, I.; Waterham, H.R.; Rijkers, G.T.; Coffer, P.J.; Frenkel, J. Statin synergizes with LPS to induce IL-1beta release by THP-1 cells through activation of caspase-1. Mol. Immunol 2008, 45, 2158–2165. [Google Scholar]
- Yu, Z.; Funayama, H.; Deng, X.; Kuroishi, T.; Sasano, T.; Sugawara, S.; Endo, Y. Comparative appraisal of clodronate, aspirin and dexamethasone as agents reducing alendronate-induced inflammation in a murine model. Basic Clin. Pharmacol. Toxicol 2005, 97, 222–229. [Google Scholar]
- Marcuzzi, A.; Crovella, S.; Monasta, L.; Vecchi Brumatti, L.; Gattorno, M.; Frenkel, J. Mevalonate kinase deficiency: Disclosing the role of mevalonate pathway modulation in inflammation. Curr. Pharm. Des 2012, 18, 5746–5752. [Google Scholar]
- Hager, E.J.; Tse, H.M.; Piganelli, J.D.; Gupta, M.; Baetscher, M.; Tse, T.E.; Pappu, A.S.; Steiner, R.D.; Hoffmann, G.F.; Gibson, K.M. Deletion of a single mevalonate kinase (Mvk) allele yields a murine model of hyper-IgD syndrome. J. Inherit. Metab. Dis 2007, 30, 888–895. [Google Scholar]
- Ohashi, K.; Osuga, J.; Tozawa, R.; Kitamine, T.; Yagyu, H.; Sekiya, M.; Tomita, S.; Okazaki, H.; Tamura, Y.; Yahagi, N.; et al. Early embryonic lethality caused by targeted disruption of the 3-hydroxy-3-methylglutaryl-CoA reductase gene. J. Biol. Chem 2003, 278, 42936–42941. [Google Scholar]
- Tozawa, R.; Ishibashi, S.; Osuga, J.; Yagyu, H.; Oka, T.; Chen, Z.; Ohashi, K.; Perrey, S.; Shionoiri, F.; Yahagi, N.; et al. Embryonic lethality and defective neural tube closure in mice lacking squalene synthase. J. Biol. Chem 1999, 274, 30843–30848. [Google Scholar]
- Marcuzzi, A.; Tricarico, P.M.; Piscianz, E.; Kleiner, G.; Brumatti, L.V.; Crovella, S. Lovastatin induces apoptosis through the mitochondrial pathway in an undifferentiated SH-SY5Y neuroblastoma cell line. Cell Death Dis 2013, 4, e585. [Google Scholar]
- Van der Burgh, R.; Ter Haar, N.M.; Boes, M.L.; Frenkel, J. Mevalonate kinase deficiency, a metabolic autoinflammatory disease. Clin. Immunol 2013, 147, 197–206. [Google Scholar]
- Brough, D.; Rothwell, N.J. Caspase-1-dependent processing of pro-interleukin-1beta is cytosolic and precedes cell death. J. Cell. Sci 2007, 120, 772–781. [Google Scholar]
- Henneman, L.; Schneiders, M.S.; Turkenburg, M.; Waterham, H.R. Compromized geranylgeranylation of RhoA and Rac1 in mevalonate kinase deficiency. J. Inherit. Metab. Dis 2010, 33, 625–632. [Google Scholar]
- Misawa, T.; Takahama, M.; Kozaki, T.; Lee, H.; Zou, J.; Saitoh, T.; Akira, S. Microtubule-driven spatial arrangement of mitochondria promotes activation of the NLRP3 inflammasome. Nat. Immunol 2013, 14, 454–460. [Google Scholar]
- Lawlor, K.E.; Vince, J.E. Ambiguities in NLRP3 inflammasome regulation: Is there a role for mitochondria? Biochim. Biophys. Acta 2013, in press. [Google Scholar]
- Liao, Y.H.; Lin, Y.C.; Tsao, S.T.; Lin, Y.C.; Yang, A.J.; Huang, C.T.; Huang, K.C.; Lin, W.W. HMG-CoA reductase inhibitors activate caspase-1 in human monocytes depending on ATP release and P2X7 activation. J. Leukoc. Biol 2013, 93, 289–299. [Google Scholar]
- Gattorno, M.; Tassi, S.; Carta, S.; Delfino, L.; Ferlito, F.; Pelagatti, M.A.; D’Osualdo, A.; Buoncompagni, A.; Alpigiani, M.G.; Alessio, M.; et al. Pattern of interleukin-1beta secretion in response to lipopolysaccharide and ATP before and after interleukin-1 blockade in patients with CIAS1 mutations. Arthritis Rheum 2007, 56, 3138–3148. [Google Scholar]
- Marcuzzi, A.; Zanin, V.; Piscianz, E.; Tricarico, P.M.; Vuch, J.; Girardelli, M.; Monasta, L.; Bianco, A.M.; Crovella, S. Lovastatin-induced apoptosis is modulated by geranylgeraniol in a neuroblastoma cell line. Int. J. Dev. Neurosci 2012, 30, 451–456. [Google Scholar]
- Fuentes-Prior, P.; Salvesen, G.S. The protein structures that shape caspase activity, specificity, activation and inhibition. Biochem. J 2004, 384, 201–232. [Google Scholar]
- Galluzzi, L.; Vitale, I.; Abrams, J.M.; Alnemri, E.S.; Baehrecke, E.H.; Blagosklonny, M.V.; Dawson, T.M.; Dawson, V.L.; El-Deiry, W.S.; Fulda, S.; et al. Molecular definitions of cell death subroutines: Recommendations of the Nomenclature Committee on Cell Death 2012. Cell Death Differ 2012, 19, 107–120. [Google Scholar]
- Herrero-Martin, G.; López-Rivas, A. Statins activate a mitochondria-operated pathway of apoptosis in breast tumor cells by a mechanism regulated by ErbB2 and dependent on the prenylation of proteins. FEBS Lett 2008, 582, 2589–2594. [Google Scholar]
- Long, A.B.; Kaiser, W.J.; Mocarski, E.S.; Caspary, T. Apaf1 apoptotic function critically limits Sonic hedgehog signaling during craniofacial development. Cell Death Differ 2013, 20, 1510–1520. [Google Scholar]
- Martinez-Caballero, S.; Dejean, L.M.; Kinnally, M.S.; Oh, K.J.; Mannella, C.A.; Kinnally, K.W. Assembly of the mitochondrial apoptosis-induced channel, MAC. J. Biol. Chem 2009, 284, 12235–12245. [Google Scholar]
- Kroemer, G.; Galluzzi, L.; Brenner, C. Mitochondrial membrane permeabilization in cell death. Physiol. Rev 2007, 87, 99–163. [Google Scholar]
- Ng, H.; Smith, D.J.; Nagley, P. Application of flow cytometry to determine differential redistribution of cytochrome c and Smac/DIABLO from mitochondria during cell death signaling. PLoS One 2012, 7, e42298. [Google Scholar]
- Simon, H.U.; Haj-Yehia, A.; Levi-Schaffer, F. Role of reactive oxygen species (ROS) in apoptosis induction. Apoptosis 2000, 5, 415–418. [Google Scholar]
- Schinzel, A.C.; Takeuchi, O.; Huang, Z.; Fisher, J.K.; Zhou, Z.; Rubens, J.; Hetz, C.; Danial, N.N.; Moskowitz, M.A.; Korsmeyer, S.J. Cyclophilin D is a component of mitochondrial permeability transition and mediates neuronal cell death after focal cerebral ischemia. Proc. Natl. Acad. Sci. USA 2005, 102, 12005–12010. [Google Scholar]
- García-Escudero, V.; Martín-Maestro, P.; Perry, G.; Avila, J. Deconstructing mitochondrial dysfunction in Alzheimer disease. Oxid. Med. Cell. Longev 2013. [Google Scholar] [CrossRef]
- Hauser, D.N.; Hastings, T.G. Mitochondrial dysfunction and oxidative stress in Parkinson’s disease and monogenic parkinsonism. Neurobiol. Dis 2013, 51, 35–42. [Google Scholar]
- Johri, A.; Beal, M.F. Mitochondrial dysfunction in neurodegenerative diseases. J. Pharmacol. Exp. Ther 2012, 342, 619–630. [Google Scholar]
- Tricarico, P.M.; Kleiner, G.; Piscianz, E.; Zanin, V.; Monasta, L.; Crovella, S.; Marcuzzi, A. Temperature and drug treatments in mevalonate kinase deficiency: An ex vivo study. BioMed Res. Int 2013, 2013, 8. [Google Scholar]
- Ruiz Gomez, A.; Couce, M.L.; Garcia-Villoria, J.; Torres, A.; Baña Souto, A.; Yagüe, J.; Vilaseca, M.A.; Ribes, A.; Aróstegui, J.I. Clinical, genetic, and therapeutic diversity in 2 patients with severe mevalonate kinase deficiency. Pediatrics 2012, 129, 535–539. [Google Scholar]
- Bodar, E.J.; van der Hilst, J.C.; van Heerde, W.; van der Meer, J.W.; Drenth, J.P.; Simon, A. Defective apoptosis of peripheral-blood lymphocytes in hyper-IgD and periodic fever syndrome. Blood 2007, 109, 2416–2418. [Google Scholar]
- Fantuzzi, G.; Dinarello, C.A. Interleukin-18 and interleukin-1 beta: Two cytokine substrates for ICE (caspase-1). J. Clin. Immunol 1999, 19, 1–11. [Google Scholar]
- Bergsbaken, T.; Fink, S.L.; Cookson, B.T. Pyroptosis: Host cell death and inflammation. Nat. Rev. Microbiol 2009, 7, 99–109. [Google Scholar]
- Denes, A.; Lopez-Castejon, G.; Brough, D. Caspase-1: Is IL-1 just the tip of the ICEberg? Cell Death Dis 2012, 3, e338. [Google Scholar]
- Byrne, B.G.; Dubuisson, J.F.; Joshi, A.D.; Persson, J.J.; Swanson, M.S. Inflammasome components coordinate autophagy and pyroptosis as macrophage responses to infection. MBio 2013, 4. [Google Scholar] [CrossRef]
- Fernandes-Alnemri, T.; Wu, J.; Yu, J.W.; Datta, P.; Miller, B.; Jankowski, W.; Rosenberg, S.; Zhang, J.; Alnemri, E.S. The pyroptosome: A supramolecular assembly of ASC dimers mediating inflammatory cell death via caspase-1 activation. Cell Death Differ 2007, 14, 1590–1604. [Google Scholar]
- Fink, S.L.; Cookson, B.T. Caspase-1-dependent pore formation during pyroptosis leads to osmotic lysis of infected host macrophages. Cell Microbiol 2006, 8, 1812–1825. [Google Scholar]
- Case, C.L. Regulating caspase-1 during infection: Roles of NLRs, AIM2, and ASC. Yale J. Biol. Med 2011, 84, 333–343. [Google Scholar]
- Miao, E.A.; Leaf, I.A.; Treuting, P.M.; Mao, D.P.; Dors, M.; Sarkar, A.; Warren, S.E.; Wewers, M.D.; Aderem, A. Caspase-1-induced pyroptosis is an innate immune effector mechanism against intracellular bacteria. Nat. Immunol 2010, 11, 1136–1142. [Google Scholar]
- Suzuki, H.; Sozen, T.; Hasegawa, Y.; Chen, W.; Zhang, J.H. Caspase-1 inhibitor prevents neurogenic pulmonary edema after subarachnoid hemorrhage in mice. Stroke 2009, 40, 3872–3875. [Google Scholar]
- Loda, E.; Balabanov, R. Interferon regulatory factor 1 regulation of oligodendrocyte injury and inflammatory demyelination. Rev. Neurosci 2012, 23, 145–152. [Google Scholar]
- Allan, S.M.; Tyrrell, P.J.; Rothwell, N.J. Interleukin-1 and neuronal injury. Nat. Rev. Immunol 2005, 5, 629–640. [Google Scholar]
- Normand, S.; Massonnet, B.; Delwail, A.; Favot, L.; Cuisset, L.; Grateau, G.; Morel, F.; Silvain, C.; Lecron, J.C. Specific increase in caspase-1 activity and secretion of IL-1 family cytokines: A putative link between mevalonate kinase deficiency and inflammation. Eur. Cytokine Netw 2009, 20, 101–107. [Google Scholar]
- Brough, D.; Tyrrell, P.J.; Allan, S.M. Regulation of interleukin-1 in acute brain injury. Trends Pharmacol. Sci 2011, 32, 617–622. [Google Scholar]
- Simi, A.; Tsakiri, N.; Wang, P.; Rothwell, N.J. Interleukin-1 and inflammatory neurodegeneration. Biochem. Soc. Trans 2007, 35, 1122–1126. [Google Scholar]
- Bulua, A.C.; Simon, A.; Maddipati, R.; Pelletier, M.; Park, H.; Kim, K.Y.; Sack, M.N.; Kastner, D.L.; Siegel, R.M. Mitochondrial reactive oxygen species promote production of proinflammatory cytokines and are elevated in TNFR1-associated periodic syndrome (TRAPS). J. Exp. Med 2011, 208, 519–533. [Google Scholar]
- Moosmann, B.; Behl, C. Selenoproteins, cholesterol-lowering drugs, and the consequences: Revisiting of the mevalonate pathway. Trends Cardiovasc. Med 2004, 14, 273–281. [Google Scholar]
- Omenetti, A.; Carta, S.; Delfino, L.; Martini, A.; Gattorno, M.; Rubartelli, A. Increased NLRP3-dependent interleukin 1β secretion in patients with familial Mediterranean fever: Correlation with MEFV genotype. Ann. Rheum. Dis 2013, in press. [Google Scholar]
- Rodríguez-Martínez, E.; Martínez, F.; Espinosa-García, M.T.; Maldonado, P.; Rivas-Arancibia, S. Mitochondrial dysfunction in the hippocampus of rats caused by chronic oxidative stress. Neuroscience 2013, 252, 384–395. [Google Scholar]
- Celec, P.; Behuliak, M. The lack of non-steroid isoprenoids causes oxidative stress in patients with mevalonic aciduria. Med. Hypotheses 2008, 70, 938–940. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Tricarico, P.M.; Marcuzzi, A.; Piscianz, E.; Monasta, L.; Crovella, S.; Kleiner, G. Mevalonate Kinase Deficiency and Neuroinflammation: Balance between Apoptosis and Pyroptosis. Int. J. Mol. Sci. 2013, 14, 23274-23288. https://doi.org/10.3390/ijms141223274
Tricarico PM, Marcuzzi A, Piscianz E, Monasta L, Crovella S, Kleiner G. Mevalonate Kinase Deficiency and Neuroinflammation: Balance between Apoptosis and Pyroptosis. International Journal of Molecular Sciences. 2013; 14(12):23274-23288. https://doi.org/10.3390/ijms141223274
Chicago/Turabian StyleTricarico, Paola Maura, Annalisa Marcuzzi, Elisa Piscianz, Lorenzo Monasta, Sergio Crovella, and Giulio Kleiner. 2013. "Mevalonate Kinase Deficiency and Neuroinflammation: Balance between Apoptosis and Pyroptosis" International Journal of Molecular Sciences 14, no. 12: 23274-23288. https://doi.org/10.3390/ijms141223274