Nonalcoholic Fatty Liver: A Possible New Target for Type 2 Diabetes Prevention and Treatment

Abstract
:1. Introduction
2. NAFLD Is Associated with Insulin Resistance
3. NAFLD, Consequence or Cause of Insulin Resistance
3.1. NAFLD: Consequence of Insulin Resistance
3.2. NAFLD: Cause of Insulin Resistance
3.3. Lipid Intermediates Are Mediators of Insulin Resistance
4. An Emerging Role of NAFLD in Diabetes Prevention and Care
4.1. NAFLD: A New Predictive Marker of Diabetes
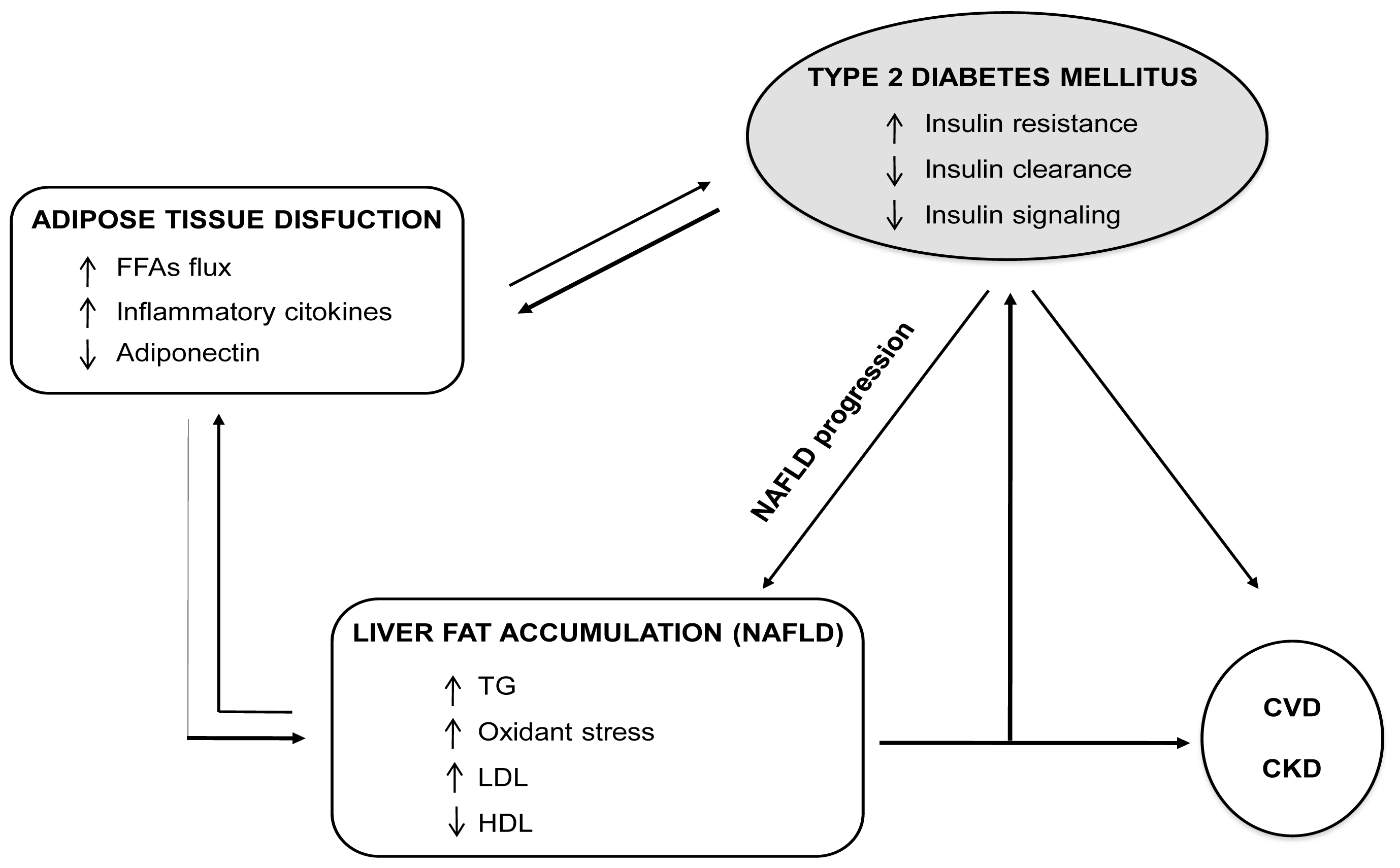
4.2. Fatty Liver and Diabetes: A Vicious Circle
4.3. NAFLD Is Associated with Cardiovascular and Kidney Disease
4.4. NAFLD, Diabetes and Cardiovascular Risk: A Molecular Link
5. Diet and Physical Exercise in NAFLD Management
6. Pharmacological Treatment of NAFLD
7. NAFLD: A Possible New Target for Antidiabetic Drugs
7.1. Metformin
7.1.1. Preclinical Studies
7.1.2. Clinical Studies
7.2. Thiazolidinediones
7.2.1. Preclinical Studies
7.2.2. Clinical Trials
7.2.2.1. Rosiglitazone
7.2.2.2. Pioglitazone
7.3. GLP-1 Analogs
7.3.1. Preclinical Studies
7.3.2. Clinical Studies
7.4. DPP-4 Inhibitors
7.4.1. Preclinical Studies
7.4.2. Clinical Studies
8. Statins
8.1. Preclinical Studies
8.2. Clinical Studies
9. Perspectives and Concluding Remarks

{kind=link}
| References | Study design | Patients number and features | Therapy and duration follow-up | Study outcomes |
|---|---|---|---|---|
| Marchesini et al. 2001 [109] | OL, SA | 20 pts (OB); NASH, elevated AMTs | Metformin 1.5 g/day; 4 months | ↓ ALT ↓ IR ↓ Liver volume |
| Nair et al. 2004 [110] | OL, SA | 28 pts (OW/OB/T2DM); NAFLD | Metformin 20 mg/kg/day; 12 months | ↓ ALT, AST ↓ IR Histology improved |
| Uygun et al. 2004 [111] | OL, RAND | 36 pts (OW/OB); NASH, elevated AMTs | Metformin 1.7 g/day + diet vs. diet; 6 months | ↓ IR ↓ ALT, AST Histology not improved |
| Bugianesi et al. 2005 [112] | OL, RAND (MC) | 110 pts (OW/OB/T2DM); NAFLD, elevated AMTs | Metformin 2 g/day + diet vs. Vit E + diet vs. diet; 12 months | ↓ AST, ALT Histology improved |
| Schwimmer et al. 2005 [120] | SA | 10 pts (OB/NT2DM children); NASH, elevated AMTs | Metformin 1 g/day; 6 months | ↓ AST, ALT ↓ Liver fat ↓ IR |
| De Oliveira et al. 2008 [113] | OL, SA | 20 pts (OW/OB/T2DM); NASH, elevated AMTs | Metformin 1 g/day; 12 months | ↓ ALT ↓ IR Histology improved |
| Idilman et al. 2008 [114] | OL, RAND | 74 pts (OW/OB/T2DM); NASH, elevated AMTs | Metformin 1.7 g/day; 12 months | ↓ ALT ↓ IR Histology not improved |
| Nobili et al. 2008 [121] | OL | 57 pts (OW/OB children); NASH/NAFLD | Metformin 1.5 g/day vs. diet; 24 months | ↓ ALT, AST ↓ IR Histology improved |
| Shields et al. 2009 [116] | RAND, PLAC | 19 pts (OW, OB, NT2DM); NASH, elevated AMTs | Metformin 1 g/day + lifestyle intervention vs. lifestyle intervention + placebo; 12 months | ALT, AST, IR and histology improved with BMI decrease (not significant, control vs. treatment) |
| Loomba et al. 2009 [108] | OL, SA | 28 pts (OW/OB/T2DM); NASH, elevated AMTs | Metformin 2 g/day; 12 months | ↓ ALT, AST ↓ IR Histology improved |
| Haukeland et al. 2009 [115] | PLAC, RAND | 48 pts (OW/OB/T2DM); NAFLD, elevated AMTs | Metformin vs. placebo; 6 months | ↓ ALT, AST ↓ IR Histology not improved |
| Nadeau et al. 2009 [122] | RAND | 50 pts (OB children); NAFLD, elevated AMTs | Metformin 1.7 g/day + diet vs. diet; 6 months | ↓ ALT, AST ↓ IR Ultrasound pattern improved |
| Garinis et al. 2010 [107] | OL, RAND | 50 pts (OW/OB); NAFLD, normal AMTs | Metformin 1 g/day + diet vs. diet; 6 months | ↓ ALT, AST ↓ IR Ultrasound pattern improved ↑ Adiponectin |
| Lavine et al. 2010 [118] | RAND (MC) | 173 pts (OW/OB children); NAFLD, elevated AMTs | Metformin 1 g/day vs. Vit E; 24 months | Not stable AST e ALT reduction Histology not improved |
| References | Study design | Patients number and features | Therapy and duration follow-up | Study outcomes |
|---|---|---|---|---|
| Wang et al. 2006 [130] | OL | 60 pts (DM2); NAFLD, elevated AMTs | Rosiglitazone 4–8 mg/day; 24 weeks | ↓ AMTs ↓ IR ↓ fasting plasma glucose and insulin ↓ HBA1C |
| Akyuz et al. 2007 [131] | OL | 11 pts NAFLD, elevated ALT | Rosiglitazone 4 mg/day vs. metformin vs. diet; 12 months | ↓ AMTs ↓ IR Histology not improved |
| Idilman et al. 2008 [114] | OL, RAND | 25 pts; NASH, elevated AMTs | Rosiglitazone 8 mg/day vs. metformin vs. diet; 12 months | ↓ AMTs Histology improved ↓ IR ↓ C-reactive protein |
| Belfort et al. 2006 [132] | PLAC, RAND | 26 pts (IGT/DM2); NASH | Pioglitazone 30 mg/day vs. placebo for the first 2 months, then 45 mg/day for 4 more months | ↓ ALT ↓ IR ↓ Liver fat ↓ Liver inflammation ↓ Ballooning necrosis Fibrosis not improved |
| Aithal et al. 2008 [133] | PLAC, RAND | 31 pts; NASH | Pioglitazone 30 mg/day vs. placebo; 12 months | ↓ ALT improved ↓ Liver inflammation Fibrosis improved |
| Ratziu V et al. 2008 [134] | OL, PLAC, RAND | 32 pts; NASH, elevated AMTs | Rosiglitazone 4 mg/day vs. placebo for the first month then 8 mg/day for 11 more months | ↓ AMTS ↓ Liver fat ↓ IR |
| Omer et al. 2010 [135] | OL, RAND | 42 pts; (IGT/DM2); NAFLD, elevated ALT | Rosiglitazone 4 mg/day alone vs. Rosiglitazone + metformin; 12 months | ↓ ALT ↓ IR ↓ Liver fat Fibrosis not improved |
| Ratziu V et al. 2010 [136] | OL extension, RAND | 18 pts; NASH | Rosiglitazone 8 mg/day, 3 years | ↓ ALT ↓ IR Histology not further improved |
| Sanyal AJ et al. 2010 [137] | PLAC, RAND | 80 pts; NASH | Pioglitazone 30 mg/day vs. Vit E vs. placebo; 96 weeks | ↓ ALT ↓ IR ↓ Liver fat ↓ Liver inflammation ↓ Ballooning necrosis Fibrosis not improved |
| References | Study design | Patients number and features | Therapy and duration follow-up | Study outcomes |
|---|---|---|---|---|
| Tushuizen et al. 2006 [158] | Case report | 1 pt (T2DM/NASH), elevated AMTs | Exenatide 10 μg plus metformin; 9 months | ↓ ALT, AST ↓ Liver fat (MRS) ↓ IR and CVD risk factors Histology not assessed |
| Klonoff et al. 2008 [159] | OL | 217 pts (OW/OB/T2DM); NAFLD elevated/normal AMTs | Exenatide 10 μg plus metformin and/or sulfanilureas; 36 months | ↓ ALT, AST ↓ IR Histology not assessed |
| Kenny et al. 2010 [160] | Case series | 8 pts (T2DM); NAFLD, elevated AMTs | Exenatide 10 μg; 6 months | ↓ AST, ALT Histology not improved |
| Iwasaki et al. 2011 [170] | OL, SA | 30 pts (T2DM); NAFLD elevated/normal AMTs | Sitagliptin 50 mg; 4 months | ↓ ALT, AST, GGT Histology not assessed |
| Itou et al. 2012 [163] | Case report | 1 pt (T2MD); NAFLD elevated AMTs | Sitagliptin 50 mg; 4 months | ↓ AST, ALT ↓ IR ↓ Liver fat (MRI) Histology not assessed |
| Ylmaz et al. 2012 [171] | OL, SA | 15 pts (T2DM); NASH elevated AMTs | Sitagliptin 100 mg; 12 months | ↓ ALT, AST Histology improved |
| Armstrong et al. 2010 [161] | RAND, PLAC, MC (meta-analys of 6 studies + sub-study) | 4442 pts (T2DM); NAFLD elevated/normal AMTs | Liraglutide 1.8 mg vs. OAD or placebo; 6 months | ↓ ALT ↓ Liver fat (CT) Histology not assessed |
| References | Study design | Patients number and features | Therapy and duration follow up | Study outcomes |
|---|---|---|---|---|
| Nelson et al. 2009 [180] | RAND PLAC | 16 pts NASH, elevated AMTs | Simvastatin 40 g/day; 12 months | AST, ALT not reduced Histology not improved |
| Athyros et al. 2010 [1] | RAND PLAC | 437 pts (OB/44%T2DM); NAFLD, elevated AMTs | Atorvastatin 24 mg/day or other statins; 36 months | ↓ AST, ALT ↓ Total cholesterol, triglycerides Histology not assessed |
| Foster et al. 2011 [179] | RAND PLAC | 80 pts (OB/51%T2DM) NAFLD | Atorvastatin 20 mg/day + Vit C + Vit E; 42 months | AST, ALT not reduced ↓ Liver fat (CT) ↓ Total cholesterol, LDL Histology not assessed |
| Pramfalk et al. 2011 [181] | RAND PLAC | 19 pts NAFLD, normocholesterolemic | Atorvastatin 80 mg/day; 1 month | AST, ALT not assesed ↓ Liver fat (biopsy) ↓ lipogenic genes expression IR not reduced |
Acknowledgments
Conflicts of Interest
References
- Athyros, V.G.; Tziomalos, K.; Gossios, T.D.; Griva, T.; Anagnostis, P.; Kargiotis, K.; Pagourelias, E.D.; Theocharidou, E.; Karagiannis, A.; Mikhailidis, D.P.; et al. Safety and efficacy of long-term statin treatment for cardiovascular events in patients with coronary heart disease and abnormal liver tests in the Greek Atorvastatin and Coronary Heart Disease Evaluation (GREACE) Study: A post-hoc analysis. Lancet 2010, 376, 1916–1922. [Google Scholar]
- Younossi, Z.M.; Stepanova, M.; Afendy, M.; Fang, Y.; Younossi, Y.; Mir, H.; Srishord, M. Changes in the prevalence of the most common causes of chronic liver diseases in the United States from 1988 to 2008. Clin. Gastroenterol. Hepatol 2011, 9, 524–530. [Google Scholar]
- Ratziu, V.; Bellentani, S.; Cortez-Pinto, H.; Day, C.; Marchesini, G. A position statement on NAFLD/NASH based on the EASL 2009 special conference. J. Hepatol 2010, 53, 372–384. [Google Scholar]
- Targher, G.; Kendrick, J.; Smits, G.; Chonchol, M. Relationship between serum gamma-glutamyltransferase and chronic kidney disease in the United States adult population. Findings from the National Health and Nutrition Examination Survey 2001–2006. Nutr. Metab. Cardiovasc. Dis 2010, 20, 583–590. [Google Scholar]
- Dowman, J.K.; Tomlinson, J.W.; Newsome, P.N. Pathogenesis of non-alcoholic fatty liver disease. QJM 2010, 103, 71–83. [Google Scholar]
- Donnelly, K.L.; Smith, C.I.; Schwarzenberg, S.J.; Jessurun, J.; Boldt, M.D.; Parks, E.J. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J. Clin. Investig 2005, 115, 1343–1351. [Google Scholar]
- Akbar, D.H.; Kawther, A.H. Nonalcoholic fatty liver disease in Saudi type 2 diabetic subjects attending a medical outpatient clinic: Prevalence and general characteristics. Diabetes Care 2003, 26, 3351–3352. [Google Scholar]
- Bellentani, S.; Saccoccio, G.; Masutti, F.; Croce, L.S.; Brandi, G.; Sasso, F.; Cristanini, G.; Tiribelli, C. Prevalence of and risk factors for hepatic steatosis in Northern Italy. Ann. Intern. Med 2000, 132, 112–117. [Google Scholar]
- De Alwis, N.M.; Day, C.P. Non-alcoholic fatty liver disease: The mist gradually clears. J. Hepatol 2008, 48, S104–S112. [Google Scholar]
- Dixon, J.B.; Bhathal, P.S.; O’Brien, P.E. Nonalcoholic fatty liver disease: Predictors of nonalcoholic steatohepatitis and liver fibrosis in the severely obese. Gastroenterology 2001, 121, 91–100. [Google Scholar]
- Garcia-Monzon, C.; Martin-Perez, E.; Iacono, O.L.; Fernandez-Bermejo, M.; Majano, P.L.; Apolinario, A.; Larranaga, E.; Moreno-Otero, R. Characterization of pathogenic and prognostic factors of nonalcoholic steatohepatitis associated with obesity. J. Hepatol 2000, 33, 716–724. [Google Scholar]
- Gupte, P.; Amarapurkar, D.; Agal, S.; Baijal, R.; Kulshrestha, P.; Pramanik, S.; Patel, N.; Madan, A.; Amarapurkar, A. Hafeezunnisa. Non-alcoholic steatohepatitis in type 2 diabetes mellitus. J. Gastroenterol. Hepatol 2004, 19, 854–858. [Google Scholar]
- Luyckx, F.H.; Desaive, C.; Thiry, A.; Dewe, W.; Scheen, A.J.; Gielen, J.E.; Lefebvre, P.J. Liver abnormalities in severely obese subjects: Effect of drastic weight loss after gastroplasty. Int. J. Obes. Relat. Metab. Disord 1998, 22, 222–226. [Google Scholar]
- Nomura, H.; Kashiwagi, S.; Hayashi, J.; Kajiyama, W.; Tani, S.; Goto, M. Prevalence of fatty liver in a general population of Okinawa, Japan. Jpn. J. Med 1988, 27, 142–149. [Google Scholar]
- Wang, Y.; Li, Y.Y.; Nie, Y.Q.; Zhou, Y.J.; Cao, C.Y.; Xu, L. Association between metabolic syndrome and the development of non-alcoholic fatty liver disease. Exp. Ther. Med 2013, 6, 77–84. [Google Scholar]
- Utzschneider, K.M.; Kahn, S.E. Review: The role of insulin resistance in nonalcoholic fatty liver disease. J. Clin. Endocrinol. Metab 2006, 91, 4753–4761. [Google Scholar]
- Marchesini, G.; Brizi, M.; Bianchi, G.; Tomassetti, S.; Bugianesi, E.; Lenzi, M.; McCullough, A.J.; Natale, S.; Forlani, G.; Melchionda, N. Nonalcoholic fatty liver disease: A feature of the metabolic syndrome. Diabetes 2001, 50, 1844–1850. [Google Scholar]
- Bugianesi, E.; Gastaldelli, A.; Vanni, E.; Gambino, R.; Cassader, M.; Baldi, S.; Ponti, V.; Pagano, G.; Ferrannini, E.; Rizzetto, M. Insulin resistance in non-diabetic patients with non-alcoholic fatty liver disease: Sites and mechanisms. Diabetologia 2005, 48, 634–642. [Google Scholar]
- Seppala-Lindroos, A.; Vehkavaara, S.; Hakkinen, A.M.; Goto, T.; Westerbacka, J.; Sovijarvi, A.; Halavaara, J.; Yki-Jarvinen, H. Fat accumulation in the liver is associated with defects in insulin suppression of glucose production and serum free fatty acids independent of obesity in normal men. J. Clin. Endocrinol. Metab 2002, 87, 3023–3028. [Google Scholar]
- Flegal, K.M.; Carroll, M.D.; Ogden, C.L.; Johnson, C.L. Prevalence and trends in obesity among US adults, 1999–2000. JAMA 2002, 288, 1723–1727. [Google Scholar]
- Moran, J.R.; Ghishan, F.K.; Halter, S.A.; Greene, H.L. Steatohepatitis in obese children: A cause of chronic liver dysfunction. Am. J. Gastroenterol 1983, 78, 374–377. [Google Scholar]
- Baldridge, A.D.; Perez-Atayde, A.R.; Graeme-Cook, F.; Higgins, L.; Lavine, J.E. Idiopathic steatohepatitis in childhood: A multicenter retrospective study. J. Pediatr 1995, 127, 700–704. [Google Scholar]
- Hebbard, L.; George, J. Animal models of nonalcoholic fatty liver disease. Nat. Rev. Gastroenterol. Hepatol 2011, 8, 35–44. [Google Scholar]
- Ota, T.; Takamura, T.; Kurita, S.; Matsuzawa, N.; Kita, Y.; Uno, M.; Akahori, H.; Misu, H.; Sakurai, M.; Zen, Y.; et al. Insulin resistance accelerates a dietary rat model of nonalcoholic steatohepatitis. Gastroenterology 2007, 132, 282–293. [Google Scholar]
- Bhargava, R.; Senior, P.A.; Ackerman, T.E.; Ryan, E.A.; Paty, B.W.; Lakey, J.R.; Shapiro, A.M. Prevalence of hepatic steatosis after islet transplantation and its relation to graft function. Diabetes 2004, 53, 1311–1317. [Google Scholar]
- Semple, R.K.; Sleigh, A.; Murgatroyd, P.R.; Adams, C.A.; Bluck, L.; Jackson, S.; Vottero, A.; Kanabar, D.; Charlton-Menys, V.; Durrington, P.; et al. Postreceptor insulin resistance contributes to human dyslipidemia and hepatic steatosis. J. Clin. Investig 2009, 119, 315–322. [Google Scholar]
- Saltiel, A.R.; Kahn, C.R. Insulin signalling and the regulation of glucose and lipid metabolism. Nature 2001, 414, 799–806. [Google Scholar]
- Brown, M.S.; Goldstein, J.L. Selective versus total insulin resistance: A pathogenic paradox. Cell Metab 2008, 7, 95–96. [Google Scholar]
- Tamura, S.; Shimomura, I. Contribution of adipose tissue and de novo lipogenesis to nonalcoholic fatty liver disease. J. Clin. Investig 2005, 115, 1139–1142. [Google Scholar]
- Wolfrum, C.; Asilmaz, E.; Luca, E.; Friedman, J.M.; Stoffel, M. Foxa2 regulates lipid metabolism and ketogenesis in the liver during fasting and in diabetes. Nature 2004, 432, 1027–1032. [Google Scholar]
- Browning, J.D.; Horton, J.D. Molecular mediators of hepatic steatosis and liver injury. J. Clin. Investig 2004, 114, 147–152. [Google Scholar]
- Nagle, C.A.; Klett, E.L.; Coleman, R.A. Hepatic triacylglycerol accumulation and insulin resistance. J. Lipid Res 2009, 50, S74–S79. [Google Scholar]
- Kim, J.; Choi, S.; Kong, B.; Oh, Y.; Shinn, S. Insulin secretion and sensitivity during oral glucose tolerance test in Korean lean elderly women. J. Korean Med. Sci 2001, 16, 592–597. [Google Scholar]
- Samuel, V.T.; Liu, Z.X.; Qu, X.; Elder, B.D.; Bilz, S.; Befroy, D.; Romanelli, A.J.; Shulman, G.I. Mechanism of hepatic insulin resistance in non-alcoholic fatty liver disease. J. Biol. Chem 2004, 279, 32345–32353. [Google Scholar]
- Dey, D.; Basu, D.; Roy, S.S.; Bandyopadhyay, A.; Bhattacharya, S. Involvement of novel PKC isoforms in FFA induced defects in insulin signaling. Mol. Cell. Endocrinol 2006, 246, 60–64. [Google Scholar]
- Considine, R.V.; Nyce, M.R.; Allen, L.E.; Morales, L.M.; Triester, S.; Serrano, J.; Colberg, J.; Lanza-Jacoby, S.; Caro, J.F. Protein kinase C is increased in the liver of humans and rats with non-insulin-dependent diabetes mellitus: An alteration not due to hyperglycemia. J. Clin. Investig 1995, 95, 2938–2944. [Google Scholar]
- Samuel, V.T.; Liu, Z.X.; Wang, A.; Beddow, S.A.; Geisler, J.G.; Kahn, M.; Zhang, X.M.; Monia, B.P.; Bhanot, S.; Shulman, G.I. Inhibition of protein kinase Cepsilon prevents hepatic insulin resistance in nonalcoholic fatty liver disease. J. Clin. Investig 2007, 117, 739–745. [Google Scholar]
- Kumashiro, N.; Erion, D.M.; Zhang, D.; Kahn, M.; Beddow, S.A.; Chu, X.; Still, C.D.; Gerhard, G.S.; Han, X.; Dziura, J.; et al. Cellular mechanism of insulin resistance in nonalcoholic fatty liver disease. Proc. Natl. Acad. Sci. USA 2011, 108, 16381–16385. [Google Scholar]
- Chen, M.T.; Kaufman, L.N.; Spennetta, T.; Shrago, E. Effects of high fat-feeding to rats on the interrelationship of body weight, plasma insulin, and fatty acyl-coenzyme A esters in liver and skeletal muscle. Metabolism 1992, 41, 564–569. [Google Scholar]
- Holland, W.L.; Brozinick, J.T.; Wang, L.P.; Hawkins, E.D.; Sargent, K.M.; Liu, Y.; Narra, K.; Hoehn, K.L.; Knotts, T.A.; Siesky, A.; et al. Inhibition of ceramide synthesis ameliorates glucocorticoid-, saturated-fat-, and obesity-induced insulin resistance. Cell Metab 2007, 5, 167–179. [Google Scholar]
- Benhamed, F.; Denechaud, P.D.; Lemoine, M.; Robichon, C.; Moldes, M.; Bertrand-Michel, J.; Ratziu, V.; Serfaty, L.; Housset, C.; Capeau, J.; et al. The lipogenic transcription factor ChREBP dissociates hepatic steatosis from insulin resistance in mice and humans. J. Clin. Investig 2012, 122, 2176–2194. [Google Scholar]
- Minehira, K.; Young, S.G.; Villanueva, C.J.; Yetukuri, L.; Oresic, M.; Hellerstein, M.K.; Farese, R.V., Jr.; Horton, J.D.; Preitner, F.; Thorens, B.; et al. Blocking VLDL secretion causes hepatic steatosis but does not affect peripheral lipid stores or insulin sensitivity in mice. J. Lipid Res 2008, 49, 2038–2044. [Google Scholar]
- Grefhorst, A.; Hoekstra, J.; Derks, T.G.; Ouwens, D.M.; Baller, J.F.; Havinga, R.; Havekes, L.M.; Romijn, J.A.; Kuipers, F. Acute hepatic steatosis in mice by blocking beta-oxidation does not reduce insulin sensitivity of very-low-density lipoprotein production. Am. J. Physiol. Gastrointest. Liver Physiol 2005, 289, G592–G598. [Google Scholar]
- Matsuzaka, T.; Shimano, H.; Yahagi, N.; Kato, T.; Atsumi, A.; Yamamoto, T.; Inoue, N.; Ishikawa, M.; Okada, S.; Ishigaki, N.; et al. Crucial role of a long-chain fatty acid elongase, Elovl6, in obesity-induced insulin resistance. Nat. Med 2007, 13, 1193–1202. [Google Scholar]
- Guerre-Millo, M.; Rouault, C.; Poulain, P.; Andre, J.; Poitout, V.; Peters, J.M.; Gonzalez, F.J.; Fruchart, J.C.; Reach, G.; Staels, B. PPAR-alpha-null mice are protected from high-fat diet-induced insulin resistance. Diabetes 2001, 50, 2809–2814. [Google Scholar]
- Heijboer, A.C.; Donga, E.; Voshol, P.J.; Dang, Z.C.; Havekes, L.M.; Romijn, J.A.; Corssmit, E.P. Sixteen hours of fasting differentially affects hepatic and muscle insulin sensitivity in mice. J. Lipid Res 2005, 46, 582–588. [Google Scholar]
- Hammond, L.E.; Neschen, S.; Romanelli, A.J.; Cline, G.W.; Ilkayeva, O.R.; Shulman, G.I.; Muoio, D.M.; Coleman, R.A. Mitochondrial glycerol-3-phosphate acyltransferase-1 is essential in liver for the metabolism of excess acyl-CoAs. J. Biol. Chem 2005, 280, 25629–25636. [Google Scholar]
- Yazdi, M.; Ahnmark, A.; William-Olsson, L.; Snaith, M.; Turner, N.; Osla, F.; Wedin, M.; Asztely, A.K.; Elmgren, A.; Bohlooly, Y.M.; et al. The role of mitochondrial glycerol-3-phosphate acyltransferase-1 in regulating lipid and glucose homeostasis in high-fat diet fed mice. Biochem. Biophys. Res. Commun 2008, 369, 1065–1070. [Google Scholar]
- Tiikkainen, M.; Bergholm, R.; Vehkavaara, S.; Rissanen, A.; Hakkinen, A.M.; Tamminen, M.; Teramo, K.; Yki-Jarvinen, H. Effects of identical weight loss on body composition and features of insulin resistance in obese women with high and low liver fat content. Diabetes 2003, 52, 701–707. [Google Scholar]
- Kotronen, A.; Juurinen, L.; Hakkarainen, A.; Westerbacka, J.; Corner, A.; Bergholm, R.; Yki-Jarvinen, H. Liver fat is increased in type 2 diabetic patients and underestimated by serum alanine aminotransferase compared with equally obese nondiabetic subjects. Diabetes Care 2008, 31, 165–169. [Google Scholar]
- Leite, N.C.; Salles, G.F.; Araujo, A.L.; Villela-Nogueira, C.A.; Cardoso, C.R. Prevalence and associated factors of non-alcoholic fatty liver disease in patients with type-2 diabetes mellitus. Liver Int 2009, 29, 113–119. [Google Scholar]
- Targher, G.; Byrne, C.D. Clinical Review: Nonalcoholic fatty liver disease: A novel cardiometabolic risk factor for type 2 diabetes and its complications. J. Clin. Endocrinol. Metab 2013, 98, 483–495. [Google Scholar]
- Younossi, Z.M.; Gramlich, T.; Matteoni, C.A.; Boparai, N.; McCullough, A.J. Nonalcoholic fatty liver disease in patients with type 2 diabetes. Clin. Gastroenterol. Hepatol 2004, 2, 262–265. [Google Scholar]
- Speliotes, E.K.; Massaro, J.M.; Hoffmann, U.; Vasan, R.S.; Meigs, J.B.; Sahani, D.V.; Hirschhorn, J.N.; O’Donnell, C.J.; Fox, C.S. Fatty liver is associated with dyslipidemia and dysglycemia independent of visceral fat: The Framingham Heart Study. Hepatology 2010, 51, 1979–1987. [Google Scholar]
- Fraser, A.; Harris, R.; Sattar, N.; Ebrahim, S.; Davey Smith, G.; Lawlor, D.A. Alanine aminotransferase, gamma-glutamyltransferase, and incident diabetes: The British Women’s Heart and Health Study and meta-analysis. Diabetes Care 2009, 32, 741–750. [Google Scholar]
- Musso, G.; Gambino, R.; Cassader, M.; Pagano, G. Meta-analysis: Natural history of non-alcoholic fatty liver disease (NAFLD) and diagnostic accuracy of non-invasive tests for liver disease severity. Ann. Med 2011, 43, 617–649. [Google Scholar]
- Targher, G.; Bertolini, L.; Padovani, R.; Rodella, S.; Tessari, R.; Zenari, L.; Day, C.; Arcaro, G. Prevalence of nonalcoholic fatty liver disease and its association with cardiovascular disease among type 2 diabetic patients. Diabetes Care 2007, 30, 1212–1218. [Google Scholar]
- Fan, J.G.; Li, F.; Cai, X.B.; Peng, Y.D.; Ao, Q.H.; Gao, Y. Effects of nonalcoholic fatty liver disease on the development of metabolic disorders. J. Gastroenterol. Hepatol 2007, 22, 1086–1091. [Google Scholar]
- Kim, C.H.; Park, J.Y.; Lee, K.U.; Kim, J.H.; Kim, H.K. Fatty liver is an independent risk factor for the development of Type 2 diabetes in Korean adults. Diabet. Med 2008, 25, 476–481. [Google Scholar]
- Sung, K.C.; Kim, S.H. Interrelationship between fatty liver and insulin resistance in the development of type 2 diabetes. J. Clin. Endocrinol. Metab 2011, 96, 1093–1097. [Google Scholar]
- Sung, K.C.; Wild, S.H.; Kwag, H.J.; Byrne, C.D. Fatty liver, insulin resistance, and features of metabolic syndrome: Relationships with coronary artery calcium in 10,153 people. Diabetes Care 2012, 35, 2359–2364. [Google Scholar]
- Yamada, T.; Fukatsu, M.; Suzuki, S.; Wada, T.; Yoshida, T.; Joh, T. Fatty liver predicts impaired fasting glucose and type 2 diabetes mellitus in Japanese undergoing a health checkup. J. Gastroenterol. Hepatol 2010, 25, 352–356. [Google Scholar]
- Loria, P.; Lonardo, A.; Anania, F. Liver and diabetes. A vicious circle. Hepatol. Res 2013, 43, 51–64. [Google Scholar]
- Smith, B.W.; Adams, L.A. Nonalcoholic fatty liver disease and diabetes mellitus: Pathogenesis and treatment. Nat. Rev. Endocrinol 2011, 7, 456–465. [Google Scholar]
- Jimba, S.; Nakagami, T.; Takahashi, M.; Wakamatsu, T.; Hirota, Y.; Iwamoto, Y.; Wasada, T. Prevalence of non-alcoholic fatty liver disease and its association with impaired glucose metabolism in Japanese adults. Diabet. Med 2005, 22, 1141–1145. [Google Scholar]
- Williamson, R.M.; Price, J.F.; Glancy, S.; Perry, E.; Nee, L.D.; Hayes, P.C.; Frier, B.M.; van Look, L.A.; Johnston, G.I.; Reynolds, R.M.; et al. Prevalence of and risk factors for hepatic steatosis and nonalcoholic Fatty liver disease in people with type 2 diabetes: The Edinburgh Type 2 Diabetes Study. Diabetes Care 2011, 34, 1139–1144. [Google Scholar]
- Zoppini, G.; Targher, G.; Trombetta, M.; Lippi, G.; Muggeo, M. Relationship of serum gamma-glutamyltransferase to atherogenic dyslipidemia and glycemic control in type 2 diabetes. Obesity 2009, 17, 370–374. [Google Scholar]
- Ryysy, L.; Hakkinen, A.M.; Goto, T.; Vehkavaara, S.; Westerbacka, J.; Halavaara, J.; Yki-Jarvinen, H. Hepatic fat content and insulin action on free fatty acids and glucose metabolism rather than insulin absorption are associated with insulin requirements during insulin therapy in type 2 diabetic patients. Diabetes 2000, 49, 749–758. [Google Scholar]
- Stefan, N.; Kantartzis, K.; Machann, J.; Schick, F.; Thamer, C.; Rittig, K.; Balletshofer, B.; Machicao, F.; Fritsche, A.; Haring, H.U. Identification and characterization of metabolically benign obesity in humans. Arch. Intern. Med 2008, 168, 1609–1616. [Google Scholar]
- Fabbrini, E.; Magkos, F.; Mohammed, B.S.; Pietka, T.; Abumrad, N.A.; Patterson, B.W.; Okunade, A.; Klein, S. Intrahepatic fat, not visceral fat, is linked with metabolic complications of obesity. Proc. Natl. Acad. Sci. USA 2009, 106, 15430–15435. [Google Scholar]
- Gastaldelli, A.; Cusi, K.; Pettiti, M.; Hardies, J.; Miyazaki, Y.; Berria, R.; Buzzigoli, E.; Sironi, A.M.; Cersosimo, E.; Ferrannini, E.; et al. Relationship between hepatic/visceral fat and hepatic insulin resistance in nondiabetic and type 2 diabetic subjects. Gastroenterology 2007, 133, 496–506. [Google Scholar]
- Hamaguchi, M.; Kojima, T.; Takeda, N.; Nagata, C.; Takeda, J.; Sarui, H.; Kawahito, Y.; Yoshida, N.; Suetsugu, A.; Kato, T.; et al. Nonalcoholic fatty liver disease is a novel predictor of cardiovascular disease. World J. Gastroenterol 2007, 13, 1579–1584. [Google Scholar]
- Schindhelm, R.K.; Dekker, J.M.; Nijpels, G.; Bouter, L.M.; Stehouwer, C.D.; Heine, R.J.; Diamant, M. Alanine aminotransferase predicts coronary heart disease events: A 10-year follow-up of the Hoorn Study. Atherosclerosis 2007, 191, 391–396. [Google Scholar]
- Mavrogiannaki, A.N.; Migdalis, I.N. Nonalcoholic Fatty liver disease, diabetes mellitus and cardiovascular disease: Newer data. Int. J. Endocrinol 2013, 2013, 450639. [Google Scholar]
- Targher, G.; Bertolini, L.; Padovani, R.; Poli, F.; Scala, L.; Zenari, L.; Zoppini, G.; Falezza, G. Non-alcoholic fatty liver disease is associated with carotid artery wall thickness in diet-controlled type 2 diabetic patients. J. Endocrinol. Investig 2006, 29, 55–60. [Google Scholar]
- Targher, G.; Bertolini, L.; Padovani, R.; Rodella, S.; Zoppini, G.; Zenari, L.; Cigolini, M.; Falezza, G.; Arcaro, G. Relations between carotid artery wall thickness and liver histology in subjects with nonalcoholic fatty liver disease. Diabetes Care 2006, 29, 1325–1330. [Google Scholar]
- Kim, H.C.; Kim, D.J.; Huh, K.B. Association between nonalcoholic fatty liver disease and carotid intima-media thickness according to the presence of metabolic syndrome. Atherosclerosis 2009, 204, 521–525. [Google Scholar]
- Sookoian, S.; Pirola, C.J. Non-alcoholic fatty liver disease is strongly associated with carotid atherosclerosis: A systematic review. J. Hepatol 2008, 49, 600–607. [Google Scholar]
- Bonapace, S.; Perseghin, G.; Molon, G.; Canali, G.; Bertolini, L.; Zoppini, G.; Barbieri, E.; Targher, G. Nonalcoholic fatty liver disease is associated with left ventricular diastolic dysfunction in patients with type 2 diabetes. Diabetes Care 2012, 35, 389–395. [Google Scholar]
- Lautamaki, R.; Borra, R.; Iozzo, P.; Komu, M.; Lehtimaki, T.; Salmi, M.; Jalkanen, S.; Airaksinen, K.E.; Knuuti, J.; Parkkola, R.; et al. Liver steatosis coexists with myocardial insulin resistance and coronary dysfunction in patients with type 2 diabetes. Am. J. Physiol. Endocrinol. Metab 2006, 291, E282–E290. [Google Scholar]
- Rijzewijk, L.J.; Jonker, J.T.; van der Meer, R.W.; Lubberink, M.; de Jong, H.W.; Romijn, J.A.; Bax, J.J.; de Roos, A.; Heine, R.J.; Twisk, J.W.; et al. Effects of hepatic triglyceride content on myocardial metabolism in type 2 diabetes. J. Am. Coll. Cardiol 2010, 56, 225–233. [Google Scholar]
- Targher, G.; Bertolini, L.; Rodella, S.; Tessari, R.; Zenari, L.; Lippi, G.; Arcaro, G. Nonalcoholic fatty liver disease is independently associated with an increased incidence of cardiovascular events in type 2 diabetic patients. Diabetes Care 2007, 30, 2119–2121. [Google Scholar]
- Targher, G.; Bertolini, L.; Rodella, S.; Zoppini, G.; Lippi, G.; Day, C.; Muggeo, M. Non-alcoholic fatty liver disease is independently associated with an increased prevalence of chronic kidney disease and proliferative/laser-treated retinopathy in type 2 diabetic patients. Diabetologia 2008, 51, 444–450. [Google Scholar]
- Hwang, S.T.; Cho, Y.K.; Yun, J.W.; Park, J.H.; Kim, H.J.; Park, D.I.; Sohn, C.I.; Jeon, W.K.; Kim, B.I.; Rhee, E.J.; et al. Impact of non-alcoholic fatty liver disease on microalbuminuria in patients with prediabetes and diabetes. Intern. Med. J 2010, 40, 437–442. [Google Scholar]
- Targher, G.; Chonchol, M.; Bertolini, L.; Rodella, S.; Zenari, L.; Lippi, G.; Franchini, M.; Zoppini, G.; Muggeo, M. Increased risk of CKD among type 2 diabetics with nonalcoholic fatty liver disease. J. Am. Soc. Nephrol 2008, 19, 1564–1570. [Google Scholar]
- Palmer, M.; Schaffner, F. Effect of weight reduction on hepatic abnormalities in overweight patients. Gastroenterology 1990, 99, 1408–1413. [Google Scholar]
- Suzuki, A.; Lindor, K.; St Saver, J.; Lymp, J.; Mendes, F.; Muto, A.; Okada, T.; Angulo, P. Effect of changes on body weight and lifestyle in nonalcoholic fatty liver disease. J. Hepatol 2005, 43, 1060–1066. [Google Scholar]
- Harrison, S.A.; Fecht, W.; Brunt, E.M.; Neuschwander-Tetri, B.A. Orlistat for overweight subjects with nonalcoholic steatohepatitis: A randomized, prospective trial. Hepatology 2009, 49, 80–86. [Google Scholar]
- Huang, M.A.; Greenson, J.K.; Chao, C.; Anderson, L.; Peterman, D.; Jacobson, J.; Emick, D.; Lok, A.S.; Conjeevaram, H.S. One-year intense nutritional counseling results in histological improvement in patients with non-alcoholic steatohepatitis: A pilot study. Am. J. Gastroenterol 2005, 100, 1072–1081. [Google Scholar]
- Park, H.S.; Kim, M.W.; Shin, E.S. Effect of weight control on hepatic abnormalities in obese patients with fatty liver. J. Korean Med. Sci 1995, 10, 414–421. [Google Scholar]
- Hickman, I.J.; Jonsson, J.R.; Prins, J.B.; Ash, S.; Purdie, D.M.; Clouston, A.D.; Powell, E.E. Modest weight loss and physical activity in overweight patients with chronic liver disease results in sustained improvements in alanine aminotransferase, fasting insulin, and quality of life. Gut 2004, 53, 413–419. [Google Scholar]
- Clark, J.M. Weight loss as a treatment for nonalcoholic fatty liver disease. J. Clin. Gastroenterol 2006, 40, S39–S43. [Google Scholar]
- Zelber-Sagi, S.; Nitzan-Kaluski, D.; Goldsmith, R.; Webb, M.; Blendis, L.; Halpern, Z.; Oren, R. Long term nutritional intake and the risk for non-alcoholic fatty liver disease (NAFLD): A population based study. J. Hepatol 2007, 47, 711–717. [Google Scholar]
- Zelber-Sagi, S.; Ratziu, V.; Oren, R. Nutrition and physical activity in NAFLD: An overview of the epidemiological evidence. World J. Gastroenterol 2011, 17, 3377–3389. [Google Scholar]
- Buechler, C.; Wanninger, J.; Neumeier, M. Adiponectin, a key adipokine in obesity related liver diseases. World J. Gastroenterol 2011, 17, 2801–2811. [Google Scholar]
- Chalasani, N.; Younossi, Z.; Lavine, J.E.; Diehl, A.M.; Brunt, E.M.; Cusi, K.; Charlton, M.; Sanyal, A.J. The diagnosis and management of non-alcoholic fatty liver disease: Practice guideline by the American Gastroenterological Association, American Association for the Study of Liver Diseases, and American College of Gastroenterology. Gastroenterology 2012, 142, 1592–1609. [Google Scholar]
- Van Wagner, L.B.; Rinella, M.E. The role of insulin-sensitizing agents in the treatment of nonalcoholic steatohepatitis. Ther. Adv. Gastroenterol 2011, 4, 249–263. [Google Scholar]
- Oakley, F.; Teoh, V.; Ching, A.S.G.; Bataller, R.; Colmenero, J.; Jonsson, J.R.; Eliopoulos, A.G.; Watson, M.R.; Manas, D.; Mann, D.A. Angiotensin II activates I kappaB kinase phosphorylation of RelA at Ser 536 to promote myofibroblast survival and liver fibrosis. Gastroenterology 2009, 136, 2334–2344. [Google Scholar]
- Kammoun, H.L.; Chabanon, H.; Hainault, I.; Luquet, S.; Magnan, C.; Koike, T.; Ferre, P.; Foufelle, F. GRP78 expression inhibits insulin and ER stress-induced SREBP-1c activation and reduces hepatic steatosis in mice. J. Clin. Investig 2009, 119, 1201–1215. [Google Scholar]
- Albanis, E.; Friedman, S.L. Antifibrotic agents for liver disease. Am. J. Transplant 2006, 6, 12–19. [Google Scholar]
- Bailey, C.J.; Turner, R.C. Metformin. N. Engl. J. Med 1996, 334, 574–579. [Google Scholar]
- El-Mir, M.Y.; Nogueira, V.; Fontaine, E.; Averet, N.; Rigoulet, M.; Leverve, X. Dimethylbiguanide inhibits cell respiration via an indirect effect targeted on the respiratory chain complex I. J. Biol. Chem 2000, 275, 223–228. [Google Scholar]
- Owen, M.R.; Doran, E.; Halestrap, A.P. Evidence that metformin exerts its anti-diabetic effects through inhibition of complex 1 of the mitochondrial respiratory chain. Biochem. J 2000, 348, 607–614. [Google Scholar]
- Mazza, A.; Fruci, B.; Garinis, G.A.; Giuliano, S.; Malaguarnera, R.; Belfiore, A. The role of metformin in the management of NAFLD. Exp. Diabetes Res 2012, 2012, 716404. [Google Scholar]
- Lin, H.Z.; Yang, S.Q.; Chuckaree, C.; Kuhajda, F.; Ronnet, G.; Diehl, A.M. Metformin reverses fatty liver disease in obese, leptin-deficient mice. Nat. Med 2000, 6, 998–1003. [Google Scholar]
- Kita, Y.; Takamura, T.; Misu, H.; Ota, T.; Kurita, S.; Takeshita, Y.; Uno, M.; Matsuzawa-Nagata, N.; Kato, K.; Ando, H.; et al. Metformin prevents and reverses inflammation in a non-diabetic mouse model of nonalcoholic steatohepatitis. PLoS One 2012, 7, e43056. [Google Scholar]
- Garinis, G.A.; Fruci, B.; Mazza, A.; de Siena, M.; Abenavoli, S.; Gulletta, E.; Ventura, V.; Greco, M.; Abenavoli, L.; Belfiore, A. Metformin versus dietary treatment in nonalcoholic hepatic steatosis: A randomized study. Int. J. Obes 2010, 34, 1255–1264. [Google Scholar]
- Loomba, R.; Lutchman, G.; Kleiner, D.E.; Ricks, M.; Feld, J.J.; Borg, B.B.; Modi, A.; Nagabhyru, P.; Sumner, A.E.; Liang, T.J.; et al. Clinical trial: Pilot study of metformin for the treatment of non-alcoholic steatohepatitis. Aliment. Pharmacol. Ther 2009, 29, 172–182. [Google Scholar]
- Marchesini, G.; Brizi, M.; Bianchi, G.; Tomassetti, S.; Zoli, M.; Melchionda, N. Metformin in non-alcoholic steatohepatitis. Lancet 2001, 358, 893–894. [Google Scholar]
- Nair, S.; Diehl, A.M.; Wiseman, M.; Farr, G.H., Jr.; Perrillo, R.P. Metformin in the treatment of non-alcoholic steatohepatitis: A pilot open label trial. Aliment. Pharmacol. Ther 2004, 20, 23–28. [Google Scholar]
- Uygun, A.; Kadayifci, A.; Isik, A.T.; Ozgurtas, T.; Deveci, S.; Tuzun, A.; Yesilova, Z.; Gulsen, M.; Dagalp, K. Metformin in the treatment of patients with non-alcoholic steatohepatitis. Aliment. Pharmacol. Ther 2004, 19, 537–544. [Google Scholar]
- Bugianesi, E.; Gentilcore, E.; Manini, R.; Natale, S.; Vanni, E.; Villanova, N.; David, E.; Rizzetto, M.; Marchesini, G. A randomized controlled trial of metformin versus vitamin E or prescriptive diet in nonalcoholic fatty liver disease. Am. J. Gastroenterol 2005, 100, 1082–1090. [Google Scholar]
- De Oliveira, C.P.; Stefano, J.T.; de Siqueira, E.R.; Silva, L.S.; de Campos Mazo, D.F.; Lima, V.M.; Furuya, C.K.; Mello, E.S.; Souza, F.G.; Rabello, F.; et al. Combination of N-acetylcysteine and metformin improves histological steatosis and fibrosis in patients with non-alcoholic steatohepatitis. Hepatol. Res 2008, 38, 159–165. [Google Scholar]
- Idilman, R.; Mizrak, D.; Corapcioglu, D.; Bektas, M.; Doganay, B.; Sayki, M.; Coban, S.; Erden, E.; Soykan, I.; Emral, R.; et al. Clinical trial: Insulin-sensitizing agents may reduce consequences of insulin resistance in individuals with non-alcoholic steatohepatitis. Aliment. Pharmacol. Ther 2008, 28, 200–208. [Google Scholar]
- Haukeland, J.W.; Konopski, Z.; Eggesbo, H.B.; von Volkmann, H.L.; Raschpichler, G.; Bjoro, K.; Haaland, T.; Loberg, E.M.; Birkeland, K. Metformin in patients with non-alcoholic fatty liver disease: A randomized, controlled trial. Scand. J. Gastroenterol 2009, 44, 853–860. [Google Scholar]
- Shields, W.W.; Thompson, K.E.; Grice, G.A.; Harrison, S.A.; Coyle, W.J. The Effect of Metformin and Standard Therapy versus Standard Therapy alone in Nondiabetic Patients with Insulin Resistance and Nonalcoholic Steatohepatitis (NASH): A Pilot Trial. Ther. Adv. Gastroenterol 2009, 2, 157–163. [Google Scholar]
- Nar, A.; Gedik, O. The effect of metformin on leptin in obese patients with type 2 diabetes mellitus and nonalcoholic fatty liver disease. Acta Diabetol 2009, 46, 113–118. [Google Scholar]
- Lavine, J.E.; Schwimmer, J.B.; Molleston, J.P.; Scheimann, A.O.; Murray, K.F.; Abrams, S.H.; Rosenthal, P.; Sanyal, A.J.; Robuck, P.R.; Brunt, E.M.; et al. Treatment of nonalcoholic fatty liver disease in children: TONIC trial design. Contemp. Clin. Trials 2010, 31, 62–70. [Google Scholar]
- Musso, G.; Cassader, M.; Rosina, F.; Gambino, R. Impact of current treatments on liver disease, glucose metabolism and cardiovascular risk in non-alcoholic fatty liver disease (NAFLD): A systematic review and meta-analysis of randomised trials. Diabetologia 2012, 55, 885–904. [Google Scholar]
- Schwimmer, J.B.; Behling, C.; Newbury, R.; Deutsch, R.; Nievergelt, C.; Schork, N.J.; Lavine, J.E. Histopathology of pediatric nonalcoholic fatty liver disease. Hepatology 2005, 42, 641–649. [Google Scholar]
- Nobili, V.; Marcellini, M.; Devito, R.; Ciampalini, P.; Piemonte, F.; Comparcola, D.; Sartorelli, M.R.; Angulo, P. NAFLD in children: A prospective clinical-pathological study and effect of lifestyle advice. Hepatology 2006, 44, 458–465. [Google Scholar]
- Nadeau, K.J.; Ehlers, L.B.; Zeitler, P.S.; Love-Osborne, K. Treatment of non-alcoholic fatty liver disease with metformin versus lifestyle intervention in insulin-resistant adolescents. Pediatr. Diabetes 2009, 10, 5–13. [Google Scholar]
- Kintscher, U.; Law, R.E. PPARgamma-mediated insulin sensitization: The importance of fat versus muscle. Am. J. Physiol. Endocrinol. Metab 2005, 288, E287–E291. [Google Scholar]
- Bourron, O.; Daval, M.; Hainault, I.; Hajduch, E.; Servant, J.M.; Gautier, J.F.; Ferre, P.; Foufelle, F. Biguanides and thiazolidinediones inhibit stimulated lipolysis in human adipocytes through activation of AMP-activated protein kinase. Diabetologia 2010, 53, 768–778. [Google Scholar]
- Berthiaume, M.; Laplante, M.; Festuccia, W.T.; Berger, J.P.; Thieringer, R.; Deshaies, Y. Additive action of 11beta-HSD1 inhibition and PPAR-gamma agonism on hepatic steatosis and triglyceridemia in diet-induced obese rats. Int. J. Obes 2009, 33, 601–604. [Google Scholar]
- Yang, S.J.; Choi, J.M.; Chae, S.W.; Kim, W.J.; Park, S.E.; Rhee, E.J.; Lee, W.Y.; Oh, K.W.; Park, S.W.; Kim, S.W.; et al. Activation of peroxisome proliferator-activated receptor gamma by rosiglitazone increases sirt6 expression and ameliorates hepatic steatosis in rats. PLoS One 2011, 6, e17057. [Google Scholar]
- Zhang, W.; Wu, R.; Zhang, F.; Xu, Y.; Liu, B.; Yang, Y.; Zhou, H.; Wang, L.; Wan, K.; Xiao, X.; et al. Thiazolidinediones improve hepatic fibrosis in rats with non-alcoholic steatohepatitis by activating the adenosine monophosphate-activated protein kinase signalling pathway. Clin. Exp. Pharmacol. Physiol 2012, 39, 1026–1033. [Google Scholar]
- Aoyama, T.; Ikejima, K.; Kon, K.; Okumura, K.; Arai, K.; Watanabe, S. Pioglitazone promotes survival and prevents hepatic regeneration failure after partial hepatectomy in obese and diabetic KK-A(y) mice. Hepatology 2009, 49, 1636–1644. [Google Scholar]
- Nan, Y.M.; Han, F.; Kong, L.B.; Zhao, S.X.; Wang, R.Q.; Wu, W.J.; Yu, J. Adenovirus-mediated peroxisome proliferator activated receptor gamma overexpression prevents nutritional fibrotic steatohepatitis in mice. Scand. J. Gastroenterol 2011, 46, 358–369. [Google Scholar]
- Wang, C.H.; Leung, C.H.; Liu, S.C.; Chung, C.H. Safety and effectiveness of rosiglitazone in type 2 diabetes patients with nonalcoholic Fatty liver disease. J. Formos. Med. Assoc 2006, 105, 743–752. [Google Scholar]
- Akyuz, F.; Demir, K.; Ozdil, S.; Aksoy, N.; Poturoglu, S.; Ibrisim, D.; Kaymakoglu, S.; Besisik, F.; Boztas, G.; Cakaloglu, Y.; et al. The effects of rosiglitazone, metformin, and diet with exercise in nonalcoholic fatty liver disease. Dig. Dis. Sci 2007, 52, 2359–2367. [Google Scholar]
- Belfort, R.; Harrison, S.A.; Brown, K.; Darland, C.; Finch, J.; Hardies, J.; Balas, B.; Gastaldelli, A.; Tio, F.; Pulcini, J.; et al. A placebo-controlled trial of pioglitazone in subjects with nonalcoholic steatohepatitis. N. Engl. J. Med 2006, 355, 2297–2307. [Google Scholar]
- Aithal, G.P.; Thomas, J.A.; Kaye, P.V.; Lawson, A.; Ryder, S.D.; Spendlove, I.; Austin, A.S.; Freeman, J.G.; Morgan, L.; Webber, J. Randomized, placebo-controlled trial of pioglitazone in nondiabetic subjects with nonalcoholic steatohepatitis. Gastroenterology 2008, 135, 1176–1184. [Google Scholar]
- Ratziu, V.; Giral, P.; Jacqueminet, S.; Charlotte, F.; Hartemann-Heurtier, A.; Serfaty, L.; Podevin, P.; Lacorte, J.M.; Bernhardt, C.; Bruckert, E.; et al. Rosiglitazone for nonalcoholic steatohepatitis: One-year results of the randomized placebo-controlled Fatty Liver Improvement with Rosiglitazone Therapy (FLIRT) Trial. Gastroenterology 2008, 135, 100–110. [Google Scholar]
- Omer, Z.; Cetinkalp, S.; Akyildiz, M.; Yilmaz, F.; Batur, Y.; Yilmaz, C.; Akarca, U. Efficacy of insulin-sensitizing agents in nonalcoholic fatty liver disease. Eur. J. Gastroenterol. Hepatol 2010, 22, 18–23. [Google Scholar]
- Ratziu, V.; Charlotte, F.; Bernhardt, C.; Giral, P.; Halbron, M.; Lenaour, G.; Hartmann-Heurtier, A.; Bruckert, E.; Poynard, T.; Group, L.S. Long-term efficacy of rosiglitazone in nonalcoholic steatohepatitis: Results of the fatty liver improvement by rosiglitazone therapy (FLIRT 2) extension trial. Hepatology 2010, 51, 445–453. [Google Scholar]
- Sanyal, A.J.; Chalasani, N.; Kowdley, K.V.; McCullough, A.; Diehl, A.M.; Bass, N.M.; Neuschwander-Tetri, B.A.; Lavine, J.E.; Tonascia, J.; Unalp, A.; et al. Pioglitazone, vitamin E, or placebo for nonalcoholic steatohepatitis. N. Engl. J. Med 2010, 362, 1675–1685. [Google Scholar]
- Promrat, K.; Lutchman, G.; Uwaifo, G.I.; Freedman, R.J.; Soza, A.; Heller, T.; Doo, E.; Ghany, M.; Premkumar, A.; Park, Y.; et al. A pilot study of pioglitazone treatment for nonalcoholic steatohepatitis. Hepatology 2004, 39, 188–196. [Google Scholar]
- Kleiner, D.E.; Brunt, E.M.; van Natta, M.; Behling, C.; Contos, M.J.; Cummings, O.W.; Ferrell, L.D.; Liu, Y.C.; Torbenson, M.S.; Unalp-Arida, A.; et al. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology 2005, 41, 1313–1321. [Google Scholar]
- Musso, G.; Gambino, R.; Cassader, M.; Pagano, G. A meta-analysis of randomized trials for the treatment of nonalcoholic fatty liver disease. Hepatology 2010, 52, 79–104. [Google Scholar]
- Rakoski, M.O.; Singal, A.G.; Rogers, M.A.; Conjeevaram, H. Meta-analysis: Insulin sensitizers for the treatment of non-alcoholic steatohepatitis. Aliment. Pharmacol. Ther 2010, 32, 1211–1221. [Google Scholar]
- Boettcher, E.; Csako, G.; Pucino, F.; Wesley, R.; Loomba, R. Meta-analysis: Pioglitazone improves liver histology and fibrosis in patients with non-alcoholic steatohepatitis. Aliment. Pharmacol. Ther 2012, 35, 66–75. [Google Scholar]
- Colmers, I.N.; Bowker, S.L.; Majumdar, S.R.; Johnson, J.A. Use of thiazolidinediones and the risk of bladder cancer among people with type 2 diabetes: A meta-analysis. CMAJ 2012, 184, E675–E683. [Google Scholar]
- Berger, J.P.; Petro, A.E.; Macnaul, K.L.; Kelly, L.J.; Zhang, B.B.; Richards, K.; Elbrecht, A.; Johnson, B.A.; Zhou, G.; Doebber, T.W.; et al. Distinct properties and advantages of a novel peroxisome proliferator-activated protein [gamma] selective modulator. Mol. Endocrinol 2003, 17, 662–676. [Google Scholar]
- Holst, J.J.; Gromada, J. Role of incretin hormones in the regulation of insulin secretion in diabetic and nondiabetic humans. Am. J. Physiol. Endocrinol. Metab 2004, 287, E199–E206. [Google Scholar]
- Baggio, L.L.; Drucker, D.J. Biology of incretins: GLP-1 and GIP. Gastroenterology 2007, 132, 2131–2157. [Google Scholar]
- Kim, W.; Egan, J.M. The role of incretins in glucose homeostasis and diabetes treatment. Pharmacol. Rev 2008, 60, 470–512. [Google Scholar]
- DeFronzo, R.A.; Ratner, R.E.; Han, J.; Kim, D.D.; Fineman, M.S.; Baron, A.D. Effects of exenatide (exendin-4) on glycemic control and weight over 30 weeks in metformin-treated patients with type 2 diabetes. Diabetes Care 2005, 28, 1092–1100. [Google Scholar]
- MacDonald, P.E.; El-Kholy, W.; Riedel, M.J.; Salapatek, A.M.; Light, P.E.; Wheeler, M.B. The multiple actions of GLP-1 on the process of glucose-stimulated insulin secretion. Diabetes 2002, 51, S434–S442. [Google Scholar]
- Ben-Shlomo, S.; Zvibel, I.; Shnell, M.; Shlomai, A.; Chepurko, E.; Halpern, Z.; Barzilai, N.; Oren, R.; Fishman, S. Glucagon-like peptide-1 reduces hepatic lipogenesis via activation of AMP-activated protein kinase. J. Hepatol 2011, 54, 1214–1223. [Google Scholar]
- Ahren, B.; Schmitz, O. GLP-1 receptor agonists and DPP-4 inhibitors in the treatment of type 2 diabetes. Horm. Metab. Res 2004, 36, 867–876. [Google Scholar]
- Ding, X.; Saxena, N.K.; Lin, S.; Gupta, N.A.; Anania, F.A. Exendin-4, a glucagon-like protein-1 (GLP-1) receptor agonist, reverses hepatic steatosis in ob/ob mice. Hepatology 2006, 43, 173–181. [Google Scholar]
- Parlevliet, E.T.; Wang, Y.; Geerling, J.J.; Schroder-van der Elst, J.P.; Picha, K.; O’Neil, K.; Stojanovic-Susulic, V.; Ort, T.; Havekes, L.M.; Romijn, J.A.; et al. GLP-1 receptor activation inhibits VLDL production and reverses hepatic steatosis by decreasing hepatic lipogenesis in high-fat-fed APOE*3-Leiden mice. PLoS One 2012, 7, e49152. [Google Scholar]
- Lee, Y.S.; Park, M.S.; Choung, J.S.; Kim, S.S.; Oh, H.H.; Choi, C.S.; Ha, S.Y.; Kang, Y.; Kim, Y.; Jun, H.S. Glucagon-like peptide-1 inhibits adipose tissue macrophage infiltration and inflammation in an obese mouse model of diabetes. Diabetologia 2012, 55, 2456–2468. [Google Scholar]
- Trevaskis, J.L.; Griffin, P.S.; Wittmer, C.; Neuschwander-Tetri, B.A.; Brunt, E.M.; Dolman, C.S.; Erickson, M.R.; Napora, J.; Parkes, D.G.; Roth, J.D. Glucagon-like peptide-1 receptor agonism improves metabolic, biochemical, and histopathological indices of nonalcoholic steatohepatitis in mice. Am. J. Physiol. Gastrointest. Liver Physiol 2012, 302, G762–G772. [Google Scholar]
- Gupta, N.A.; Mells, J.; Dunham, R.M.; Grakoui, A.; Handy, J.; Saxena, N.K.; Anania, F.A. Glucagon-like peptide-1 receptor is present on human hepatocytes and has a direct role in decreasing hepatic steatosis in vitro by modulating elements of the insulin signaling pathway. Hepatology 2010, 51, 1584–1592. [Google Scholar]
- Sharma, S.; Mells, J.E.; Fu, P.P.; Saxena, N.K.; Anania, F.A. GLP-1 analogs reduce hepatocyte steatosis and improve survival by enhancing the unfolded protein response and promoting macroautophagy. PLoS One 2011, 6, e25269. [Google Scholar]
- Tushuizen, M.E.; Bunck, M.C.; Pouwels, P.J.; van Waesberghe, J.H.; Diamant, M.; Heine, R.J. Incretin mimetics as a novel therapeutic option for hepatic steatosis. Liver Int 2006, 26, 1015–1017. [Google Scholar]
- Klonoff, D.C.; Buse, J.B.; Nielsen, L.L.; Guan, X.; Bowlus, C.L.; Holcombe, J.H.; Wintle, M.E.; Maggs, D.G. Exenatide effects on diabetes, obesity, cardiovascular risk factors and hepatic biomarkers in patients with type 2 diabetes treated for at least 3 years. Curr. Med. Res. Opin 2008, 24, 275–286. [Google Scholar]
- Kenny, P.R.; Brady, D.E.; Torres, D.M.; Ragozzino, L.; Chalasani, N.; Harrison, S.A. Exenatide in the treatment of diabetic patients with non-alcoholic steatohepatitis: A case series. Am. J. Gastroenterol 2010, 105, 2707–2709. [Google Scholar]
- Armstrong, M.J.; Houlihan, D.D.; Rowe, I.A.; Clausen, W.H.; Elbrond, B.; Gough, S.C.; Tomlinson, J.W.; Newsome, P.N. Safety and efficacy of liraglutide in patients with type 2 diabetes and elevated liver enzymes: Individual patient data meta-analysis of the LEAD program. Aliment. Pharmacol. Ther 2013, 37, 234–242. [Google Scholar]
- Barnett, A. DPP-4 inhibitors and their potential role in the management of type 2 diabetes. Int. J. Clin. Pract 2006, 60, 1454–1470. [Google Scholar]
- Itou, M.; Kawaguchi, T.; Taniguchi, E.; Oriishi, T.; Sata, M. Dipeptidyl peptidase IV inhibitor improves insulin resistance and steatosis in a refractory nonalcoholic fatty liver disease patient: A case report. Case Rep. Gastroenterol 2012, 6, 538–544. [Google Scholar]
- McIntosh, C.H.; Demuth, H.U.; Pospisilik, J.A.; Pederson, R. Dipeptidyl peptidase IV inhibitors: How do they work as new antidiabetic agents? Regul. Pept 2005, 128, 159–165. [Google Scholar]
- Miyazaki, M.; Kato, M.; Tanaka, K.; Tanaka, M.; Kohjima, M.; Nakamura, K.; Enjoji, M.; Nakamuta, M.; Kotoh, K.; Takayanagi, R. Increased hepatic expression of dipeptidyl peptidase-4 in non-alcoholic fatty liver disease and its association with insulin resistance and glucose metabolism. Mol. Med. Rep 2012, 5, 729–733. [Google Scholar]
- Balaban, Y.H.; Korkusuz, P.; Simsek, H.; Gokcan, H.; Gedikoglu, G.; Pinar, A.; Hascelik, G.; Asan, E.; Hamaloglu, E.; Tatar, G. Dipeptidyl peptidase IV (DDP IV) in NASH patients. Ann. Hepatol 2007, 6, 242–250. [Google Scholar]
- Shirakawa, J.; Fujii, H.; Ohnuma, K.; Sato, K.; Ito, Y.; Kaji, M.; Sakamoto, E.; Koganei, M.; Sasaki, H.; Nagashima, Y.; et al. Diet-induced adipose tissue inflammation and liver steatosis are prevented by DPP-4 inhibition in diabetic mice. Diabetes 2011, 60, 1246–1257. [Google Scholar]
- Kern, M.; Kloting, N.; Niessen, H.G.; Thomas, L.; Stiller, D.; Mark, M.; Klein, T.; Bluher, M. Linagliptin improves insulin sensitivity and hepatic steatosis in diet-induced obesity. PLoS One 2012, 7, e38744. [Google Scholar]
- Klein, T.; Fujii, M.; Sandel, J.; Shibazaki, Y.; Wakamatsu, K.; Mark, M.; Yoneyama, H. Linagliptin alleviates hepatic steatosis and inflammation in a mouse model of non-alcoholic steatohepatitis. Med. Mol. Morphol 2013. [Google Scholar] [CrossRef]
- Iwasaki, T.; Yoneda, M.; Inamori, M.; Shirakawa, J.; Higurashi, T.; Maeda, S.; Terauchi, Y.; Nakajima, A. Sitagliptin as a novel treatment agent for non-alcoholic Fatty liver disease patients with type 2 diabetes mellitus. Hepatogastroenterology 2011, 58, 2103–2105. [Google Scholar]
- Yilmaz, Y.; Yonal, O.; Deyneli, O.; Celikel, C.A.; Kalayci, C.; Duman, D.G. Effects of sitagliptin in diabetic patients with nonalcoholic steatohepatitis. Acta Gastroenterol. Belg 2012, 75, 240–244. [Google Scholar]
- Blum, A.; Shamburek, R. The pleiotropic effects of statins on endothelial function, vascular inflammation, immunomodulation and thrombogenesis. Atherosclerosis 2009, 203, 325–330. [Google Scholar]
- Fraulob, J.C.; Souza-Mello, V.; Aguila, M.B.; Mandarim-de-Lacerda, C.A. Beneficial effects of rosuvastatin on insulin resistance, adiposity, inflammatory markers and non-alcoholic fatty liver disease in mice fed on a high-fat diet. Clin. Sci 2012, 123, 259–270. [Google Scholar]
- Ji, G.; Zhao, X.; Leng, L.; Liu, P.; Jiang, Z. Comparison of dietary control and atorvastatin on high fat diet induced hepatic steatosis and hyperlipidemia in rats. Lipids Health Dis 2011, 10, 23. [Google Scholar]
- Van Rooyen, D.M.; Gan, L.T.; Yeh, M.M.; Haigh, W.G.; Larter, C.Z.; Ioannou, G.; Teoh, N.C.; Farrell, G.C. Pharmacological cholesterol lowering reverses fibrotic NASH in obese, diabetic mice with metabolic syndrome. J. Hepatol 2013, 59, 144–152. [Google Scholar]
- Okada, Y.; Yamaguchi, K.; Nakajima, T.; Nishikawa, T.; Jo, M.; Mitsumoto, Y.; Kimura, H.; Nishimura, T.; Tochiki, N.; Yasui, K.; et al. Rosuvastatin ameliorates high-fat and high-cholesterol diet-induced nonalcoholic steatohepatitis in rats. Liver Int 2013, 33, 301–311. [Google Scholar]
- Gomez-Dominguez, E.; Gisbert, J.P.; Moreno-Monteagudo, J.A.; Garcia-Buey, L.; Moreno-Otero, R. A pilot study of atorvastatin treatment in dyslipemid, non-alcoholic fatty liver patients. Aliment. Pharmacol. Ther 2006, 23, 1643–1647. [Google Scholar]
- Nakahara, T.; Hyogo, H.; Kimura, Y.; Ishitobi, T.; Arihiro, K.; Aikata, H.; Takahashi, S.; Chayama, K. Efficacy of rosuvastatin for the treatment of non-alcoholic steatohepatitis with dyslipidemia: An open-label, pilot study. Hepatol. Res 2012, 42, 1065–1072. [Google Scholar]
- Foster, T.; Budoff, M.J.; Saab, S.; Ahmadi, N.; Gordon, C.; Guerci, A.D. Atorvastatin and antioxidants for the treatment of nonalcoholic fatty liver disease: The St Francis Heart Study randomized clinical trial. Am. J. Gastroenterol 2011, 106, 71–77. [Google Scholar]
- Nelson, A.; Torres, D.M.; Morgan, A.E.; Fincke, C.; Harrison, S.A. A pilot study using simvastatin in the treatment of nonalcoholic steatohepatitis: A randomized placebo-controlled trial. J. Clin. Gastroenterol 2009, 43, 990–994. [Google Scholar]
- Pramfalk, C.; Parini, P.; Gustafsson, U.; Sahlin, S.; Eriksson, M. Effects of high-dose statin on the human hepatic expression of genes involved in carbohydrate and triglyceride metabolism. J. Intern. Med 2011, 269, 333–339. [Google Scholar]
- Musso, G.; Cassader, M.; Gambino, R. Cholesterol-lowering therapy for the treatment of nonalcoholic fatty liver disease: An update. Curr. Opin. Lipidol 2011, 22, 489–496. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Fruci, B.; Giuliano, S.; Mazza, A.; Malaguarnera, R.; Belfiore, A. Nonalcoholic Fatty Liver: A Possible New Target for Type 2 Diabetes Prevention and Treatment. Int. J. Mol. Sci. 2013, 14, 22933-22966. https://doi.org/10.3390/ijms141122933
Fruci B, Giuliano S, Mazza A, Malaguarnera R, Belfiore A. Nonalcoholic Fatty Liver: A Possible New Target for Type 2 Diabetes Prevention and Treatment. International Journal of Molecular Sciences. 2013; 14(11):22933-22966. https://doi.org/10.3390/ijms141122933
Chicago/Turabian StyleFruci, Barbara, Stefania Giuliano, Angela Mazza, Roberta Malaguarnera, and Antonino Belfiore. 2013. "Nonalcoholic Fatty Liver: A Possible New Target for Type 2 Diabetes Prevention and Treatment" International Journal of Molecular Sciences 14, no. 11: 22933-22966. https://doi.org/10.3390/ijms141122933