Hepatic Adverse Effects of Fructose Consumption Independent of Overweight/Obesity

Abstract
:1. Introduction
2. Results and Discussion
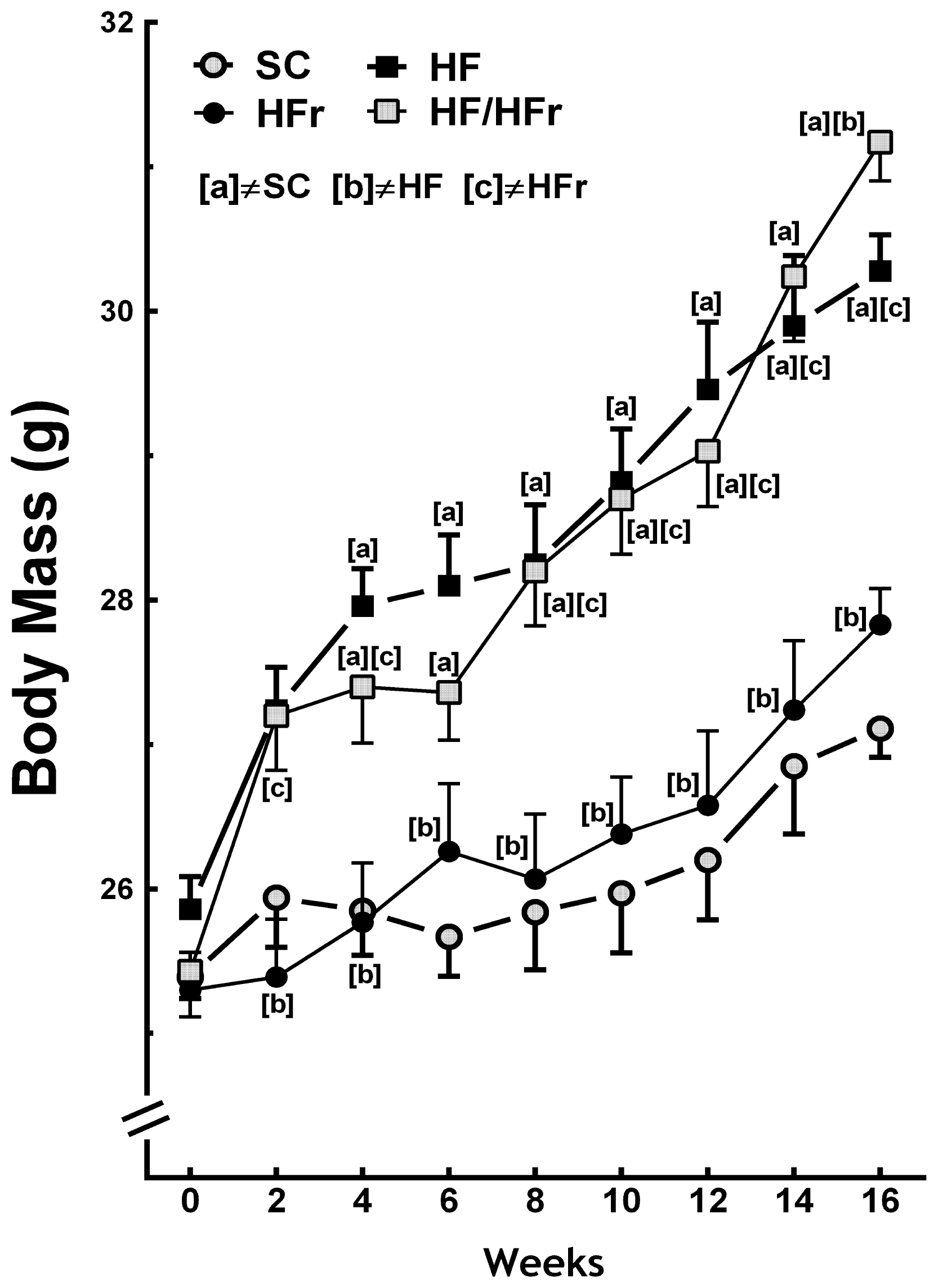
2.1. Body Mass and Intake of Food
2.2. OGTT (Oral Glucose Tolerance Test), IPITT (Intraperitoneal Insulin Tolerance Test) and Insulin
2.3. Plasma Total Cholesterol, Triglycerides and Glucose
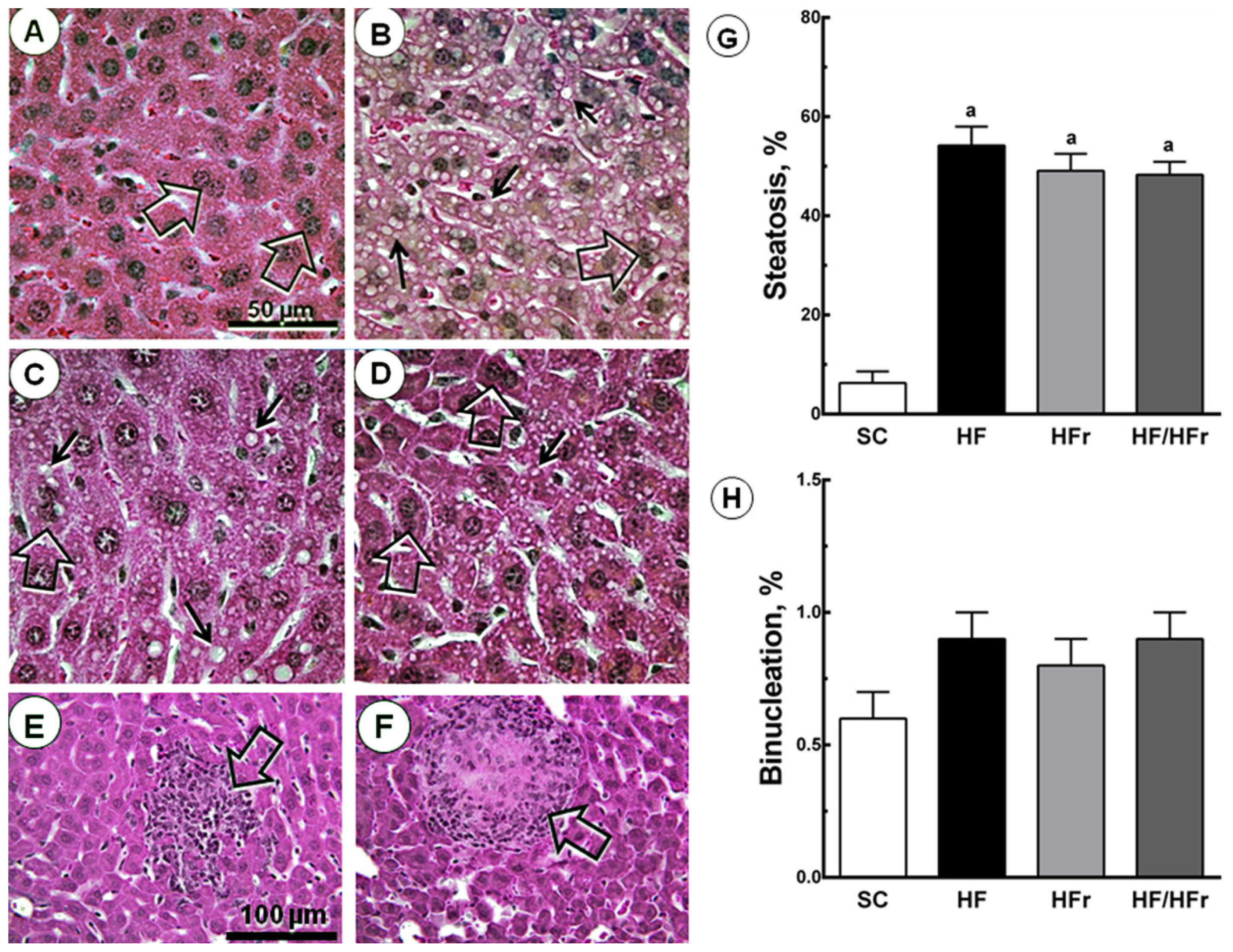
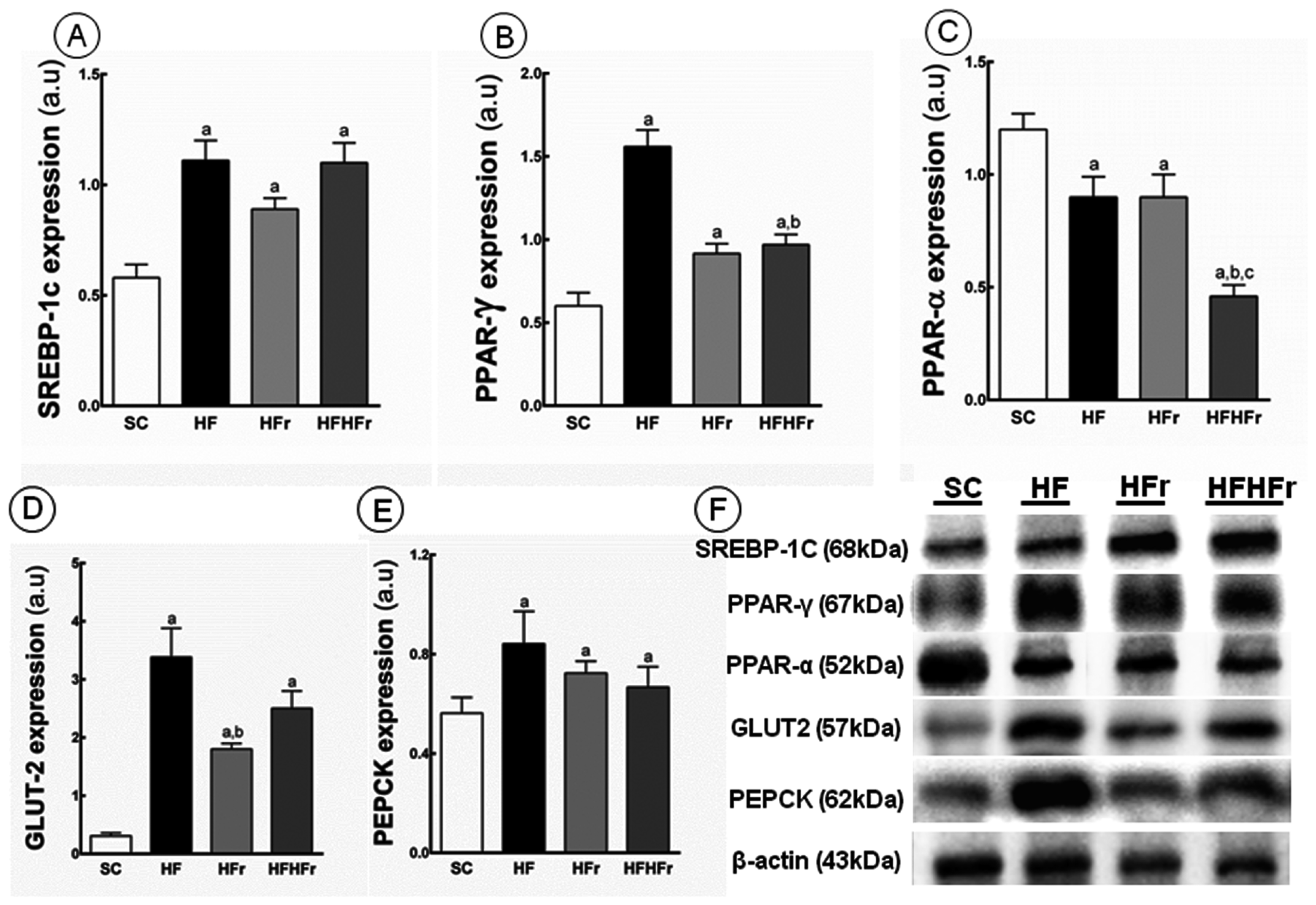
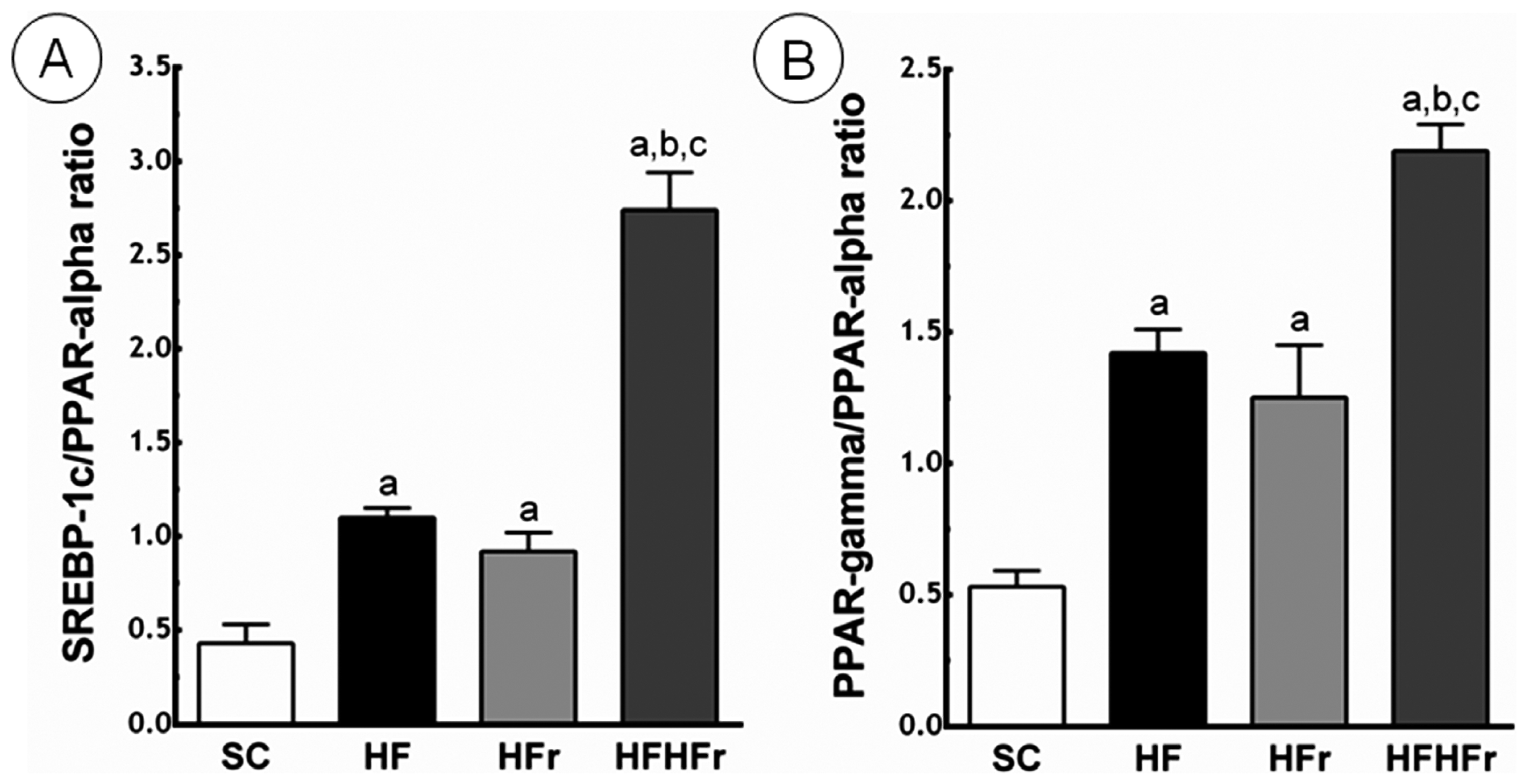
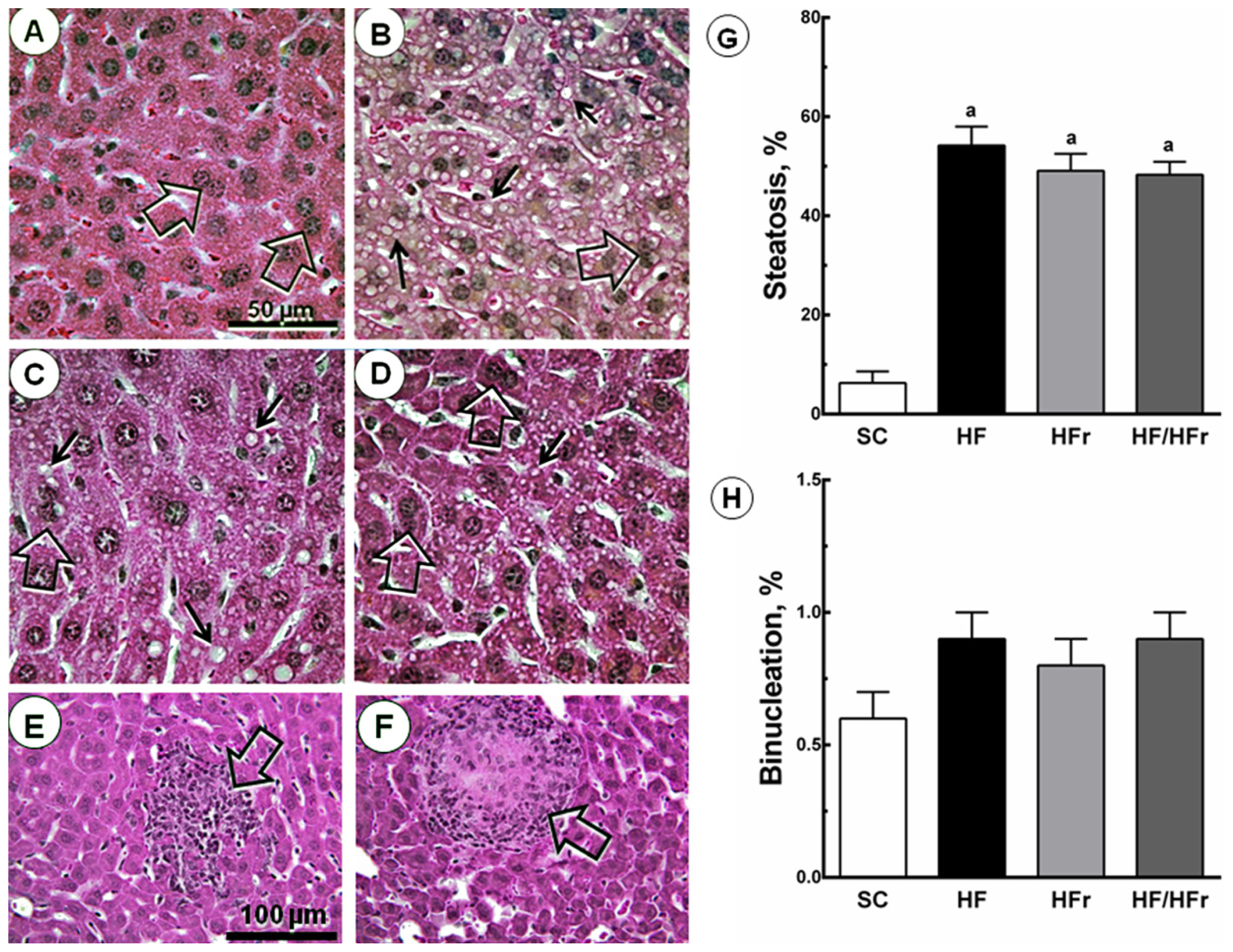
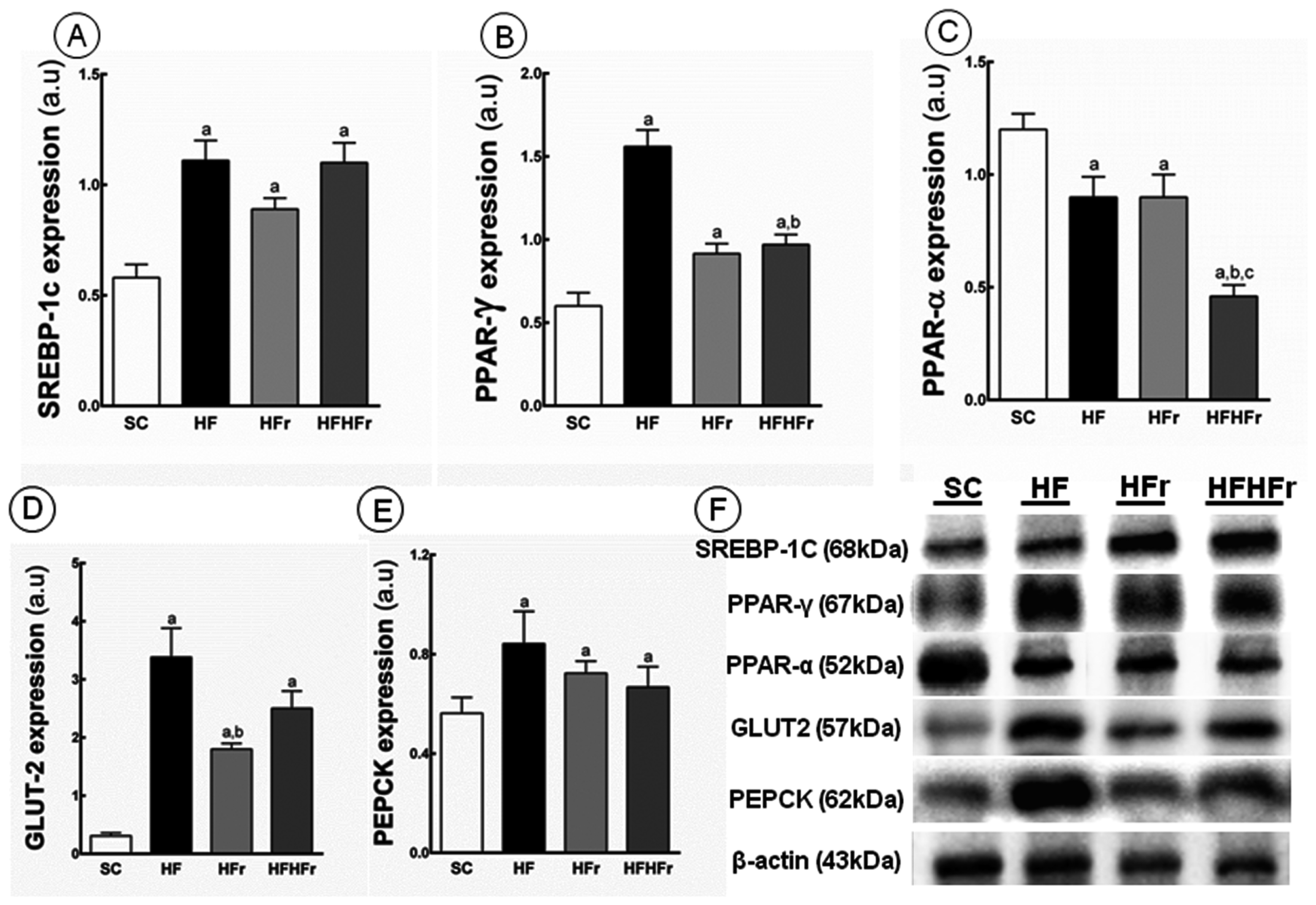
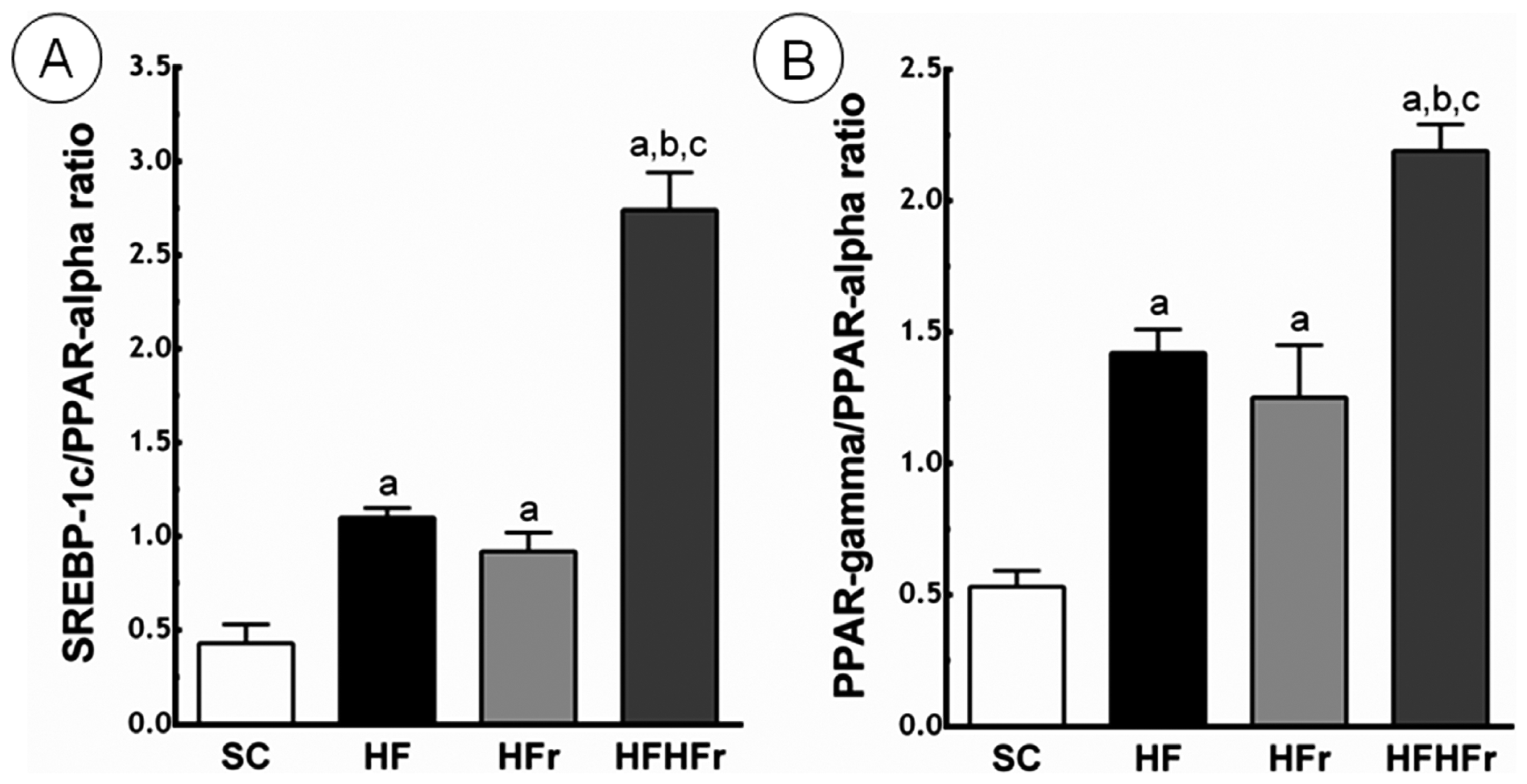
2.4. Liver
3. Experimental Section
3.1. Animal and Diets
3.2. Body Mass and Food Intake
3.3. Oral Glucose Tolerance Test (OGTT) and Intraperitoneal Insulin Tolerance Test (IPITT)
3.4. Euthanasia
3.5. Plasma Analysis
3.6. Liver
3.7. Immunoblotting
3.8. Data Analysis
4. Conclusions


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Data | Groups | |||
|---|---|---|---|---|
| SC | HF | HFr | HF/HFr | |
| Alanine aminotransferase (mg/dL) | 35.05 ± 7.81 | 81.93 ± 6.53 a | 75.20 ± 5.66 a | 70.97 ± 4.92 a |
| Energy intake (kcal/animal/week) | 12330 ± 204 | 13741 ± 348 a | 12126 ± 427 b | 13565 ± 216 a,c |
| Food Intake (g/animal/week) | 3.24 ± 0.40 | 2.92 ± 0.37 | 3.18 ± 0.56 | 2.88 ± 0.23 |
| Glucose (mmol/L) | 5.08 ± 0.70 | 12.31 ± 1.14 a | 9.98 ±0.88 a | 10.20 ± 1.04 a |
| Hepatic TG (mg/dL) | 74.8 ± 1.4 | 126.2 ± 4.6 a | 93.0 ± 1.8 a,b | 110.9 ± 6.1 a,b,c |
| HOMA-IR | 1.0 ± 0.14 | 4.1 ± 0.35 a | 3.1 ± 0.32 a | 3.1 ± 0.31 a |
| Insulin (μIU/mL) | 4.6 ± 0.11 | 7.8 ± 0.50 a | 6.9 ± 0.36 a | 6.8 ± 0.49 a |
| IPITT (AUC, a.u.) | 601.7 ± 59.9 | 817.0 ± 23.4 a | 896.9 ± 22.3 a | 829.8 ± 73.0 a |
| OGTT (AUC, a.u.) | 1086.2 ± 21.2 | 1249.2 ± 24.5 a | 1261.8 ± 42.8 a | 1214.8 ± 32.5 b,c |
| TC (mg/dL) | 74.2 ± 5.6 | 110.8 ± 5.9 a | 101.8 ± 3.8 a | 124.5 ± 8.4 a |
| TG (mg/dL) | 58.0 ± 1.4 | 66.0 ± 0.5 a | 66.8 ± 0.9 a | 66.4 ± 0.7 a |
| Nutrient (g/Kg) | Diets | |||
|---|---|---|---|---|
| SC | HF | HFr | HF/HFr | |
| Casein (≥85% protein) | 140.0 | 160.0 | 140.0 | 160.0 |
| Cornstarch | 620.7 | 420.69 | 296.8 | 20.7 |
| Sucrose | 100.0 | 100.0 | 100.0 | 100.0 |
| Fructose | – | – | 323.85 | 400.0 |
| Soybean Oil | 40.0 | 40.0 | 40.0 | 40.0 |
| Lard | – | 180.0 | – | 180.0 |
| Fiber | 50.0 | 50.0 | 50.0 | 50.0 |
| Vitamin Mix | 10.0 | 10.0 | 10.0 | 10.0 |
| Mineral Mix | 35.0 | 35.0 | 35.0 | 35.0 |
| Cystine | 1.8 | 1.8 | 1.8 | 1.8 |
| Coline | 2.5 | 2.5 | 2.5 | 2.5 |
| Antioxidant | 0.008 | 0.008 | 0.008 | 0.008 |
| Total (g) | 1.000 | 1.000 | 1.000 | 1.000 |
| Energy (kcal/Kg) | 3802.8 | 4702.8 | 3802.8 | 4702.8 |
| Carbohydrate (%) | 76 | 44 | 76 | 44 |
| Fructose (%) | – | – | 34 | 34 |
| Protein (%) | 15 | 14 | 15 | 14 |
| Lipid (%) | 9 | 42 | 9 | 42 |
Acknowledgments
Conflicts of Interest
References
- Murphy, S.P.; Johnson, R.K. The scientific basis of recent US guidance on sugars intake. Am. J. Clin. Nutr 2003, 78, 827S–833S. [Google Scholar]
- Lim, J.S.; Mietus-Snyder, M.; Valente, A.; Schwarz, J.M.; Lustig, R.H. The role of fructose in the pathogenesis of NAFLD and the metabolic syndrome. Nat. Rev. Gastroenterol. Hepatol 2010, 7, 251–264. [Google Scholar]
- Nomura, K.; Yamanouchi, T. The role of fructose-enriched diets in mechanisms of nonalcoholic fatty liver disease. J. Nutr. Biochem 2012, 23, 203–208. [Google Scholar]
- Aeberli, I.; Hochuli, M.; Gerber, P.A.; Sze, L.; Murer, S.B.; Tappy, L.; Spinas, G.A.; Berneis, K. Moderate amounts of fructose consumption impair insulin sensitivity in healthy young men: A randomized controlled trial. Diabetes Care 2013, 36, 150–156. [Google Scholar]
- Lustig, R.H.; Schmidt, L.A.; Brindis, C.D. Public health: The toxic truth about sugar. Nature 2012, 482, 27–29. [Google Scholar]
- Tsuchiya, H.; Ebata, Y.; Sakabe, T.; Hama, S.; Kogure, K.; Shiota, G. High-fat, high-fructose diet induces hepatic iron overload via a hepcidin-independent mechanism prior to the onset of liver steatosis and insulin resistance in mice. Metabolism 2013, 62, 62–69. [Google Scholar]
- Kim, C.H.; Younossi, Z.M. Nonalcoholic fatty liver disease: A manifestation of the metabolic syndrome. Clevel. Clin. J. Med 2008, 75, 721–728. [Google Scholar]
- Schultz, A.; Mendonca, L.S.; Aguila, M.B.; Mandarim-de-Lacerda, C.A. Swimming training beneficial effects in a mice model of nonalcoholic fatty liver disease. Exp. Toxicol. Pathol 2012, 64, 273–282. [Google Scholar]
- Tappy, L.; Le, K.A.; Tran, C.; Paquot, N. Fructose and metabolic diseases: New findings, new questions. Nutrition 2010, 26, 1044–1049. [Google Scholar]
- Abdelmalek, M.F.; Lazo, M.; Horska, A.; Bonekamp, S.; Lipkin, E.W.; Balasubramanyam, A.; Bantle, J.P.; Johnson, R.J.; Diehl, A.M.; Clark, J.M.; et al. Higher dietary fructose is associated with impaired hepatic adenosine triphosphate homeostasis in obese individuals with type 2 diabetes. Hepatology 2012, 56, 952–960. [Google Scholar]
- Janevski, M.; Ratnayake, S.; Siljanovski, S.; McGlynn, M.A.; Cameron-Smith, D.; Lewandowski, P. Fructose containing sugars modulate mRNA of lipogenic genes ACC and FAS and protein levels of transcription factors ChREBP and SREBP1c with no effect on body weight or liver fat. Food Funct 2012, 3, 141–149. [Google Scholar]
- Coate, K.C.; Smith, M.S.; Shiota, M.; Irimia, J.M.; Roach, P.J.; Farmer, B.; Williams, P.E.; Moore, M.C. Hepatic glucose metabolism in late pregnancy: Normal versus high-fat and -fructose diet. Diabetes 2013, 62, 753–761. [Google Scholar]
- Bezerra, R.M.; Ueno, M.; Silva, M.S.; Tavares, D.Q.; Carvalho, C.R.; Saad, M.J. A high fructose diet affects the early steps of insulin action in muscle and liver of rats. J. Nutr 2000, 130, 1531–1535. [Google Scholar]
- Delarue, J.; Normand, S.; Couet, C.; Pachiaudi, C.; Urbain, C.; Lamisse, F.; Riou, J.P. Effects of free fatty acids on the metabolic response to oral fructose in lean healthy humans. Int. J. Obes. Relat. Metab. Disord 1996, 20, 130–136. [Google Scholar]
- McGarry, J.D. Malonyl-CoA and carnitine palmitoyltransferase I: An expanding partnership. Biochem. Soc. Trans 1995, 23, 481–485. [Google Scholar]
- Kawasaki, T.; Igarashi, K.; Koeda, T.; Sugimoto, K.; Nakagawa, K.; Hayashi, S.; Yamaji, R.; Inui, H.; Fukusato, T.; Yamanouchi, T. Rats fed fructose-enriched diets have characteristics of nonalcoholic hepatic steatosis. J. Nutr 2009, 139, 2067–2071. [Google Scholar]
- Moran-Salvador, E.; Lopez-Parra, M.; Garcia-Alonso, V.; Titos, E.; Martinez-Clemente, M.; Gonzalez-Periz, A.; Lopez-Vicario, C.; Barak, Y.; Arroyo, V.; Claria, J. Role for PPAR-γ in obesity-induced hepatic steatosis as determined by hepatocyte- and macrophage-specific conditional knockouts. FASEB J 2011, 25, 2538–2550. [Google Scholar]
- Schadinger, S.E.; Bucher, N.L.; Schreiber, B.M.; Farmer, S.R. PPAR-γ2 regulates lipogenesis and lipid accumulation in steatotic hepatocytes. Am. J. Physiol. Endocrinol. Metab 2005, 288, E1195–E1205. [Google Scholar]
- Caton, P.W.; Nayuni, N.K.; Khan, N.Q.; Wood, E.G.; Corder, R. Fructose induces gluconeogenesis and lipogenesis through a SIRT1-dependent mechanism. J. Endocrinol 2011, 208, 273–283. [Google Scholar]
- Valera, A.; Pujol, A.; Pelegrin, M.; Bosch, F. Transgenic mice overexpressing phosphoenolpyruvate carboxykinase develop non-insulin-dependent diabetes mellitus. Proc. Natl. Acad. Sci. USA 1994, 91, 9151–9154. [Google Scholar]
- Guillemain, G.; Loizeau, M.; Pincon-Raymond, M.; Girard, J.; Leturque, A. The large intracytoplasmic loop of the glucose transporter GLUT2 is involved in glucose signaling in hepatic cells. J. Cell. Sci 2000, 113, 841–847. [Google Scholar]
- Okamoto, Y.; Tanaka, S.; Haga, Y. Enhanced GLUT 2 gene expression in an oleic acid-induced in vitro fatty liver model. Hepatol. Res 2002, 23, 138–144. [Google Scholar]
- Yamamoto, T.; Fukumoto, H.; Koh, G.; Yano, H.; Yasuda, K.; Masuda, K.; Ikeda, H.; Imura, H.; Seino, Y. Liver and muscle-fat type glucose transporter gene expression in obese and diabetic rats. Biochem. Biophys. Res. Commun 1991, 175, 995–1002. [Google Scholar]
- Alisi, A.; Manco, M.; Pezzullo, M.; Nobili, V. Fructose at the center of necroinflammation and fibrosis in nonalcoholic steatohepatitis. Hepatology 2011, 53, 372–373. [Google Scholar]
- Francini, F.; Castro, M.C.; Schinella, G.; Garcia, M.E.; Maiztegui, B.; Raschia, M.A.; Gagliardino, J.J.; Massa, M.L. Changes induced by a fructose-rich diet on hepatic metabolism and the antioxidant system. Life Sci 2010, 86, 965–971. [Google Scholar]
- Farrell, G.C.; Larter, C.Z. Nonalcoholic fatty liver disease: From steatosis to cirrhosis. Hepatology 2006, 43, S99–S112. [Google Scholar]
- Yoshiike, N.; Lwin, H. Epidemiological aspects of obesity and NASH/NAFLD in Japan. Hepatol. Res 2005, 33, 77–82. [Google Scholar]
- Malhi, H.; Gores, G.J. Molecular mechanisms of lipotoxicity in nonalcoholic fatty liver disease. Semin. Liver Dis 2008, 28, 360–369. [Google Scholar]
- Day, C.P. Pathogenesis of steatohepatitis. Best Pract. Res. Clin. Gastroenterol 2002, 16, 663–678. [Google Scholar]
- Day, C.P.; James, O.F. Steatohepatitis: A tale of two “hits”? Gastroenterology 1998, 114, 842–845. [Google Scholar]
- Kohli, R.; Kirby, M.; Xanthakos, S.A.; Softic, S.; Feldstein, A.E.; Saxena, V.; Tang, P.H.; Miles, L.; Miles, M.V.; Balistreri, W.F.; et al. High-fructose, medium chain trans fat diet induces liver fibrosis and elevates plasma coenzyme Q9 in a novel murine model of obesity and nonalcoholic steatohepatitis. Hepatology 2010, 52, 934–944. [Google Scholar]
- Takahashi, Y.; Soejima, Y.; Fukusato, T. Animal models of nonalcoholic fatty liver disease/nonalcoholic steatohepatitis. World J. Gastroenterol 2012, 18, 2300–2308. [Google Scholar]
- Institute of Laboratory Animal Resources (US), Guide for the Care and Use of Laboratory Animals; National Academy Press: Washington, DC, USA, 1996.
- Reeves, P.G.; Nielsen, F.H.; Fahey, G.C., Jr. AIN-93 purified diets for laboratory rodents: Final report of the American Institute of Nutrition ad hoc writing committee on the reformulation of the AIN-76A rodent diet. J. Nutr 1993, 123, 1939–1951. [Google Scholar]
- Matthews, D.R.; Hosker, J.P.; Rudenski, A.S.; Naylor, B.A.; Treacher, D.F.; Turner, R.C. Homeostasis model assessment: Insulin resistance and β-cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia 1985, 28, 412–419. [Google Scholar]
- Catta-Preta, M.; Mendonca, L.S.; Fraulob-Aquino, J.; Aguila, M.B.; Mandarim-de-Lacerda, C.A. A critical analysis of three quantitative methods of assessment of hepatic steatosis in liver biopsies. Virchows Arch 2011, 459, 477–485. [Google Scholar]
- Aguila, M.B.; Pinheiro, A.R.; Parente, L.B.; Mandarim-de-Lacerda, C.A. Dietary effect of different high-fat diet on rat liver stereology. Liver Int 2003, 23, 363–370. [Google Scholar]
- Santos, C.F.; Souza-Mello, V.; Faria, T.S.; Mandarim-de-Lacerda, C.A. Quantitative morphology update: Image analysis. Int. J. Morphol 2013, 31, 23–30. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Schultz, A.; Neil, D.; Aguila, M.B.; Mandarim-de-Lacerda, C.A. Hepatic Adverse Effects of Fructose Consumption Independent of Overweight/Obesity. Int. J. Mol. Sci. 2013, 14, 21873-21886. https://doi.org/10.3390/ijms141121873
Schultz A, Neil D, Aguila MB, Mandarim-de-Lacerda CA. Hepatic Adverse Effects of Fructose Consumption Independent of Overweight/Obesity. International Journal of Molecular Sciences. 2013; 14(11):21873-21886. https://doi.org/10.3390/ijms141121873
Chicago/Turabian StyleSchultz, Alini, Debora Neil, Marcia B. Aguila, and Carlos A. Mandarim-de-Lacerda. 2013. "Hepatic Adverse Effects of Fructose Consumption Independent of Overweight/Obesity" International Journal of Molecular Sciences 14, no. 11: 21873-21886. https://doi.org/10.3390/ijms141121873