Systems Biology Approach to the Dissection of the Complexity of Regulatory Networks in the S. scrofa Cardiocirculatory System

Abstract
:1. Introduction
1.1. Supervised Approaches: Pathway Analysis
1.2. Unsupervised Approaches: Reverse Engineering Approach
1.3. The Missing Element: MicroRNAs (miRNAs)
1.4. Case Study: The Pig as a Model Organism
2. Results and Discussion
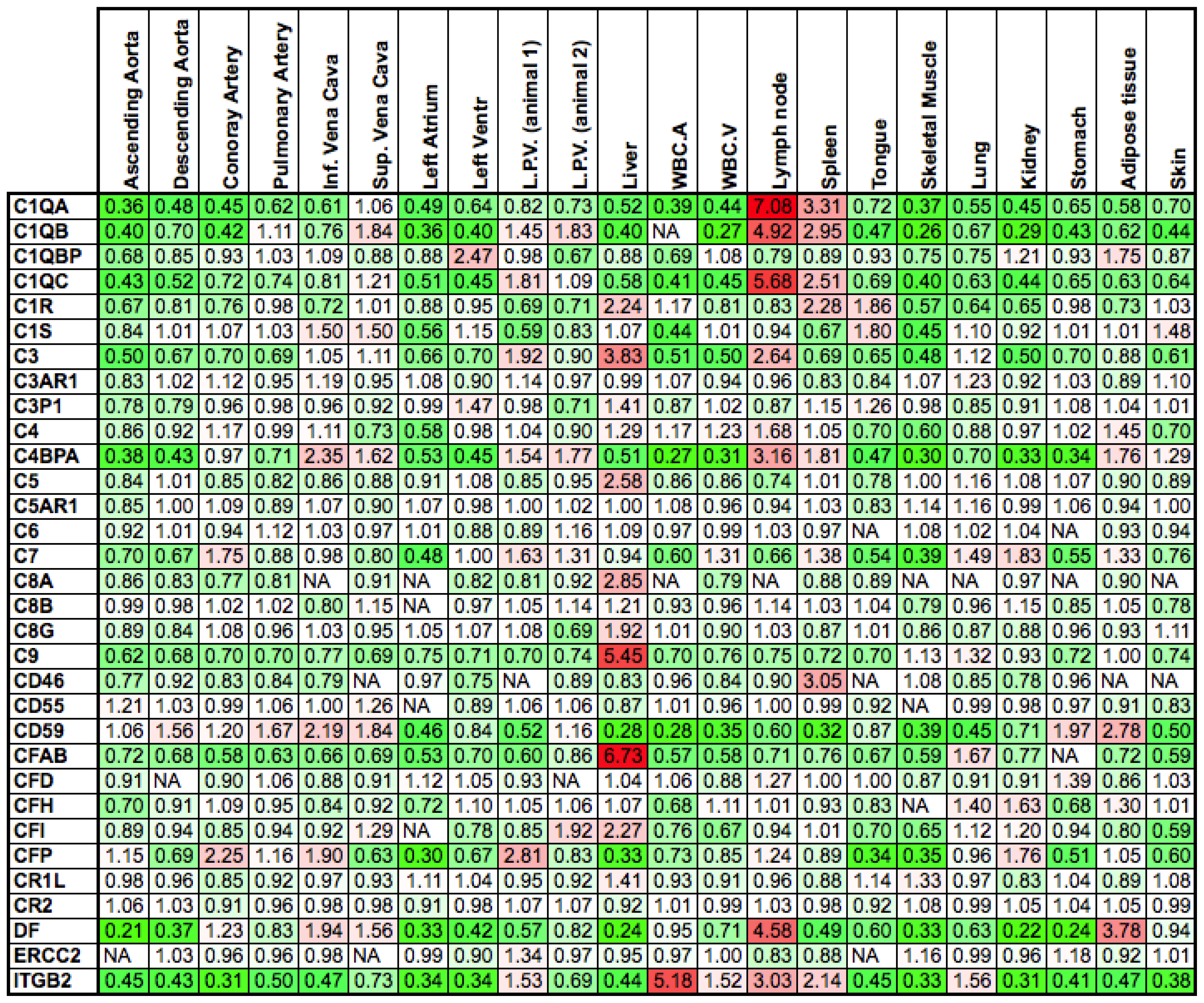
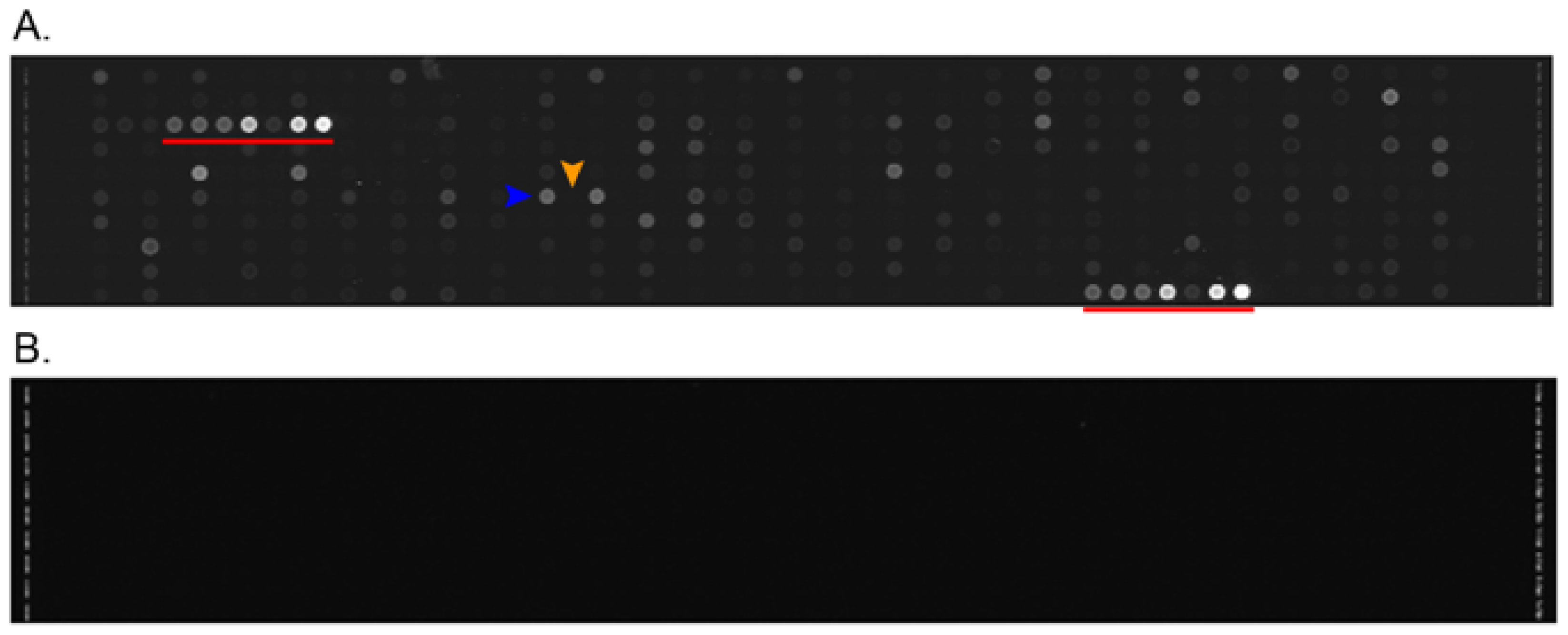
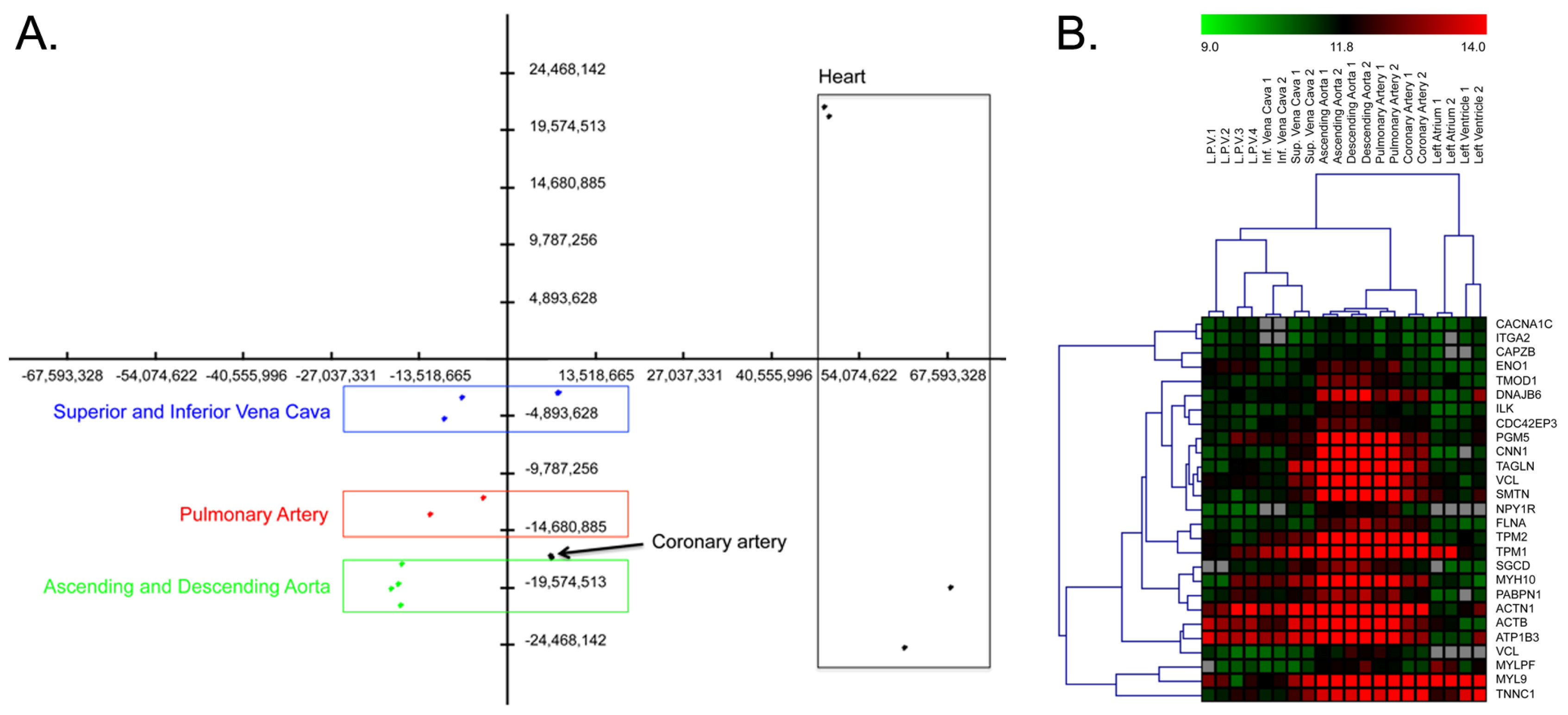
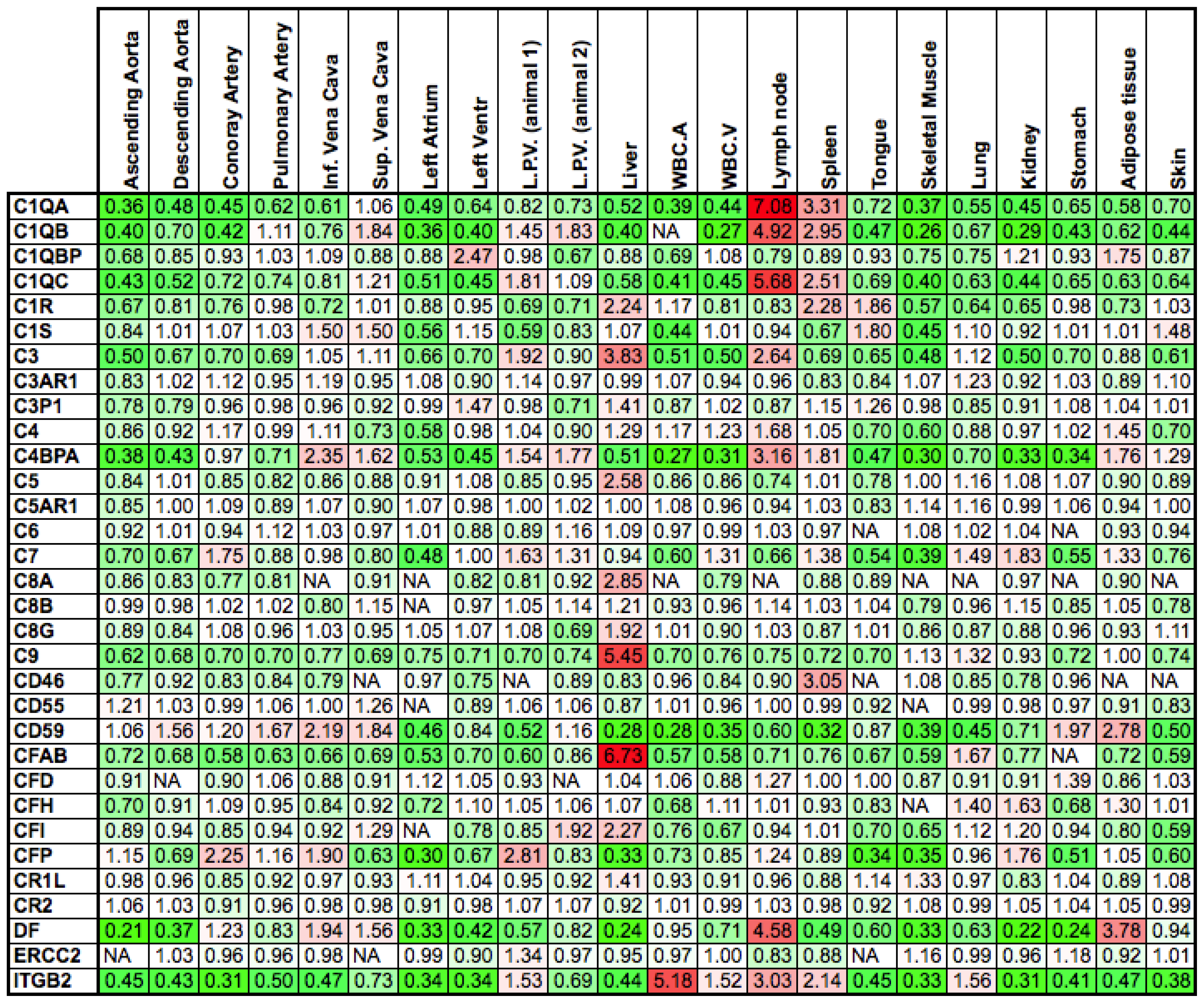
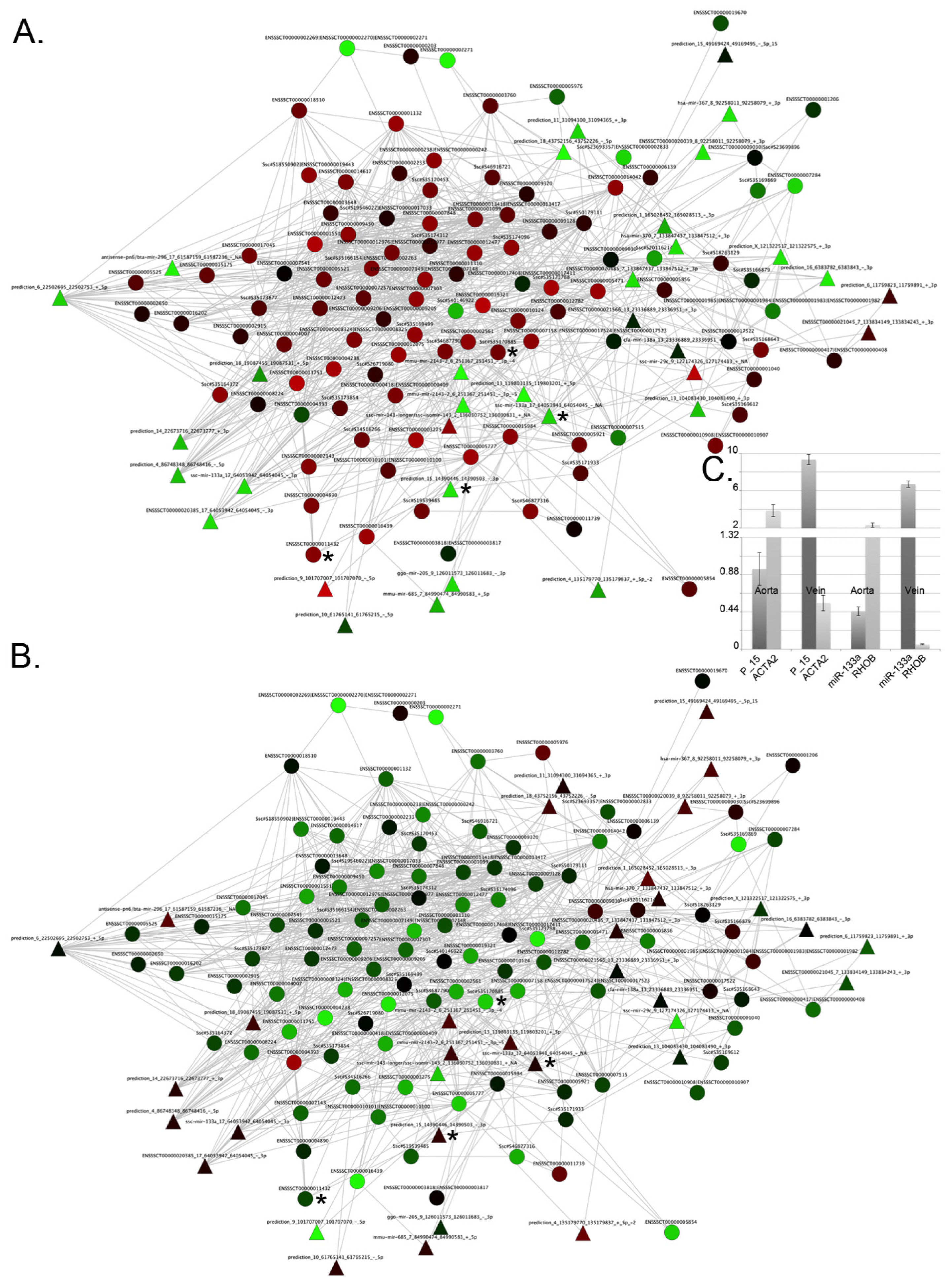
2.1. Differences between Arteries and Veins
2.2. Pathway Analysis
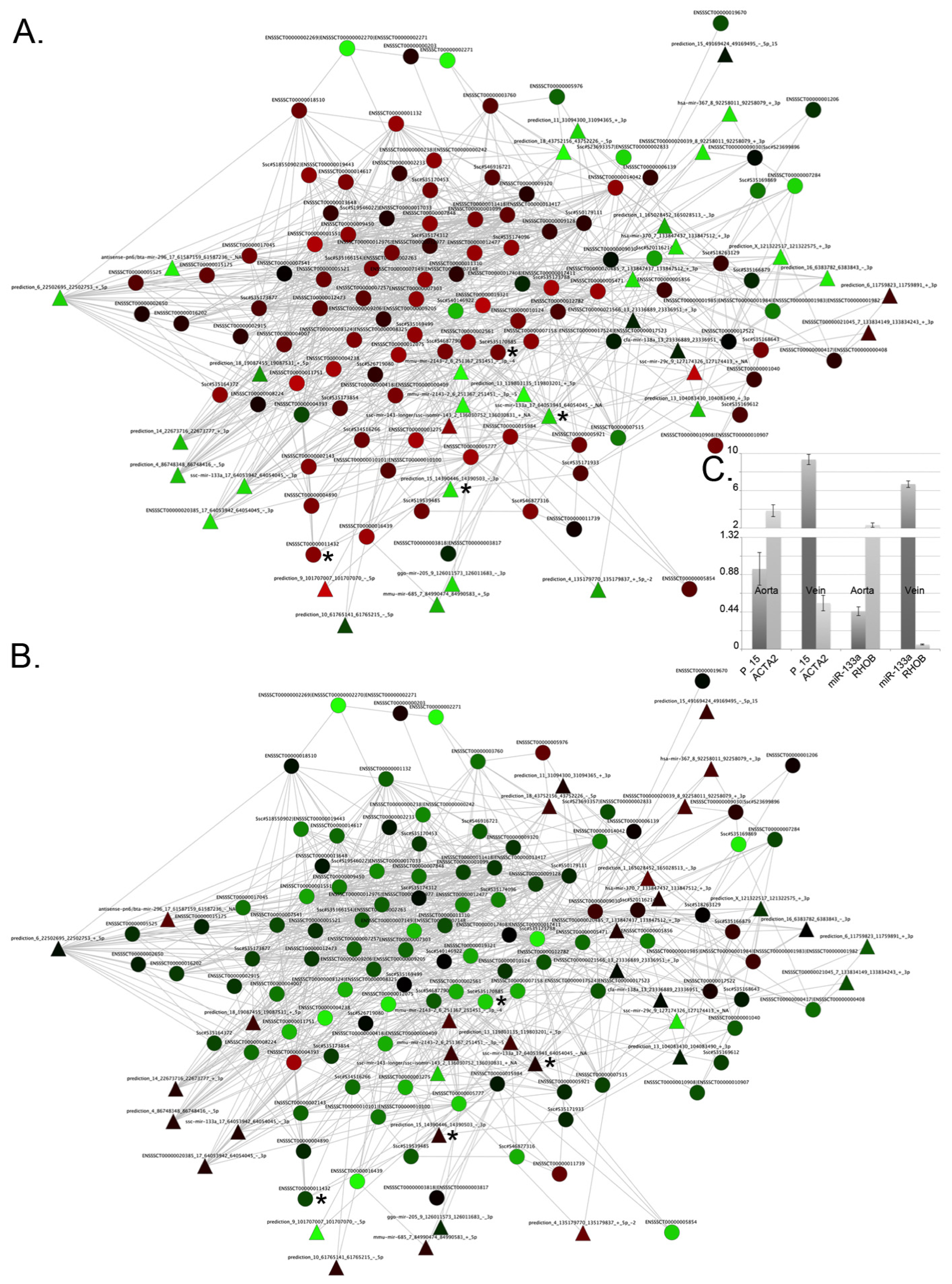
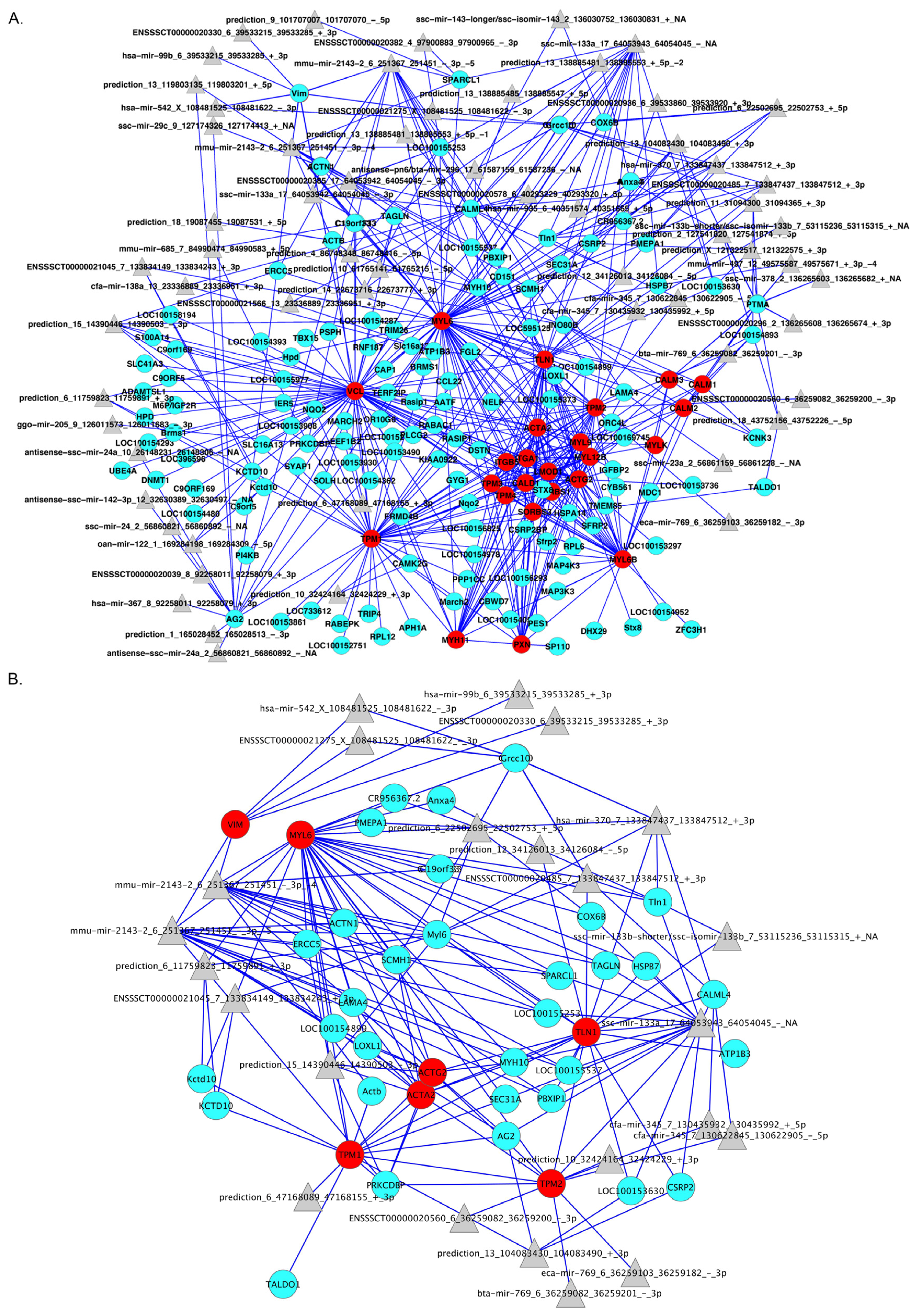
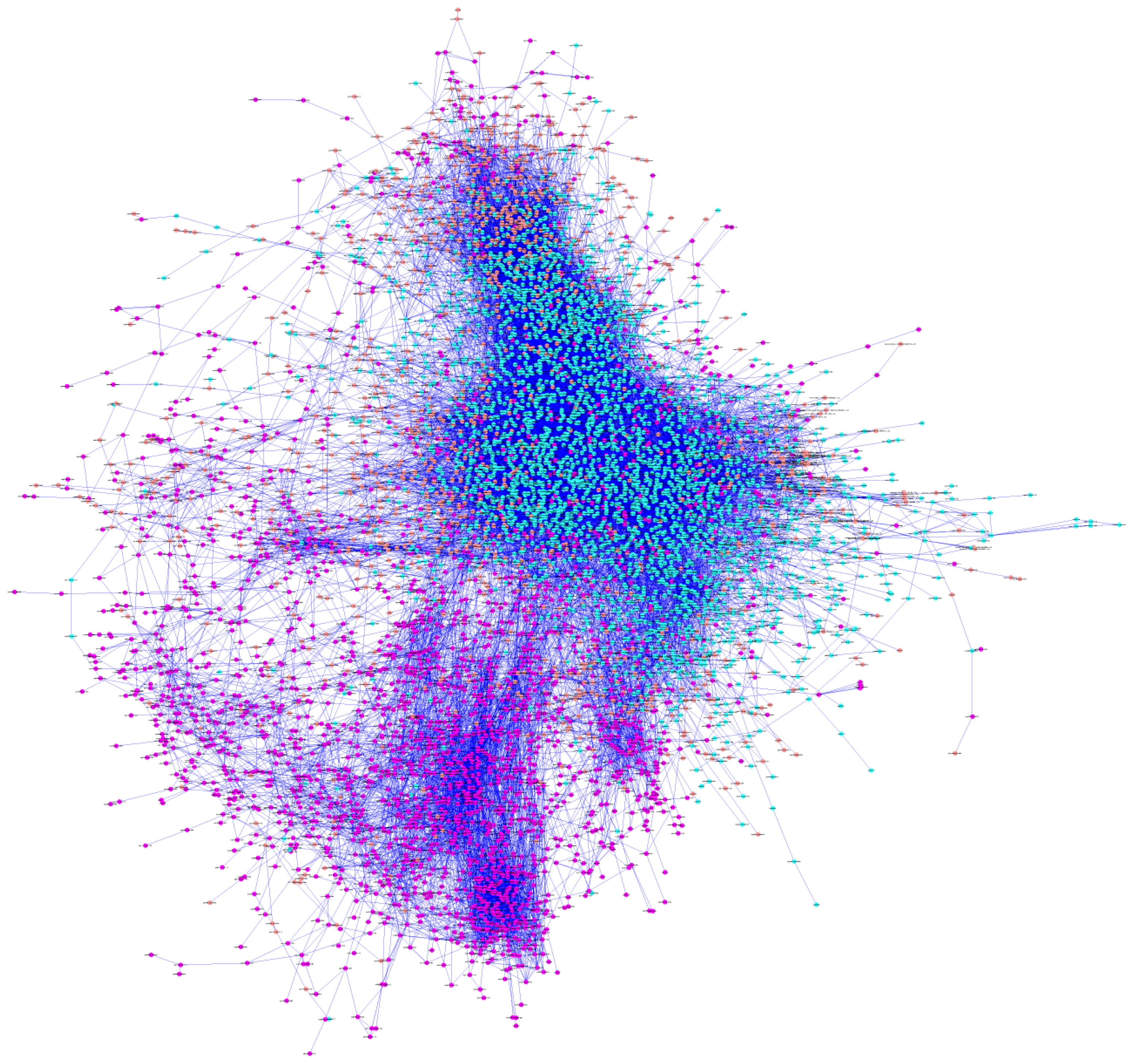
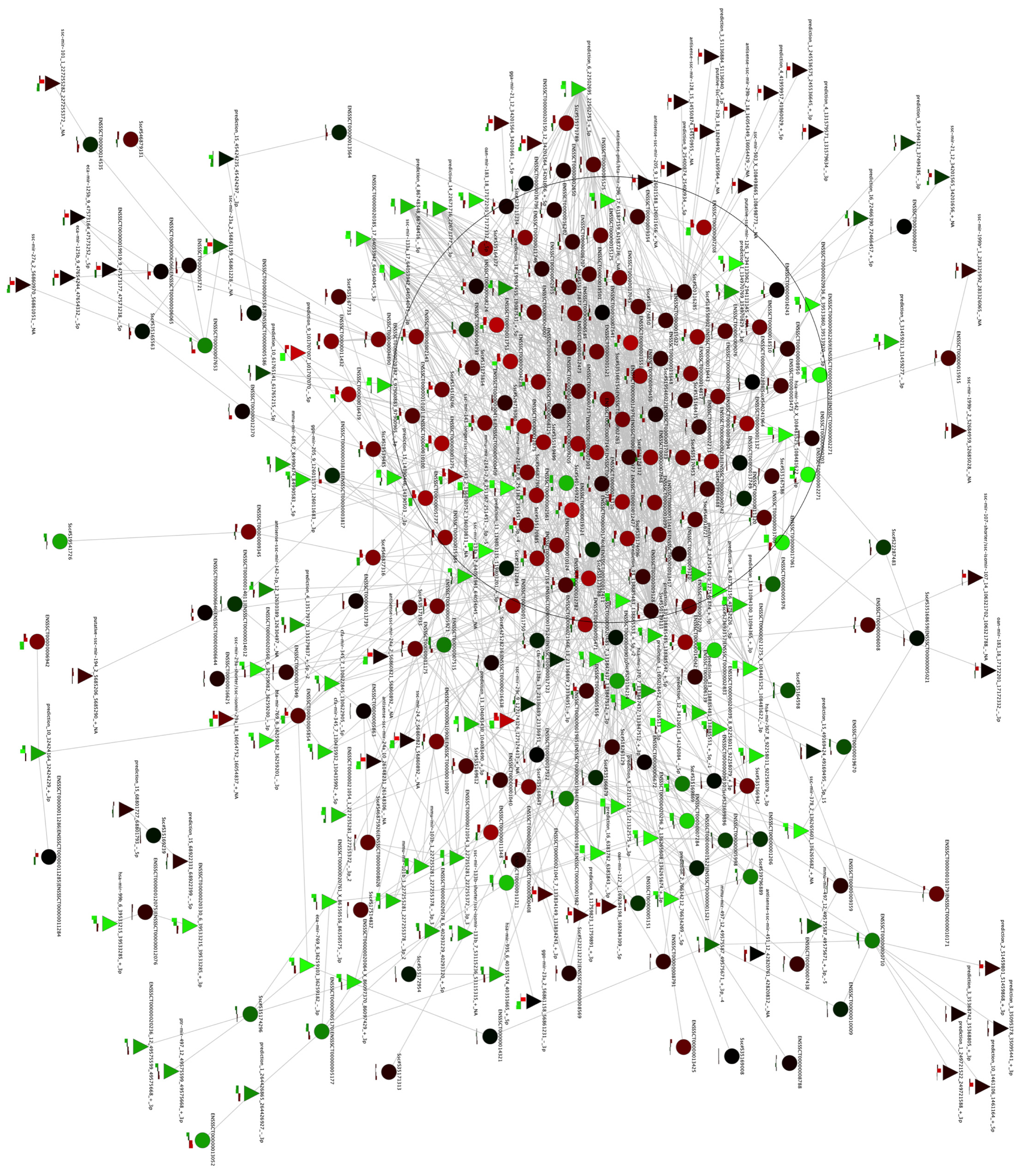
2.3. De Novo Pathway Reconstruction: Topological Parameters
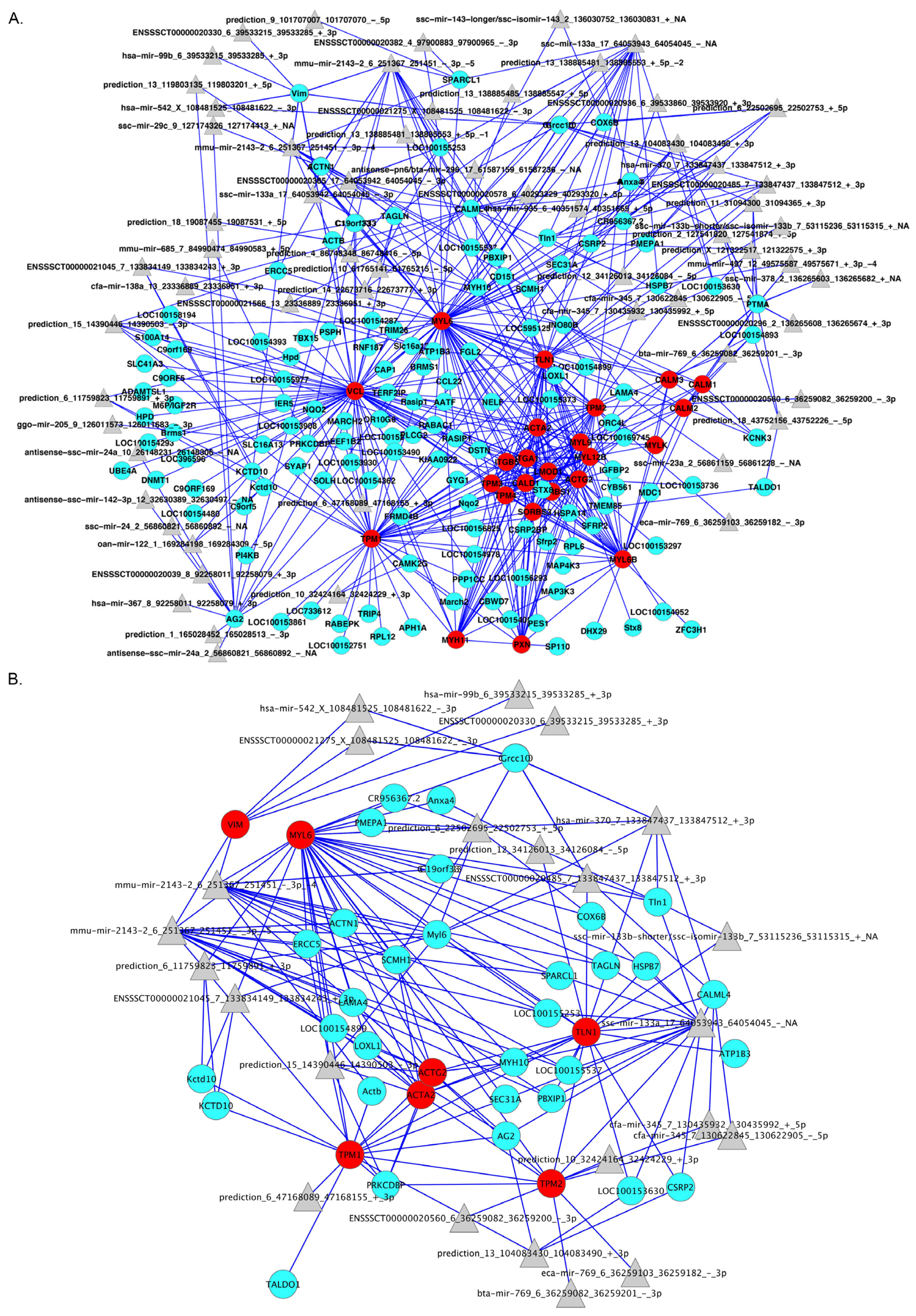
2.4. Integration of Supervised and Unsupervised Approaches
3. Experimental Section
3.1. Sample Preparation
3.2. Microarray Platforms
3.3. Microarray mRNA and miRNA Gene Expression and qRT-PCR
3.3.1. mRNA
- 6× SSPET (SSPE added with 0.05% of Tween-20) preheated at 42 °C for 5 min;
- 3× SSPET for 1 min at room temperature;
- 0.5× SSPET for 1 min at room temperature; and
- PBST for 1 min at room temperature.
3.3.2. miRNA
- 1 min at room temperature with 6× SSPET (SSPE containing 0.05% Tween-20);
- 1 min at room temperature with 3× SSPET;
- 1 min at room temperature with 2× PBS;
- 1 min at room temperature with 1× Buffer 2 (the buffer for the Klenow enzyme).
3.4. Data Analysis
4. Conclusions
Supplementary Information
ijms-14-23160-s001.pdf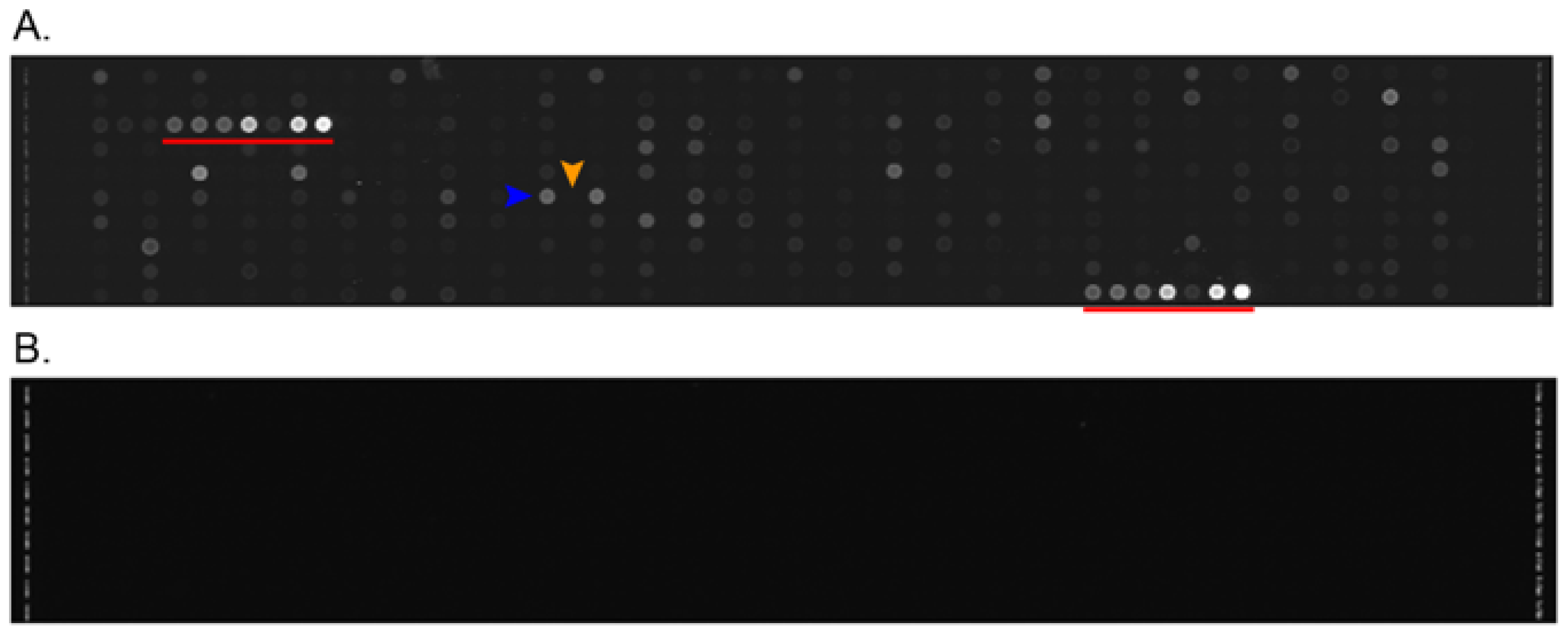




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pathway | Set size | NTk | Q-Value |
|---|---|---|---|
| Complement cascade | 18 | −5.29 | 0 |
| Arachidonic acid metabolism | 11 | −3.09 | 0.044912281 |
| Glycosaminoglycan metabolism | 54 | −3.09 | 0.044912281 |
| MPS I—Hurler syndrome | 54 | −3.09 | 0.044912281 |
| MPS II—Hunter syndrome | 54 | −3.09 | 0.044912281 |
| MPS IIIA—Sanfilippo syndrome A | 54 | −3.09 | 0.044912281 |
| MPS IIIB—Sanfilippo syndrome B | 54 | −3.09 | 0.044912281 |
| MPS IIIC—Sanfilippo syndrome C | 54 | −3.09 | 0.044912281 |
| MPS IIID—Sanfilippo syndrome D | 54 | −3.09 | 0.044912281 |
| MPS IV—Morquio syndrome A | 54 | −3.09 | 0.044912281 |
| MPS IV—Morquio syndrome B | 54 | −3.09 | 0.044912281 |
| Biological oxidations | 56 | −2.75 | 0.106666667 |
| Cell surface interactions at the vascular wall | 54 | −2.75 | 0.106666667 |
| Keratan sulfate/keratin metabolism | 20 | −2.46 | 0.205977011 |
| G α (12/13) signaling events | 35 | −2.37 | 0.24 |
| Antigen presentation: Folding, assembly and peptide loading of class I MHC | 11 | −2.33 | 0.250980392 |
| Golgi associated vesicle biogenesis | 29 | −2.29 | 0.247017544 |
| Glutathione conjugation | 10 | −2.26 | 0.249756098 |
| Phase II conjugation | 23 | −2.26 | 0.249756098 |
| EGFR interacts with phospholipase C-γ | 17 | 2.12 | 0.273710692 |
| Ca-dependent events | 14 | 2.14 | 0.262564103 |
| Calmodulin induced events | 14 | 2.14 | 0.262564103 |
| CaM pathway | 14 | 2.14 | 0.262564103 |
| Cell-extracellular matrix interactions | 15 | 2.2 | 0.254184397 |
| PLCG1 events in ERBB2 signaling | 18 | 2.23 | 0.252121212 |
| DARPP-32 events | 12 | 2.26 | 0.249756098 |
| DAG and IP3 signaling | 15 | 2.29 | 0.247017544 |
| PLC-γ1 signaling | 15 | 2.29 | 0.247017544 |
| Amyloids | 18 | 2.33 | 0.250980392 |
| Telomere Maintenance | 31 | 2.46 | 0.192688172 |
| RNA polymerase I promoter opening | 18 | 2.65 | 0.131282051 |
| Chromosome maintenance | 53 | 2.75 | 0.1024 |
| Meiotic synapsis | 24 | 2.88 | 0.077575758 |
| Deposition of new CENPA-containing nucleosomes at the centromere | 21 | 2.88 | 0.077575758 |
| Nucleosome assembly | 21 | 2.88 | 0.077575758 |
| Packaging of telomere ends | 12 | 3.09 | 0.044912281 |
| Striated muscle contraction | 21 | 4.76 | 0 |
| Smooth muscle contraction | 19 | 6.13 | 0 |
| Muscle contraction | 36 | 7.25 | 0 |
| Topological parameters | Heart network | Vessels network |
|---|---|---|
| Average clustering coefficient | 0.195 | 0.234 |
| Connected components | 237 | 86 |
| Avg. number of neighbors | 6.329 | 15.611 |
| Network radius | 1 | 1 |
| Network diameter | 36 | 16 |
| Network centralization | 0.020 | 0.036 |
| Network density | 0.002 | 0.005 |
| Network heterogeneity | 1.198 | 1.183 |
Acknowledgments
Conflicts of Interest
References
- Smyth, G.K. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat. Appl. Genet. Mol. Biol 2004, 3, 1–28. [Google Scholar]
- Tusher, V.G.; Tibshirani, R.; Chu, G. Significance analysis of microarrays applied to the ionizing radiation response. Proc. Natl. Acad. Sci. USA 2001, 98, 5116–5121. [Google Scholar]
- Shen, K.; Tseng, G.C. Meta-analysis for pathway enrichment analysis when combining multiple genomic studies. Bioinformatics 2010, 26, 1316–1323. [Google Scholar]
- Callegaro, A.; Basso, D.; Bicciato, S. A locally adaptive statistical procedure (LAP) to identify differentially expressed chromosomal regions. Bioinformatics 2006, 22, 2658–2666. [Google Scholar]
- Toedling, J.; Schmeier, S.; Heinig, M.; Georgi, B.; Roepcke, S. MACAT—Microarray chromosome analysis tool. Bioinformatics 2005, 21, 2112–2113. [Google Scholar]
- Turkheimer, F.E.; Roncaroli, F.; Hennuy, B.; Herens, C.; Nguyen, M.; Martin, D.; Evrard, A.; Bours, V.; Boniver, J.; Deprez, M. Chromosomal patterns of gene expression from microarray data: Methodology, validation and clinical relevance in gliomas. BMC Bioinform 2006, 7, 526. [Google Scholar]
- Barry, W.T.; Nobel, A.B.; Wright, F.A. Significance analysis of functional categories in gene expression studies: A structured permutation approach. Bioinformatics 2005, 21, 1943–1949. [Google Scholar]
- Goeman, J.J.; van de Geer, S.A.; de Kort, F.; van Houwelingen, H.C. A global test for groups of genes: Testing association with a clinical outcome. Bioinformatics 2004, 20, 93–99. [Google Scholar]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar]
- Tian, L.; Greenberg, S.A.; Kong, S.W.; Altschuler, J.; Kohane, I.S.; Park, P.J. Discovering statistically significant pathways in expression profiling studies. Proc. Natl. Acad. Sci. USA 2005, 102, 13544–13549. [Google Scholar]
- Levin, A.M.; Ghosh, D.; Cho, K.R.; Kardia, S.L. A model-based scan statistic for identifying extreme chromosomal regions of gene expression in human tumors. Bioinformatics 2005, 21, 2867–2874. [Google Scholar]
- Efron, B.; Tibshirani, R. On testing the significance of sets of genes. Ann. Appl. Stat 2007, 1, 107–129. [Google Scholar]
- Tarca, A.L.; Draghici, S.; Khatri, P.; Hassan, S.S.; Mittal, P.; Kim, J.S.; Kim, C.J.; Kusanovic, J.P.; Romero, R. A novel signaling pathway impact analysis. Bioinformatics 2009, 25, 75–82. [Google Scholar]
- Martini, P.; Sales, G.; Massa, M.S.; Chiogna, M.; Romualdi, C. Along signal paths: An empirical gene set approach exploiting pathway topology. Nucleic Acids Res 2013, 41, e19. [Google Scholar]
- Ackermann, M.; Strimmer, K. A general modular framework for gene set enrichment analysis. BMC Bioinform 2009, 10, 47. [Google Scholar]
- Nam, D.; Kim, S.Y. Gene-set approach for expression pattern analysis. Brief. Bioinform 2008, 9, 189–197. [Google Scholar]
- Sales, G.; Calura, E.; Cavalieri, D.; Romualdi, C. Graphite—A Bioconductor package to convert pathway topology to gene network. BMC Bioinform 2012, 13, 20. [Google Scholar]
- Martini, P.; Risso, D.; Sales, G.; Romualdi, C.; Lanfranchi, G.; Cagnin, S. Statistical Test of Expression Pattern (STEPath): A new strategy to integrate gene expression data with genomic information in individual and meta-analysis studies. BMC Bioinform 2011, 12, 92. [Google Scholar]
- Massa, M.S.; Chiogna, M.; Romualdi, C. Gene set analysis exploiting the topology of a pathway. BMC Syst. Biol 2010, 4, 121. [Google Scholar]
- Sales, G.; Calura, E.; Martini, P.; Romualdi, C. Graphite Web: Web tool for gene set analysis exploiting pathway topology. Nucleic Acids Res 2013, 41, W89–W97. [Google Scholar]
- Morgat, A.; Coissac, E.; Coudert, E.; Axelsen, K.B.; Keller, G.; Bairoch, A.; Bridge, A.; Bougueleret, L.; Xenarios, I.; Viari, A. UniPathway: A resource for the exploration and annotation of metabolic pathways. Nucleic Acids Res 2012, 40, D761–D769. [Google Scholar]
- Kanehisa, M.; Goto, S.; Furumichi, M.; Tanabe, M.; Hirakawa, M. KEGG for representation and analysis of molecular networks involving diseases and drugs. Nucleic Acids Res 2010, 38, D355–D360. [Google Scholar]
- Caspi, R.; Foerster, H.; Fulcher, C.A.; Kaipa, P.; Krummenacker, M.; Latendresse, M.; Paley, S.; Rhee, S.Y.; Shearer, A.G.; Tissier, C.; et al. The MetaCyc Database of metabolic pathways and enzymes and the BioCyc collection of Pathway/Genome Databases. Nucleic Acids Res 2008, 36, D623–D631. [Google Scholar]
- Joshi-Tope, G.; Gillespie, M.; Vastrik, I.; D’Eustachio, P.; Schmidt, E.; de Bono, B.; Jassal, B.; Gopinath, G.R.; Wu, G.R.; Matthews, L.; et al. Reactome: A knowledgebase of biological pathways. Nucleic Acids Res 2005, 33, D428–D432. [Google Scholar]
- Friedman, N. Inferring cellular networks using probabilistic graphical models. Science 2004, 303, 799–805. [Google Scholar]
- Butte, A.J.; Tamayo, P.; Slonim, D.; Golub, T.R.; Kohane, I.S. Discovering functional relationships between RNA expression and chemotherapeutic susceptibility using relevance networks. Proc. Natl. Acad. Sci. USA 2000, 97, 12182–12186. [Google Scholar]
- Schafer, J.; Strimmer, K. An empirical Bayes approach to inferring large-scale gene association networks. Bioinformatics 2005, 21, 754–764. [Google Scholar]
- Markowetz, F.; Spang, R. Inferring cellular networks—A review. BMC Bioinform 2007, 8, S5. [Google Scholar]
- Basso, K.; Margolin, A.A.; Stolovitzky, G.; Klein, U.; Dalla-Favera, R.; Califano, A. Reverse engineering of regulatory networks in human B cells. Nat. Genet 2005, 37, 382–390. [Google Scholar]
- Margolin, A.A.; Nemenman, I.; Basso, K.; Wiggins, C.; Stolovitzky, G.; Dalla Favera, R.; Califano, A. ARACNE: An algorithm for the reconstruction of gene regulatory networks in a mammalian cellular context. BMC Bioinform 2006, 7, S7. [Google Scholar]
- Sales, G.; Romualdi, C. Parmigene—A parallel R package for mutual information estimation and gene network reconstruction. Bioinformatics 2011, 27, 1876–1877. [Google Scholar]
- Marbach, D.; Costello, J.C.; Kuffner, R.; Vega, N.M.; Prill, R.J.; Camacho, D.M.; Allison, K.R.; Consortium, D.; Kellis, M.; Collins, J.J.; et al. Wisdom of crowds for robust gene network inference. Nat. Methods 2012, 9, 796–804. [Google Scholar]
- Macias, S.; Plass, M.; Stajuda, A.; Michlewski, G.; Eyras, E.; Caceres, J.F. DGCR8 HITS-CLIP reveals novel functions for the Microprocessor. Nat. Struct. Mol. Biol 2012, 19, 760–766. [Google Scholar]
- Thomson, D.W.; Bracken, C.P.; Goodall, G.J. Experimental strategies for microRNA target identification. Nucleic Acids Res 2011, 39, 6845–6853. [Google Scholar]
- Hafner, M.; Landthaler, M.; Burger, L.; Khorshid, M.; Hausser, J.; Berninger, P.; Rothballer, A.; Ascano, M., Jr.; Jungkamp, A.C.; Munschauer, M.; et al. Transcriptome-wide identification of RNA-binding protein and microRNA target sites by PAR-CLIP. Cell 2010, 141, 129–141. [Google Scholar]
- Chi, S.W.; Zang, J.B.; Mele, A.; Darnell, R.B. Argonaute HITS-CLIP decodes microRNA-mRNA interaction maps. Nature 2009, 460, 479–486. [Google Scholar]
- Yousef, M.; Showe, L.; Showe, M. A study of microRNAs in silico and in vivo: Bioinformatics approaches to microRNA discovery and target identification. FEBS J 2009, 276, 2150–2156. [Google Scholar]
- Witkos, T.M.; Koscianska, E.; Krzyzosiak, W.J. Practical aspects of microRNA target prediction. Curr. Mol. Med 2011, 11, 93–109. [Google Scholar]
- Sales, G.; Coppe, A.; Bisognin, A.; Biasiolo, M.; Bortoluzzi, S.; Romualdi, C. MAGIA, a web-based tool for miRNA and Genes Integrated Analysis. Nucleic Acids Res 2010, 38, W352–W359. [Google Scholar]
- Bisognin, A.; Sales, G.; Coppe, A.; Bortoluzzi, S.; Romualdi, C. MAGIA2: From miRNA and genes expression data integrative analysis to microRNA-transcription factor mixed regulatory circuits (2012 update). Nucleic Acids Res 2012, 40, W13–W21. [Google Scholar]
- Nam, S.; Li, M.; Choi, K.; Balch, C.; Kim, S.; Nephew, K.P. MicroRNA and mRNA integrated analysis (MMIA): A web tool for examining biological functions of microRNA expression. Nucleic Acids Res 2009, 37, W356–W362. [Google Scholar]
- Ross, J.W.; Fernandez de Castro, J.P.; Zhao, J.; Samuel, M.; Walters, E.; Rios, C.; Bray-Ward, P.; Jones, B.W.; Marc, R.E.; Wang, W.; et al. Generation of an inbred miniature pig model of retinitis pigmentosa. Investig. Ophthalmol. Vis. Sci 2012, 53, 501–507. [Google Scholar]
- Maxmen, A. Model pigs face messy path. Nature 2012, 486, 453. [Google Scholar]
- Sandrin, M.S.; Loveland, B.E.; McKenzie, I.F. Genetic engineering for xenotransplantation. J. Card. Surg 2001, 16, 448–457. [Google Scholar]
- Ekser, B.; Rigotti, P.; Gridelli, B.; Cooper, D.K. Xenotransplantation of solid organs in the pig-to-primate model. Transpl. Immunol 2009, 21, 87–92. [Google Scholar]
- Zhang, Q.; Widmer, G.; Tzipori, S. A pig model of the human gastrointestinal tract. Gut Microbes 2013, 4, 193–200. [Google Scholar]
- Kragh, P.M.; Nielsen, A.L.; Li, J.; Du, Y.; Lin, L.; Schmidt, M.; Bogh, I.B.; Holm, I.E.; Jakobsen, J.E.; Johansen, M.G.; et al. Hemizygous minipigs produced by random gene insertion and handmade cloning express the Alzheimer’s disease-causing dominant mutation APPsw. Transgenic Res 2009, 18, 545–558. [Google Scholar]
- Granada, J.F.; Kaluza, G.L.; Wilensky, R.L.; Biedermann, B.C.; Schwartz, R.S.; Falk, E. Porcine models of coronary atherosclerosis and vulnerable plaque for imaging and interventional research. EuroIntervention 2009, 5, 140–148. [Google Scholar]
- Groenen, M.A.; Archibald, A.L.; Uenishi, H.; Tuggle, C.K.; Takeuchi, Y.; Rothschild, M.F.; Rogel-Gaillard, C.; Park, C.; Milan, D.; Megens, H.J.; et al. Analyses of pig genomes provide insight into porcine demography and evolution. Nature 2012, 491, 393–398. [Google Scholar]
- Li, M.; Wu, H.; Luo, Z.; Xia, Y.; Guan, J.; Wang, T.; Gu, Y.; Chen, L.; Zhang, K.; Ma, J.; et al. An atlas of DNA methylomes in porcine adipose and muscle tissues. Nat. Commun 2012, 3, 850. [Google Scholar] [Green Version]
- Fairbairn, L.; Kapetanovic, R.; Beraldi, D.; Sester, D.P.; Tuggle, C.K.; Archibald, A.L.; Hume, D.A. Comparative analysis of monocyte subsets in the pig. J. Immunol 2013, 190, 6389–6396. [Google Scholar]
- Martins, R.P.; Lorenzi, V.; Arce, C.; Lucena, C.; Carvajal, A.; Garrido, J.J. Innate and adaptive immune mechanisms are effectively induced in ileal Peyer’s patches of Salmonella typhimurium infected pigs. Dev. Comp. Immunol 2013, 41, 100–104. [Google Scholar]
- Hulst, M.; Smits, M.; Vastenhouw, S.; de Wit, A.; Niewold, T.; van der Meulen, J. Transcription networks responsible for early regulation of Salmonella-induced inflammation in the jejunum of pigs. J. Inflamm 2013, 10, 18. [Google Scholar]
- Adler, M.; Murani, E.; Brunner, R.; Ponsuksili, S.; Wimmers, K. Transcriptomic response of porcine PBMCs to vaccination with tetanus toxoid as a model antigen. PLoS One 2013, 8, e58306. [Google Scholar]
- Freeman, T.C.; Ivens, A.; Baillie, J.K.; Beraldi, D.; Barnett, M.W.; Dorward, D.; Downing, A.; Fairbairn, L.; Kapetanovic, R.; Raza, S.; et al. A gene expression atlas of the domestic pig. BMC Biol 2012, 10, 90. [Google Scholar]
- McDaneld, T.G.; Smith, T.P.; Harhay, G.P.; Wiedmann, R.T. Next-generation sequencing of the porcine skeletal muscle transcriptome for computational prediction of microRNA gene targets. PLoS One 2012, 7, e42039. [Google Scholar]
- Zhou, B.; Liu, H.L.; Shi, F.X.; Wang, J.Y. MicroRNA expression profiles of porcine skeletal muscle. Anim. Genet 2010, 41, 499–508. [Google Scholar]
- Liu, Y.; Li, M.; Ma, J.; Zhang, J.; Zhou, C.; Wang, T.; Gao, X.; Li, X. Identification of differences in microRNA transcriptomes between porcine oxidative and glycolytic skeletal muscles. BMC Mol. Biol 2013, 14, 7. [Google Scholar]
- Siengdee, P.; Trakooljul, N.; Murani, E.; Schwerin, M.; Wimmers, K.; Ponsuksili, S. Transcriptional profiling and miRNA-dependent regulatory network analysis of longissimus dorsi muscle during prenatal and adult stages in two distinct pig breeds. Anim. Genet 2013, 44, 398–407. [Google Scholar]
- McDaneld, T.G.; Smith, T.P.; Doumit, M.E.; Miles, J.R.; Coutinho, L.L.; Sonstegard, T.S.; Matukumalli, L.K.; Nonneman, D.J.; Wiedmann, R.T. MicroRNA transcriptome profiles during swine skeletal muscle development. BMC Genomics 2009, 10, 77. [Google Scholar]
- Huang, T.H.; Zhu, M.J.; Li, X.Y.; Zhao, S.H. Discovery of porcine microRNAs and profiling from skeletal muscle tissues during development. PLoS One 2008, 3, e3225. [Google Scholar]
- Shen, H.; Liu, T.; Fu, L.; Zhao, S.; Fan, B.; Cao, J.; Li, X. Identification of microRNAs involved in dexamethasone-induced muscle atrophy. Mol. Cell. Biochem 2013, 381, 105–113. [Google Scholar]
- Timoneda, O.; Balcells, I.; Nunez, J.I.; Egea, R.; Vera, G.; Castello, A.; Tomas, A.; Sanchez, A. miRNA expression profile analysis in kidney of different porcine breeds. PLoS One 2013, 8, e55402. [Google Scholar]
- Li, A.; Song, T.; Wang, F.; Liu, D.; Fan, Z.; Zhang, C.; He, J.; Wang, S. MicroRNAome and expression profile of developing tooth germ in miniature pigs. PLoS One 2012, 7, e52256. [Google Scholar]
- Sharbati, S.; Friedlander, M.R.; Sharbati, J.; Hoeke, L.; Chen, W.; Keller, A.; Stahler, P.F.; Rajewsky, N.; Einspanier, R. Deciphering the porcine intestinal microRNA transcriptome. BMC Genomics 2010, 11, 275. [Google Scholar]
- Podolska, A.; Kaczkowski, B.; Kamp Busk, P.; Sokilde, R.; Litman, T.; Fredholm, M.; Cirera, S. MicroRNA expression profiling of the porcine developing brain. PLoS One 2011, 6, e14494. [Google Scholar]
- Zhou, Y.; Tang, X.; Song, Q.; Ji, Y.; Wang, H.; Jiao, H.; Ouyang, H.; Pang, D. Identification and characterization of pig embryo microRNAs by Solexa sequencing. Reprod. Domest. Anim 2013, 48, 112–120. [Google Scholar]
- Lian, C.; Sun, B.; Niu, S.; Yang, R.; Liu, B.; Lu, C.; Meng, J.; Qiu, Z.; Zhang, L.; Zhao, Z. A comparative profile of the microRNA transcriptome in immature and mature porcine testes using Solexa deep sequencing. FEBS J 2012, 279, 964–975. [Google Scholar]
- Li, M.; Liu, Y.; Wang, T.; Guan, J.; Luo, Z.; Chen, H.; Wang, X.; Chen, L.; Ma, J.; Mu, Z.; et al. Repertoire of porcine microRNAs in adult ovary and testis by deep sequencing. Int. J. Biol. Sci 2011, 7, 1045–1055. [Google Scholar]
- Curry, E.; Safranski, T.J.; Pratt, S.L. Differential expression of porcine sperm microRNAs and their association with sperm morphology and motility. Theriogenology 2011, 76, 1532–1539. [Google Scholar]
- Luo, L.; Ye, L.; Liu, G.; Shao, G.; Zheng, R.; Ren, Z.; Zuo, B.; Xu, D.; Lei, M.; Jiang, S.; et al. Microarray-based approach identifies differentially expressed microRNAs in porcine sexually immature and mature testes. PLoS One 2010, 5, e11744. [Google Scholar]
- Li, H.; Xi, Q.; Xiong, Y.; Cheng, X.; Qi, Q.; Yang, L.; Shu, G.; Wang, S.; Wang, L.; Gao, P.; et al. A comprehensive expression profile of microRNAs in porcine pituitary. PLoS One 2011, 6, e24883. [Google Scholar]
- Li, H.Y.; Xi, Q.Y.; Xiong, Y.Y.; Liu, X.L.; Cheng, X.; Shu, G.; Wang, S.B.; Wang, L.N.; Gao, P.; Zhu, X.T.; et al. Identification and comparison of microRNAs from skeletal muscle and adipose tissues from two porcine breeds. Anim. Genet 2012, 43, 704–713. [Google Scholar]
- Martini, P.; Sales, G.; Brugiolo, M.; Gandaglia, A.; Naso, F.; De Pitta’, C.; Spina, M.; Gerosa, G.; Romualdi, C.; Cagnin, S.; et al. Tissue-specific expression and regulatory networks of pig microRNAome. PLoS One 2013, unpublished work. [Google Scholar]
- Nelson, P.T.; Baldwin, D.A.; Kloosterman, W.P.; Kauppinen, S.; Plasterk, R.H.; Mourelatos, Z. RAKE and LNA-ISH reveal microRNA expression and localization in archival human brain. RNA 2006, 12, 187–191. [Google Scholar]
- Nelson, P.T.; Baldwin, D.A.; Scearce, L.M.; Oberholtzer, J.C.; Tobias, J.W.; Mourelatos, Z. Microarray-based, high-throughput gene expression profiling of microRNAs. Nat. Methods 2004, 1, 155–161. [Google Scholar]
- Kronick, M.N. Creation of the whole human genome microarray. Expert Rev. Proteomics 2004, 1, 19–28. [Google Scholar]
- Wong, A.P.; Nili, N.; Strauss, B.H. In vitro differences between venous and arterial-derived smooth muscle cells: Potential modulatory role of decorin. Cardiovasc. Res 2005, 65, 702–710. [Google Scholar]
- Ross, J.M.; McIntire, L.V.; Moake, J.L.; Rand, J.H. Platelet adhesion and aggregation on human type VI collagen surfaces under physiological flow conditions. Blood 1995, 85, 1826–1835. [Google Scholar]
- Smedegard, G.; Hedqvist, P.; Dahlen, S.E.; Revenas, B.; Hammarstrom, S.; Samuelsson, B. Leukotriene C4 affects pulmonary and cardiovascular dynamics in monkey. Nature 1982, 295, 327–329. [Google Scholar]
- Pawloski, J.R.; Chapnick, B.M. Antagonism of LTD4-evoked relaxation in canine renal artery and vein. Am. J. Physiol 1993, 265, H980–H985. [Google Scholar]
- Brink, C.; Dahlen, S.E.; Drazen, J.; Evans, J.F.; Hay, D.W.; Nicosia, S.; Serhan, C.N.; Shimizu, T.; Yokomizo, T. International Union of Pharmacology XXXVII. Nomenclature for leukotriene and lipoxin receptors. Pharmacol. Rev 2003, 55, 195–227. [Google Scholar]
- Inoue, H.; Taba, Y.; Miwa, Y.; Yokota, C.; Miyagi, M.; Sasaguri, T. Transcriptional and posttranscriptional regulation of cyclooxygenase-2 expression by fluid shear stress in vascular endothelial cells. Arterioscler. Thromb. Vasc. Biol 2002, 22, 1415–1420. [Google Scholar]
- Dahboul, F.; Leroy, P.; Maguin Gate, K.; Boudier, A.; Gaucher, C.; Liminana, P.; Lartaud, I.; Pompella, A.; Perrin-Sarrado, C. Endothelial γ-glutamyltransferase contributes to the vasorelaxant effect of S-nitrosoglutathione in rat aorta. PLoS One 2012, 7, e43190. [Google Scholar]
- Yousaf, N.; Howard, J.C.; Williams, B.D. Studies in the rat of antibody-coated and N-ethylmaleimide-treated erythrocyte clearance by the spleen. I. Effects of in vivo complement activation. Immunology 1986, 59, 75–79. [Google Scholar]
- Ravingerova, T.; Barancik, M.; Strniskova, M. Mitogen-activated protein kinases: A new therapeutic target in cardiac pathology. Mol. Cell. Biochem 2003, 247, 127–138. [Google Scholar]
- Howe, G.A.; Addison, C.L. RhoB controls endothelial cell morphogenesis in part via negative regulation of RhoA. Vasc. Cell 2012, 4, 1. [Google Scholar]
- Torella, D.; Iaconetti, C.; Catalucci, D.; Ellison, G.M.; Leone, A.; Waring, C.D.; Bochicchio, A.; Vicinanza, C.; Aquila, I.; Curcio, A.; et al. MicroRNA-133 controls vascular smooth muscle cell phenotypic switch in vitro and vascular remodeling in vivo. Circ. Res. 2011, 109, 880–893. [Google Scholar]
- Kobayashi, R.; Kubota, T.; Hidaka, H. Purification, characterization, and partial sequence analysis of a new 25-kDa actin-binding protein from bovine aorta: A SM22 homolog. Biochem. Biophys. Res. Commun 1994, 198, 1275–1280. [Google Scholar]
- Rhodes, L.V.; Tilghman, S.L.; Boue, S.M.; Wang, S.; Khalili, H.; Muir, S.E.; Bratton, M.R.; Zhang, Q.; Wang, G.; Burow, M.E.; et al. Glyceollins as novel targeted therapeutic for the treatment of triple-negative breast cancer. Oncol. Lett 2012, 3, 163–171. [Google Scholar]
- Cordes, K.R.; Sheehy, N.T.; White, M.P.; Berry, E.C.; Morton, S.U.; Muth, A.N.; Lee, T.H.; Miano, J.M.; Ivey, K.N.; Srivastava, D. miR-145 and miR-143 regulate smooth muscle cell fate and plasticity. Nature 2009, 460, 705–710. [Google Scholar]
- Ikeda, S.; Pu, W.T. Expression and function of microRNAs in heart disease. Curr. Drug Targets 2010, 11, 913–925. [Google Scholar]
- Chhabra, R.; Dubey, R.; Saini, N. Cooperative and individualistic functions of the microRNAs in the miR-23a~27a~24-2 cluster and its implication in human diseases. Mol. Cancer 2010, 9, 232. [Google Scholar]
- Li, S.; Ran, Y.; Zhang, D.; Chen, J.; Zhu, D. MicroRNA-138 plays a role in hypoxic pulmonary vascular remodelling by targeting Mst1. Biochem. J 2013, 452, 281–291. [Google Scholar]
- Park, C.; Yan, W.; Ward, S.M.; Hwang, S.J.; Wu, Q.; Hatton, W.J.; Park, J.K.; Sanders, K.M.; Ro, S. MicroRNAs dynamically remodel gastrointestinal smooth muscle cells. PLoS One 2011, 6, e18628. [Google Scholar]
- Talasila, A.; Yu, H.; Ackers-Johnson, M.; Bot, M.; van Berkel, T.; Bennett, M.; Bot, I.; Sinha, S. Myocardin regulates vascular response to injury through miR-24/-29a and platelet-derived growth factor recepto-β. Arterioscler. Thromb. Vasc. Biol 2013, 33, 2355–2365. [Google Scholar]
- Saeed, A.I.; Sharov, V.; White, J.; Li, J.; Liang, W.; Bhagabati, N.; Braisted, J.; Klapa, M.; Currier, T.; Thiagarajan, M.; et al. TM4: A free, open-source system for microarray data management and analysis. Biotechniques 2003, 34, 374–378. [Google Scholar]
- Raychaudhuri, S.; Stuart, J.M.; Altman, R.B. Principal components analysis to summarize microarray experiments: Application to sporulation time series. Pac. Symp. Biocomput 2000, 5, 455–466. [Google Scholar]
- Fellenberg, K.; Hauser, N.C.; Brors, B.; Neutzner, A.; Hoheisel, J.D.; Vingron, M. Correspondence analysis applied to microarray data. Proc. Natl. Acad. Sci. USA 2001, 98, 10781–10786. [Google Scholar]
- Huang da, W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc 2009, 4, 44–57. [Google Scholar]
- Risso, D.; Massa, M.S.; Chiogna, M.; Romualdi, C. A modified LOESS normalization applied to microRNA arrays: A comparative evaluation. Bioinformatics 2009, 25, 2685–2691. [Google Scholar]
- Spath, H. Two Dimensional Spline Interpolation Algorithms; A K Peters/CRC Press: Wellesley, MA, USA, 1995. [Google Scholar]
- Kanehisa, M.; Goto, S.; Kawashima, S.; Okuno, Y.; Hattori, M. The KEGG resource for deciphering the genome. Nucleic Acids Res 2004, 32, D277–D280. [Google Scholar]
- Saito, R.; Smoot, M.E.; Ono, K.; Ruscheinski, J.; Wang, P.L.; Lotia, S.; Pico, A.R.; Bader, G.D.; Ideker, T. A travel guide to Cytoscape plugins. Nat. Methods 2012, 9, 1069–1076. [Google Scholar]
- Doncheva, N.T.; Assenov, Y.; Domingues, F.S.; Albrecht, M. Topological analysis and interactive visualization of biological networks and protein structures. Nat. Protoc 2012, 7, 670–685. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Martini, P.; Sales, G.; Calura, E.; Brugiolo, M.; Lanfranchi, G.; Romualdi, C.; Cagnin, S. Systems Biology Approach to the Dissection of the Complexity of Regulatory Networks in the S. scrofa Cardiocirculatory System. Int. J. Mol. Sci. 2013, 14, 23160-23187. https://doi.org/10.3390/ijms141123160
Martini P, Sales G, Calura E, Brugiolo M, Lanfranchi G, Romualdi C, Cagnin S. Systems Biology Approach to the Dissection of the Complexity of Regulatory Networks in the S. scrofa Cardiocirculatory System. International Journal of Molecular Sciences. 2013; 14(11):23160-23187. https://doi.org/10.3390/ijms141123160
Chicago/Turabian StyleMartini, Paolo, Gabriele Sales, Enrica Calura, Mattia Brugiolo, Gerolamo Lanfranchi, Chiara Romualdi, and Stefano Cagnin. 2013. "Systems Biology Approach to the Dissection of the Complexity of Regulatory Networks in the S. scrofa Cardiocirculatory System" International Journal of Molecular Sciences 14, no. 11: 23160-23187. https://doi.org/10.3390/ijms141123160