Cancer Development, Progression, and Therapy: An Epigenetic Overview


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
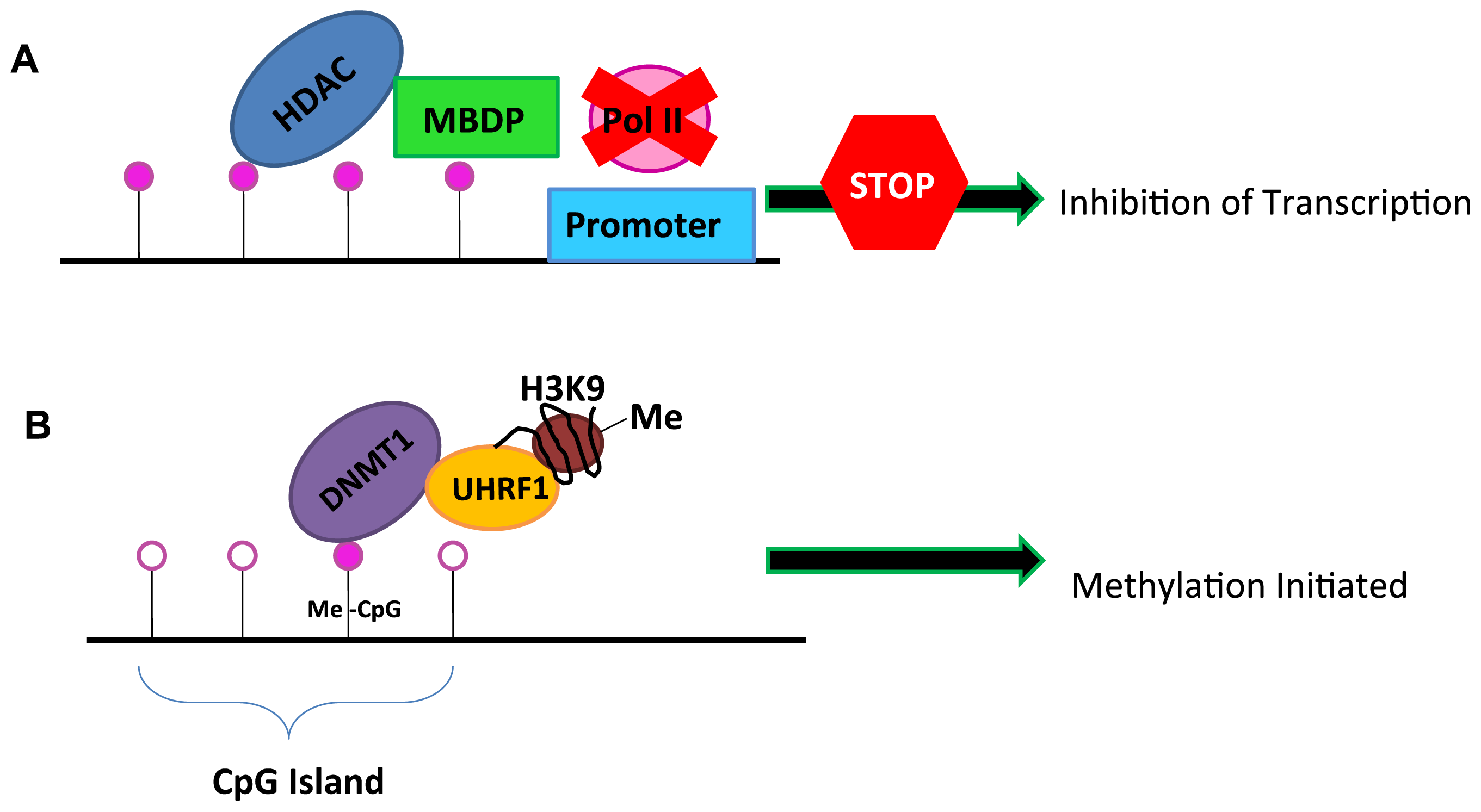
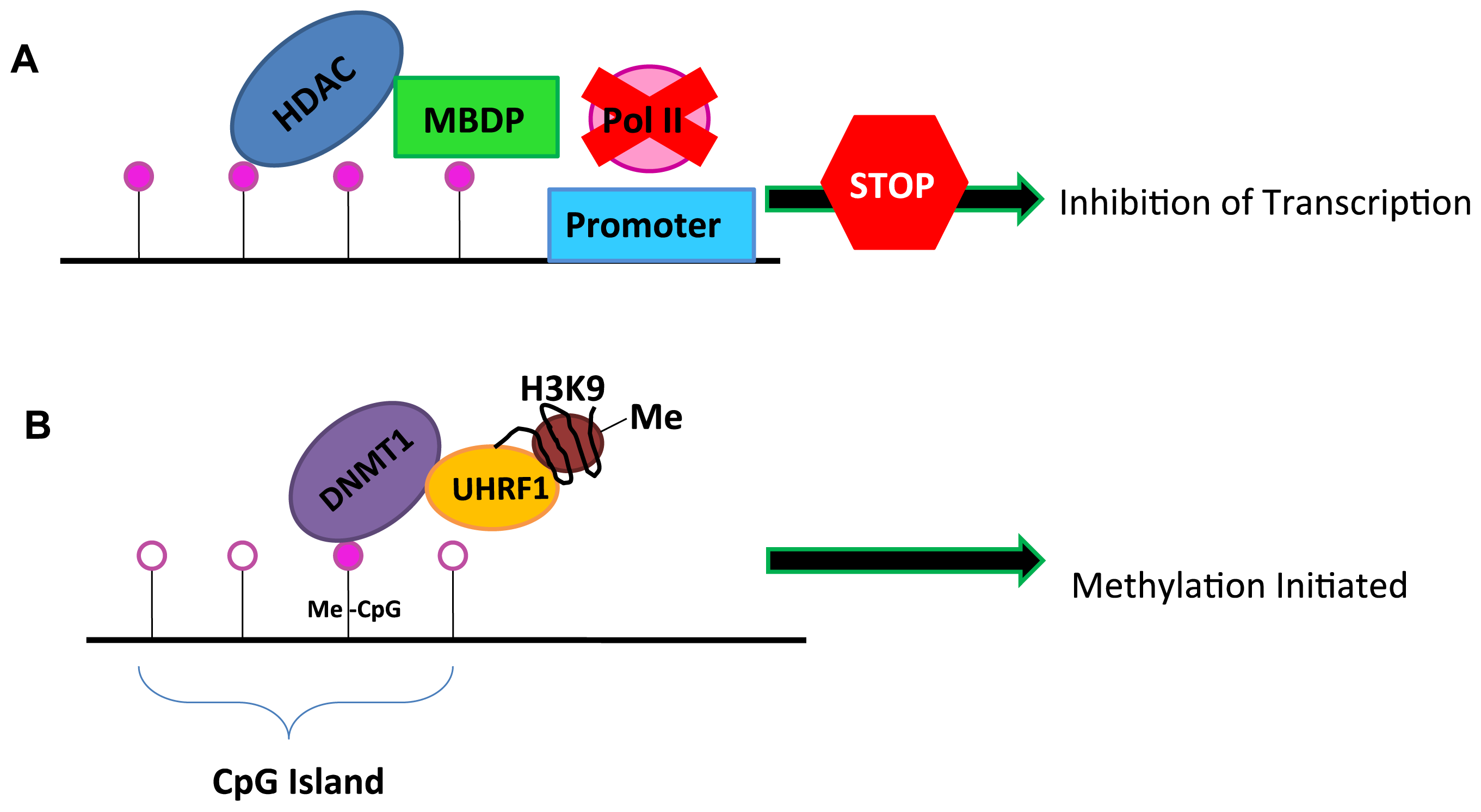
2. DNA Methylation
3. Hydroxymethylation
4. Apoptosis and Autophagy
5. MicroRNA
6. Epithelial-Mesenchymal Transition
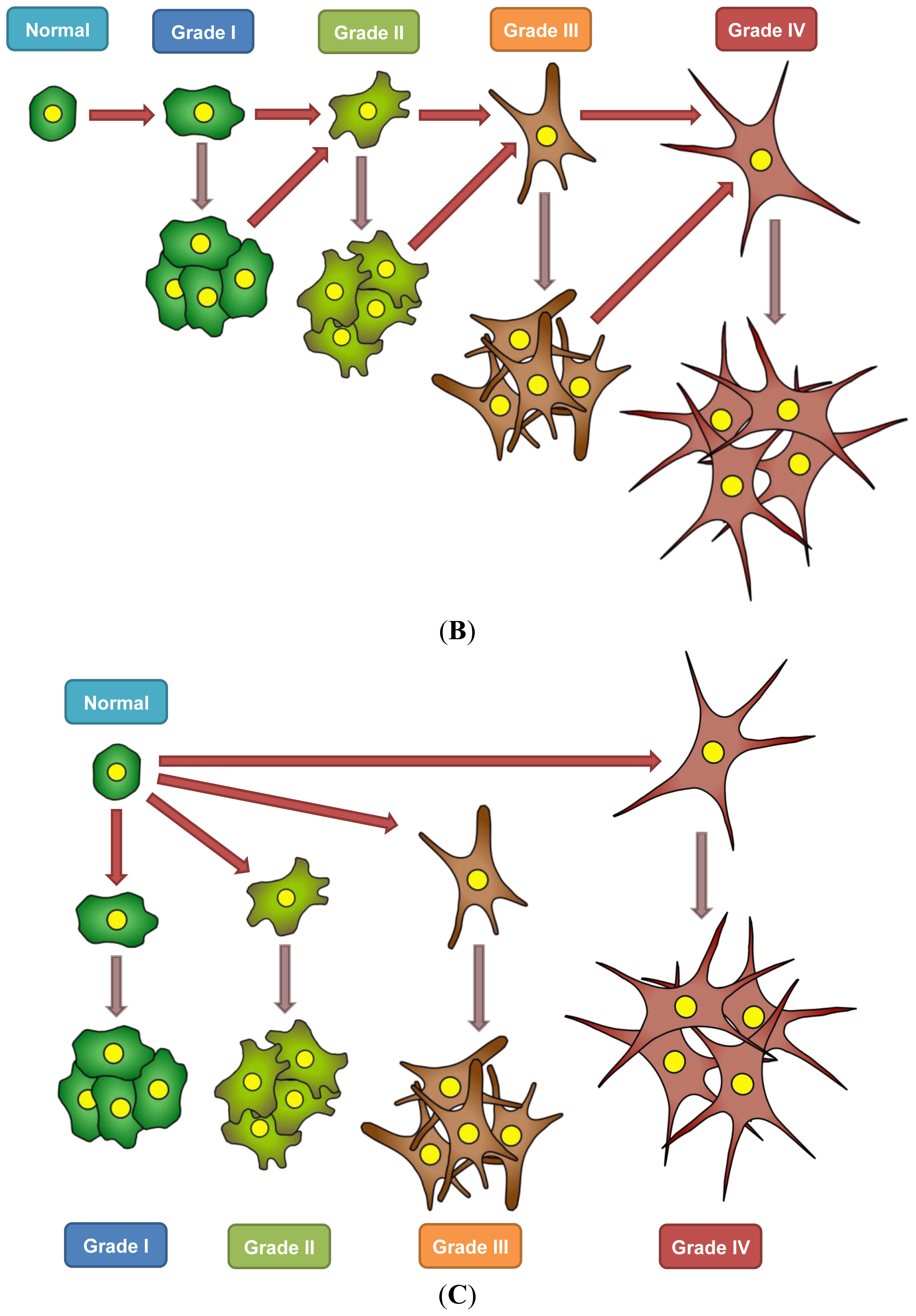
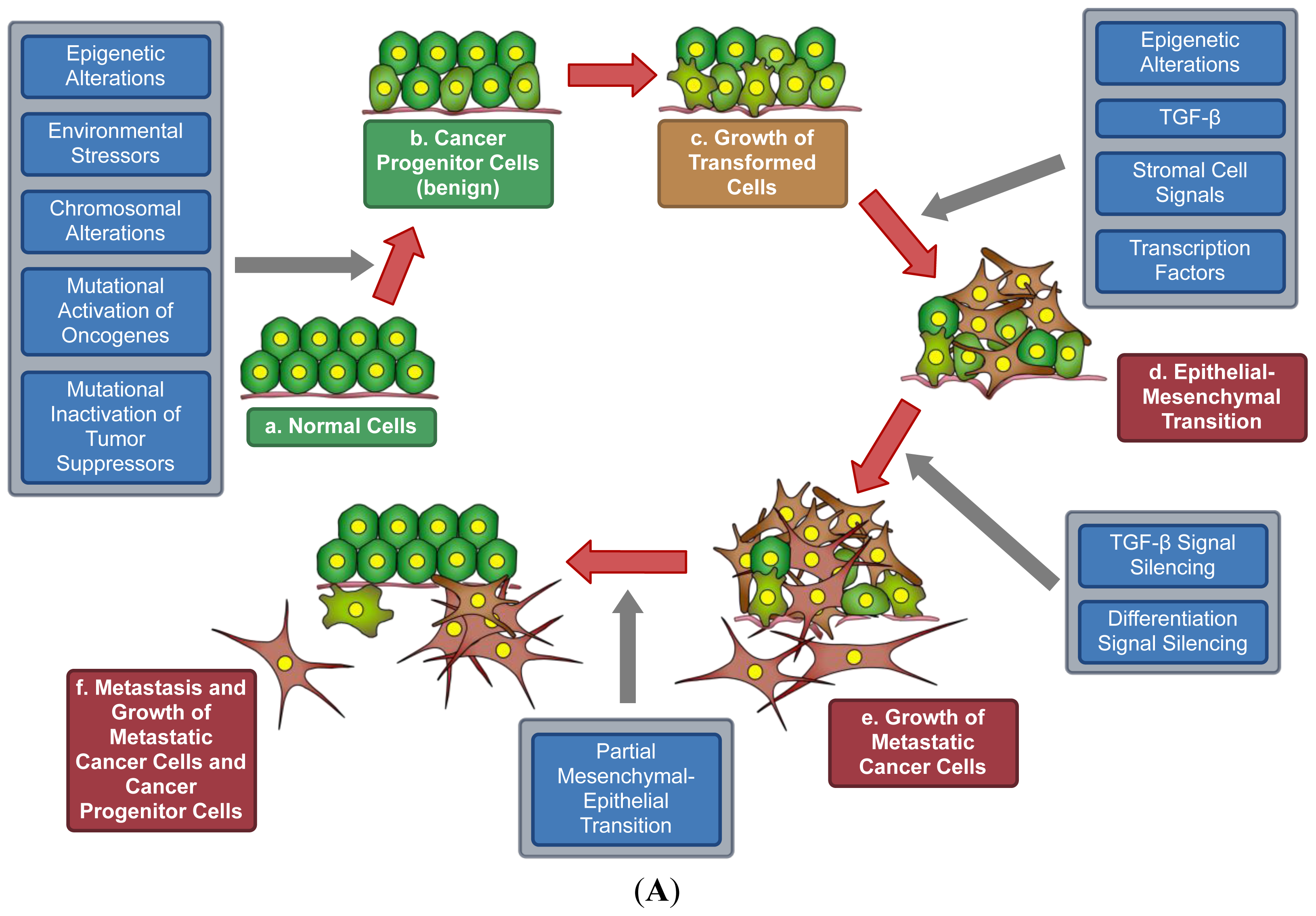
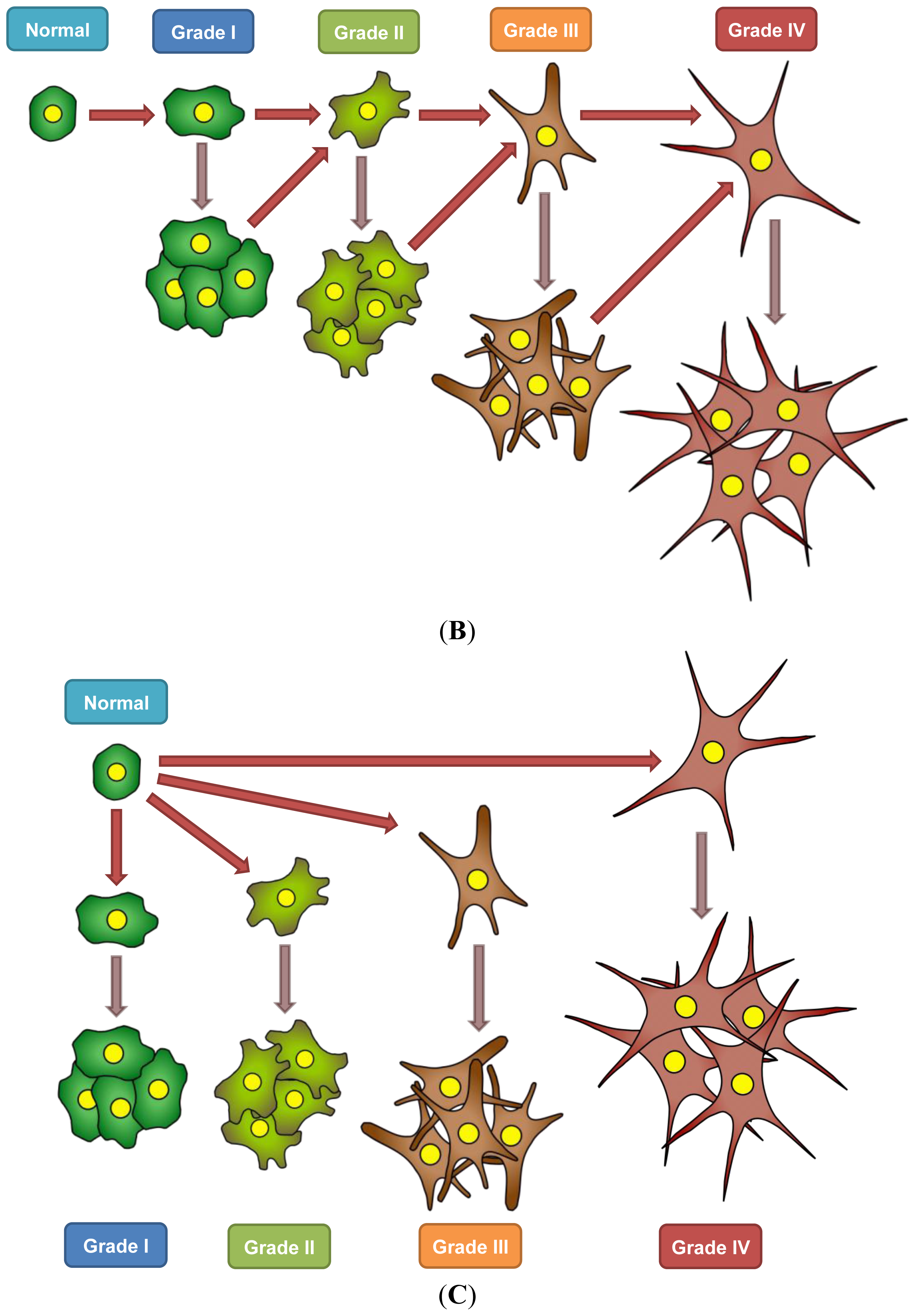
7. A Model for Epigenetics in Carcinogenesis, Progression, and Metastasis
8. Clinical Aspects of Cancer and Therapeutics
9. Conclusions
Acknowledgments
Conflicts of Interest
References
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar]
- Fearon, E.R.; Vogelstein, B. A genetic model for colorectal tumorigenesis. Cell 1990, 61, 759–767. [Google Scholar]
- Vogelstein, B.; Kinzler, K.W. Cancer genes and the pathways they control. Nat. Med 2004, 10, 789–799. [Google Scholar]
- Sarkar, S.; Goldgar, S.; Byler, S.; Rosenthal, S.; Heerboth, S. Demethylation and re-expression of epigenetically silenced tumor suppressor genes: Sensitization of cancer cells by combination therapy. Epigenomics 2013, 5, 87–94. [Google Scholar]
- Gal-Yam, E.N.; Saito, Y.; Egger, G.; Jones, P.A. Cancer epigenetics: Modifications, screening, and therapy. Annu. Rev. Med 2008, 59, 267–280. [Google Scholar]
- Bird, A.P. CpG-rich islands and the function of DNA methylation. Nature 1986, 321, 209–213. [Google Scholar]
- Merlo, A.; Herman, J.G.; Mao, L.; Lee, D.J.; Gabrielson, E.; Burger, P.C.; Baylin, S.B.; Sidransky, D. 5′ CpG island methylation is associated with transcriptional silencing of the tumour suppressor p16/CDKN2/MTS1 in human cancers. Nat. Med 1995, 1, 686–692. [Google Scholar]
- Taby, R.; Issa, J.P. Cancer epigenetics. CA Cancer J. Clin 2010, 60, 376–392. [Google Scholar]
- Balch, C.; Huang, T.H.; Brown, R.; Nephew, K.P. The epigenetics of ovarian cancer drug resistance and resensitization. Am. J. Obstet. Gynecol 2004, 191, 1552–1572. [Google Scholar]
- Denissenko, M.F.; Chen, J.X.; Tang, M.S.; Pfeifer, G.P. Cytosine methylation determines hot spots of DNA damage in the human P53 gene. Proc. Natl. Acad. Sci. USA 1997, 94, 3893–3898. [Google Scholar]
- Neureiter, D.; Zopf, S.; Leu, T.; Dietze, O.; Hauser-Kronberger, C.; Hahn, E.G.; Herold, C.; Ocker, M. Apoptosis, proliferation and differentiation patterns are influenced by Zebularine and SAHA in pancreatic cancer models. Scand. J. Gastroenterol 2007, 42, 103–116. [Google Scholar]
- Issa, J.P. Cancer prevention: Epigenetics steps up to the plate. Cancer Prev. Res. (Phila.) 2008, 1, 219–222. [Google Scholar]
- Ren, M.; Pozzi, S.; Bistulfi, G.; Somenzi, G.; Rossetti, S.; Sacchi, N. Impaired retinoic acid (RA) signal leads to RARβ2 epigenetic silencing and RA resistance. Mol. Cell. Biol 2005, 25, 10591–10603. [Google Scholar]
- Jones, P.A.; Baylin, S.B. The fundamental role of epigenetic events in cancer. Nat. Rev. Genet 2002, 3, 415–428. [Google Scholar]
- Yu, Y.; Fujii, S.; Yuan, J.; Luo, R.Z.; Wang, L.; Bao, J.; Kadota, M.; Oshimura, M.; Dent, S.R.; Issa, J.P.; et al. Epigenetic regulation of ARHI in breast and ovarian cancer cells. Ann. N. Y. Acad. Sci 2003, 983, 268–277. [Google Scholar]
- Mataga, M.A.; Rosenthal, S.; Heerboth, S.; Devalapalli, A.; Kokolus, S.; Evans, L.R.; Longacre, M.; Housman, G.; Sarkar, S. Anti-breast cancer effects of histone deacetylase inhibitors and calpain inhibitor. Anticancer Res 2012, 32, 2523–2529. [Google Scholar]
- Esteller, M.; Hamilton, S.R.; Burger, P.C.; Baylin, S.B.; Herman, J.G. Inactivation of the DNA repair gene O6-methylguanine-DNA methyltransferase by promoter hypermethylation is a common event in primary human neoplasia. Cancer Res 1999, 59, 793–797. [Google Scholar]
- Hegi, M.E.; Diserens, A.C.; Gorlia, T.; Hamou, M.F.; de Tribolet, N.; Weller, M.; Kros, J.M.; Hainfellner, J.A.; Mason, W.; Mariani, L.; et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N. Engl. J. Med 2005, 352, 997–1003. [Google Scholar]
- Chen, Y.; Hu, F.; Zhou, Y.; Chen, W.; Shao, H.; Zhang, Y. MGMT promoter methylation and glioblastoma prognosis: A systematic review and meta-analysis. Arch. Med. Res 2013, 44, 281–290. [Google Scholar]
- Robertson, K.D.; Keyomarsi, K.; Gonzales, F.A.; Velicescu, M.; Jones, P.A. Differential mRNA expression of the human DNA methyltransferases (DNMTs) 1, 3a and 3b during the G0/G1 to S phase transition in normal and tumor cells. Nucleic Acids Res 2000, 28, 2108–2113. [Google Scholar]
- Bolden, J.E.; Peart, M.J.; Johnstone, R.W. Anticancer activities of histone deacetylase inhibitors. Nat. Rev. Drug Discov 2006, 5, 769–784. [Google Scholar]
- Rodriguez-Paredes, M.; Esteller, M. Cancer epigenetics reaches mainstream oncology. Nat. Med 2011, 17, 330–339. [Google Scholar]
- Rothbart, S.B.; Krajewski, K.; Nady, N.; Tempel, W.; Xue, S.; Badeaux, A.I.; Barsyte-Lovejoy, D.; Martinez, J.Y.; Bedford, M.T.; Fuchs, S.M.; et al. Association of UHRF1 with methylated H3K9 directs the maintenance of DNA methylation. Nat. Struct. Mol. Biol 2012, 19, 1155–1160. [Google Scholar]
- Kriaucionis, S.; Heintz, N. The nuclear DNA base 5-hydroxymethylcytosine is present in Purkinje neurons and the brain. Science 2009, 324, 929–930. [Google Scholar]
- Tahiliani, M.; Koh, K.P.; Shen, Y.; Pastor, W.A.; Bandukwala, H.; Brudno, Y.; Agarwal, S.; Iyer, L.M.; Liu, D.R.; Aravind, L.; et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science 2009, 324, 930–935. [Google Scholar]
- Ko, M.; An, J.; Bandukwala, H.S.; Chavez, L.; Aijo, T.; Pastor, W.A.; Segal, M.F.; Li, H.; Koh, K.P.; Lahdesmaki, H.; et al. Modulation of TET2 expression and 5-methylcytosine oxidation by the CXXC domain protein IDAX. Nature 2013, 497, 122–126. [Google Scholar]
- Piccolo, F.M.; Bagci, H.; Brown, K.E.; Landeira, D.; Soza-Ried, J.; Feytout, A.; Mooijman, D.; Hajkova, P.; Leitch, H.G.; Tada, T.; et al. Different roles for Tet1 and Tet2 proteins in reprogramming-mediated erasure of imprints induced by EGC fusion. Mol. Cell 2013, 49, 1023–1033. [Google Scholar]
- Gu, T.P.; Guo, F.; Yang, H.; Wu, H.P.; Xu, G.F.; Liu, W.; Xie, Z.G.; Shi, L.; He, X.; Jin, S.G.; et al. The role of Tet3 DNA dioxygenase in epigenetic reprogramming by oocytes. Nature 2011, 477, 606–610. [Google Scholar]
- Iqbal, K.; Jin, S.G.; Pfeifer, G.P.; Szabo, P.E. Reprogramming of the paternal genome upon fertilization involves genome-wide oxidation of 5-methylcytosine. Proc. Natl. Acad. Sci. USA 2011, 108, 3642–3647. [Google Scholar]
- Wossidlo, M.; Nakamura, T.; Lepikhov, K.; Marques, C.J.; Zakhartchenko, V.; Boiani, M.; Arand, J.; Nakano, T.; Reik, W.; Walter, J. 5-Hydroxymethylcytosine in the mammalian zygote is linked with epigenetic reprogramming. Nat. Commun 2011, 2. [Google Scholar] [CrossRef]
- Haffner, M.C.; Chaux, A.; Meeker, A.K.; Esopi, D.M.; Gerber, J.; Pellakuru, L.G.; Toubaji, A.; Argani, P.; Iacobuzio-Donahue, C.; Nelson, W.G.; et al. Global 5-hydroxymethylcytosine content is significantly reduced in tissue stem/progenitor cell compartments and in human cancers. Oncotarget 2011, 2, 627–637. [Google Scholar]
- Delhommeau, F.; Dupont, S.; Della Valle, V.; James, C.; Trannoy, S.; Masse, A.; Kosmider, O.; Le Couedic, J.P.; Robert, F.; Alberdi, A.; et al. Mutation in TET2 in myeloid cancers. N. Engl. J. Med 2009, 360, 2289–2301. [Google Scholar]
- Langemeijer, S.M.; Kuiper, R.P.; Berends, M.; Knops, R.; Aslanyan, M.G.; Massop, M.; Stevens-Linders, E.; van Hoogen, P.; van Kessel, A.G.; Raymakers, R.A.; et al. Acquired mutations in TET2 are common in myelodysplastic syndromes. Nat. Genet 2009, 41, 838–842. [Google Scholar]
- Ono, R.; Taki, T.; Taketani, T.; Taniwaki, M.; Kobayashi, H.; Hayashi, Y. LCX, leukemia-associated protein with a CXXC domain, is fused to MLL in acute myeloid leukemia with trilineage dysplasia having t(10;11)(q22;q23). Cancer Res 2002, 62, 4075–4080. [Google Scholar]
- Lorsbach, R.B.; Moore, J.; Mathew, S.; Raimondi, S.C.; Mukatira, S.T.; Downing, J.R. TET1, a member of a novel protein family, is fused to MLL in acute myeloid leukemia containing the t(10;11)(q22;q23). Leukemia 2003, 17, 637–641. [Google Scholar]
- Yang, H.; Liu, Y.; Bai, F.; Zhang, J.Y.; Ma, S.H.; Liu, J.; Xu, Z.D.; Zhu, H.G.; Ling, Z.Q.; Ye, D.; et al. Tumor development is associated with decrease of TET gene expression and 5-methylcytosine hydroxylation. Oncogene 2013, 32, 663–669. [Google Scholar]
- Liu, C.; Liu, L.; Chen, X.; Shen, J.; Shan, J.; Xu, Y.; Yang, Z.; Wu, L.; Xia, F.; Bie, P.; et al. Decrease of 5-hydroxymethylcytosine is associated with progression of hepatocellular carcinoma through downregulation of TET1. PLoS One 2013, 8, e62828. [Google Scholar]
- Lian, C.G.; Xu, Y.; Ceol, C.; Wu, F.; Larson, A.; Dresser, K.; Xu, W.; Tan, L.; Hu, Y.; Zhan, Q.; et al. Loss of 5-hydroxymethylcytosine is an epigenetic hallmark of melanoma. Cell 2012, 150, 1135–1146. [Google Scholar]
- Dang, L.; White, D.W.; Gross, S.; Bennett, B.D.; Bittinger, M.A.; Driggers, E.M.; Fantin, V.R.; Jang, H.G.; Jin, S.; Keenan, M.C.; et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 2009, 462, 739–744. [Google Scholar]
- Zhao, S.; Lin, Y.; Xu, W.; Jiang, W.; Zha, Z.; Wang, P.; Yu, W.; Li, Z.; Gong, L.; Peng, Y.; et al. Glioma-derived mutations in IDH1 dominantly inhibit IDH1 catalytic activity and induce HIF-1α. Science 2009, 324, 261–265. [Google Scholar]
- Amary, M.F.; Damato, S.; Halai, D.; Eskandarpour, M.; Berisha, F.; Bonar, F.; McCarthy, S.; Fantin, V.R.; Straley, K.S.; Lobo, S.; et al. Ollier disease and Maffucci syndrome are caused by somatic mosaic mutations of IDH1 and IDH2. Nat. Genet 2011, 43, 1262–1265. [Google Scholar]
- Pansuriya, T.C.; van Eijk, R.; d’Adamo, P.; van Ruler, M.A.; Kuijjer, M.L.; Oosting, J.; Cleton-Jansen, A.M.; van Oosterwijk, J.G.; Verbeke, S.L.; Meijer, D.; et al. Somatic mosaic IDH1 and IDH2 mutations are associated with enchondroma and spindle cell hemangioma in Ollier disease and Maffucci syndrome. Nat. Genet 2011, 43, 1256–1261. [Google Scholar]
- Parsons, D.W.; Jones, S.; Zhang, X.; Lin, J.C.; Leary, R.J.; Angenendt, P.; Mankoo, P.; Carter, H.; Siu, I.M.; Gallia, G.L.; et al. An integrated genomic analysis of human glioblastoma multiforme. Science 2008, 321, 1807–1812. [Google Scholar]
- Hemerly, J.P.; Bastos, A.U.; Cerutti, J.M. Identification of several novel non-p.R132 IDH1 variants in thyroid carcinomas. Eur. J. Endocrinol 2010, 163, 747–755. [Google Scholar]
- Murugan, A.K.; Bojdani, E.; Xing, M. Identification and functional characterization of isocitrate dehydrogenase 1 (IDH1) mutations in thyroid cancer. Biochem. Biophys. Res. Commun 2010, 393, 555–559. [Google Scholar]
- Ito, S.; Shen, L.; Dai, Q.; Wu, S.C.; Collins, L.B.; Swenberg, J.A.; He, C.; Zhang, Y. Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science 2011, 333, 1300–1303. [Google Scholar]
- Xu, S.; Li, W.; Zhu, J.; Wang, R.; Li, Z.; Xu, G.L.; Ding, J. Crystal structures of isoorotate decarboxylases reveal a novel catalytic mechanism of 5-carboxyl-uracil decarboxylation and shed light on the search for DNA decarboxylase. Cell Res 2013. [Google Scholar] [CrossRef]
- He, Y.F.; Li, B.Z.; Li, Z.; Liu, P.; Wang, Y.; Tang, Q.; Ding, J.; Jia, Y.; Chen, Z.; Li, L.; et al. Tet-mediated formation of 5-carboxylcytosine and its excision by TDG in mammalian DNA. Science 2011, 333, 1303–1307. [Google Scholar]
- Maiti, A.; Drohat, A.C. Thymine DNA glycosylase can rapidly excise 5-formylcytosine and 5-carboxylcytosine: Potential implications for active demethylation of CpG sites. J. Biol. Chem 2011, 286, 35334–35338. [Google Scholar]
- Zhang, L.; Lu, X.; Lu, J.; Liang, H.; Dai, Q.; Xu, G.L.; Luo, C.; Jiang, H.; He, C. Thymine DNA glycosylase specifically recognizes 5-carboxylcytosine-modified DNA. Nat. Chem. Biol 2012, 8, 328–330. [Google Scholar]
- Guo, J.U.; Su, Y.; Zhong, C.; Ming, G.L.; Song, H. Hydroxylation of 5-methylcytosine by TET1 promotes active DNA demethylation in the adult brain. Cell 2011, 145, 423–434. [Google Scholar]
- Cortellino, S.; Xu, J.; Sannai, M.; Moore, R.; Caretti, E.; Cigliano, A.; Le Coz, M.; Devarajan, K.; Wessels, A.; Soprano, D.; et al. Thymine DNA glycosylase is essential for active DNA demethylation by linked deamination-base excision repair. Cell 2011, 146, 67–79. [Google Scholar]
- Frauer, C.; Hoffmann, T.; Bultmann, S.; Casa, V.; Cardoso, M.C.; Antes, I.; Leonhardt, H. Recognition of 5-hydroxymethylcytosine by the Uhrf1 SRA domain. PLoS One 2011, 6, e21306. [Google Scholar]
- Yildirim, O.; Li, R.; Hung, J.H.; Chen, P.B.; Dong, X.; Ee, L.S.; Weng, Z.; Rando, O.J.; Fazzio, T.G. Mbd3/NURD complex regulates expression of 5-hydroxymethylcytosine marked genes in embryonic stem cells. Cell 2011, 147, 1498–1510. [Google Scholar]
- Adams, J.M.; Cory, S. The Bcl-2 protein family: Arbiters of cell survival. Science 1998, 281, 1322–1326. [Google Scholar]
- Kaufmann, T.; Strasser, A.; Jost, P.J. Fas death receptor signalling: Roles of Bid and XIAP. Cell Death Differ 2012, 19, 42–50. [Google Scholar]
- Subramaniam, K.; Hirpara, J.L.; Tucker-Kellogg, L.; Tucker-Kellogg, G.; Pervaiz, S. FLIP: A flop for execution signals. Cancer Lett 2013, 332, 151–155. [Google Scholar]
- Horak, P.; Pils, D.; Haller, G.; Pribill, I.; Roessler, M.; Tomek, S.; Horvat, R.; Zeillinger, R.; Zielinski, C.; Krainer, M. Contribution of epigenetic silencing of tumor necrosis factor-related apoptosis inducing ligand receptor 1 (DR4) to TRAIL resistance and ovarian cancer. Mol. Cancer Res 2005, 3, 335–343. [Google Scholar]
- Sarkar, S.; Faller, D.V. Telomere-homologous G-rich oligonucleotides sensitize human ovarian cancer cells to TRAIL-induced growth inhibition and apoptosis. Nucleic Acid Ther 2013, 23, 167–174. [Google Scholar]
- Morselli, E.; Galluzzi, L.; Kepp, O.; Vicencio, J.M.; Criollo, A.; Maiuri, M.C.; Kroemer, G. Anti- and pro-tumor functions of autophagy. Biochim. Biophys. Acta 2009, 1793, 1524–1532. [Google Scholar]
- Lee, J.H.; Khor, T.O.; Shu, L.; Su, Z.Y.; Fuentes, F.; Kong, A.N. Dietary phytochemicals and cancer prevention: Nrf2 signaling, epigenetics, and cell death mechanisms in blocking cancer initiation and progression. Pharmacol. Ther 2013, 137, 153–171. [Google Scholar]
- Fang, M.Z.; Chen, D.; Sun, Y.; Jin, Z.; Christman, J.K.; Yang, C.S. Reversal of hypermethylation and reactivation of p16INK4a, RARβ, and MGMT genes by genistein and other isoflavones from soy. Clin. Cancer Res 2005, 11, 7033–7041. [Google Scholar]
- Tan, J.; Yang, X.; Zhuang, L.; Jiang, X.; Chen, W.; Lee, P.L.; Karuturi, R.K.; Tan, P.B.; Liu, E.T.; Yu, Q. Pharmacologic disruption of Polycomb-repressive complex 2-mediated gene repression selectively induces apoptosis in cancer cells. Genes Dev 2007, 21, 1050–1063. [Google Scholar]
- Young, A.R.; Narita, M.; Ferreira, M.; Kirschner, K.; Sadaie, M.; Darot, J.F.; Tavare, S.; Arakawa, S.; Shimizu, S.; Watt, F.M.; et al. Autophagy mediates the mitotic senescence transition. Genes Dev 2009, 23, 798–803. [Google Scholar]
- Krek, A.; Grun, D.; Poy, M.N.; Wolf, R.; Rosenberg, L.; Epstein, E.J.; MacMenamin, P.; da Piedade, I.; Gunsalus, K.C.; Stoffel, M.; et al. Combinatorial microRNA target predictions. Nat. Genet 2005, 37, 495–500. [Google Scholar]
- Walter, B.A.; Valera, V.A.; Pinto, P.A.; Merino, M.J. Comprehensive microRNA profiling of prostate cancer. J. Cancer 2013, 4, 350–357. [Google Scholar]
- Leite, K.R.; Morais, D.R.; Reis, S.T.; Viana, N.; Moura, C.; Florez, M.G.; Silva, I.A.; Dip, N.; Srougi, M. MicroRNA 100: A context dependent miRNA in prostate cancer. Clinics (Sao Paulo) 2013, 68, 797–802. [Google Scholar]
- Calin, G.A.; Croce, C.M. MicroRNA signatures in human cancers. Nat. Rev. Cancer 2006, 6, 857–866. [Google Scholar]
- Van Jaarsveld, M.T.; Helleman, J.; Berns, E.M.; Wiemer, E.A. MicroRNAs in ovarian cancer biology and therapy resistance. Int. J. Biochem. Cell Biol 2010, 42, 1282–1290. [Google Scholar]
- Lu, J.; Getz, G.; Miska, E.A.; Alvarez-Saavedra, E.; Lamb, J.; Peck, D.; Sweet-Cordero, A.; Ebert, B.L.; Mak, R.H.; Ferrando, A.A.; et al. MicroRNA expression profiles classify human cancers. Nature 2005, 435, 834–838. [Google Scholar]
- Zhang, Y.; Li, M.; Wang, H.; Fisher, W.E.; Lin, P.H.; Yao, Q.; Chen, C. Profiling of 95 microRNAs in pancreatic cancer cell lines and surgical specimens by real-time PCR analysis. World J. Surg 2009, 33, 698–709. [Google Scholar]
- Nakata, K.; Ohuchida, K.; Mizumoto, K.; Kayashima, T.; Ikenaga, N.; Sakai, H.; Lin, C.; Fujita, H.; Otsuka, T.; Aishima, S.; et al. MicroRNA-10b is overexpressed in pancreatic cancer, promotes its invasiveness, and correlates with a poor prognosis. Surgery 2011, 150, 916–922. [Google Scholar]
- Nagaraja, A.K.; Creighton, C.J.; Yu, Z.; Zhu, H.; Gunaratne, P.H.; Reid, J.G.; Olokpa, E.; Itamochi, H.; Ueno, N.T.; Hawkins, S.M.; et al. A link between miR-100 and FRAP1/mTOR in clear cell ovarian cancer. Mol. Endocrinol 2010, 24, 447–463. [Google Scholar]
- Catto, J.W.; Miah, S.; Owen, H.C.; Bryant, H.; Myers, K.; Dudziec, E.; Larre, S.; Milo, M.; Rehman, I.; Rosario, D.J.; et al. Distinct microRNA alterations characterize high- and low-grade bladder cancer. Cancer Res 2009, 69, 8472–8481. [Google Scholar]
- Neely, L.A.; Rieger-Christ, K.M.; Neto, B.S.; Eroshkin, A.; Garver, J.; Patel, S.; Phung, N.A.; McLaughlin, S.; Libertino, J.A.; Whitney, D.; et al. A microRNA expression ratio defining the invasive phenotype in bladder tumors. Urol. Oncol 2010, 28, 39–48. [Google Scholar]
- Hannafon, B.N.; Sebastiani, P.; de las Morenas, A.; Lu, J.; Rosenberg, C.L. Expression of microRNA and their gene targets are dysregulated in preinvasive breast cancer. Breast Cancer Res 2011, 13. [Google Scholar] [CrossRef]
- Yanaihara, N.; Caplen, N.; Bowman, E.; Seike, M.; Kumamoto, K.; Yi, M.; Stephens, R.M.; Okamoto, A.; Yokota, J.; Tanaka, T.; et al. Unique microRNA molecular profiles in lung cancer diagnosis and prognosis. Cancer Cell 2006, 9, 189–198. [Google Scholar]
- Papagiannakopoulos, T.; Shapiro, A.; Kosik, K.S. MicroRNA-21 targets a network of key tumor-suppressive pathways in glioblastoma cells. Cancer Res 2008, 68, 8164–8172. [Google Scholar]
- Huang, Q.; Gumireddy, K.; Schrier, M.; le Sage, C.; Nagel, R.; Nair, S.; Egan, D.A.; Li, A.; Huang, G.; Klein-Szanto, A.J.; et al. The microRNAs miR-373 and miR-520c promote tumour invasion and metastasis. Nat. Cell Biol 2008, 10, 202–210. [Google Scholar]
- Lujambio, A.; Ropero, S.; Ballestar, E.; Fraga, M.F.; Cerrato, C.; Setien, F.; Casado, S.; Suarez-Gauthier, A.; Sanchez-Cespedes, M.; Git, A.; et al. Genetic unmasking of an epigenetically silenced microRNA in human cancer cells. Cancer Res 2007, 67, 1424–1429. [Google Scholar]
- Brzezianska, E.; Dutkowska, A.; Antczak, A. The significance of epigenetic alterations in lung carcinogenesis. Mol. Biol. Rep 2013, 40, 309–325. [Google Scholar]
- Diederichs, S.; Haber, D.A. Sequence variations of microRNAs in human cancer: Alterations in predicted secondary structure do not affect processing. Cancer Res 2006, 66, 6097–6104. [Google Scholar]
- Shen, J.; Xia, W.; Khotskaya, Y.B.; Huo, L.; Nakanishi, K.; Lim, S.O.; Du, Y.; Wang, Y.; Chang, W.C.; Chen, C.H.; et al. EGFR modulates microRNA maturation in response to hypoxia through phosphorylation of AGO2. Nature 2013, 497, 383–387. [Google Scholar]
- Blick, C.; Ramachandran, A.; Wigfield, S.; McCormick, R.; Jubb, A.; Buffa, F.M.; Turley, H.; Knowles, M.A.; Cranston, D.; Catto, J.; et al. Hypoxia regulates FGFR3 expression via HIF-1α and miR-100 and contributes to cell survival in non-muscle invasive bladder cancer. Br. J. Cancer 2013, 109, 50–59. [Google Scholar]
- Thiery, J.P. Epithelial-mesenchymal transitions in tumour progression. Nat. Rev. Cancer 2002, 2, 442–454. [Google Scholar]
- Wendt, M.K.; Allington, T.M.; Schiemann, W.P. Mechanisms of the epithelial-mesenchymal transition by TGF-β. Future Oncol 2009, 5, 1145–1168. [Google Scholar]
- Graff, J.R.; Gabrielson, E.; Fujii, H.; Baylin, S.B.; Herman, J.G. Methylation patterns of the E-cadherin 5′ CpG island are unstable and reflect the dynamic, heterogeneous loss of E-cadherin expression during metastatic progression. J. Biol. Chem 2000, 275, 2727–2732. [Google Scholar]
- Lombaerts, M.; van Wezel, T.; Philippo, K.; Dierssen, J.W.; Zimmerman, R.M.; Oosting, J.; van Eijk, R.; Eilers, P.H.; van de Water, B.; Cornelisse, C.J.; et al. E-cadherin transcriptional downregulation by promoter methylation but not mutation is related to epithelial-to-mesenchymal transition in breast cancer cell lines. Br. J. Cancer 2006, 94, 661–671. [Google Scholar]
- Nass, S.J.; Herman, J.G.; Gabrielson, E.; Iversen, P.W.; Parl, F.F.; Davidson, N.E.; Graff, J.R. Aberrant methylation of the estrogen receptor and E-cadherin 5′ CpG islands increases with malignant progression in human breast cancer. Cancer Res 2000, 60, 4346–4348. [Google Scholar]
- Wendt, M.K.; Smith, J.A.; Schiemann, W.P. Transforming growth factor-β-induced epithelial-mesenchymal transition facilitates epidermal growth factor-dependent breast cancer progression. Oncogene 2010, 29, 6485–6498. [Google Scholar]
- Witz, I.P. The selectin-selectin ligand axis in tumor progression. Cancer Metastasis Rev 2008, 27, 19–30. [Google Scholar]
- Paschos, K.A.; Canovas, D.; Bird, N.C. The role of cell adhesion molecules in the progression of colorectal cancer and the development of liver metastasis. Cell. Signal 2009, 21, 665–674. [Google Scholar]
- Laubli, H.; Borsig, L. Selectins promote tumor metastasis. Semin. Cancer Biol 2010, 20, 169–177. [Google Scholar] [Green Version]
- Desgrosellier, J.S.; Cheresh, D.A. Integrins in cancer: Biological implications and therapeutic opportunities. Nat. Rev. Cancer 2010, 10, 9–22. [Google Scholar]
- Bendas, G.; Borsig, L. Cancer cell adhesion and metastasis: Selectins, integrins, and the inhibitory potential of heparins. Int. J. Cell Biol 2012, 2012, 676731:1–676731:10. [Google Scholar]
- Barkan, D.; Kleinman, H.; Simmons, J.L.; Asmussen, H.; Kamaraju, A.K.; Hoenorhoff, M.J.; Liu, Z.Y.; Costes, S.V.; Cho, E.H.; Lockett, S.; et al. Inhibition of metastatic outgrowth from single dormant tumor cells by targeting the cytoskeleton. Cancer Res 2008, 68, 6241–6250. [Google Scholar]
- Shibue, T.; Weinberg, R.A. Integrin β1-focal adhesion kinase signaling directs the proliferation of metastatic cancer cells disseminated in the lungs. Proc. Natl. Acad. Sci. USA 2009, 106, 10290–10295. [Google Scholar]
- Bhowmick, N.A.; Zent, R.; Ghiassi, M.; McDonnell, M.; Moses, H.L. Integrin β1 signaling is necessary for transforming growth factor-β activation of p38MAPK and epithelial plasticity. J. Biol. Chem 2001, 276, 46707–46713. [Google Scholar]
- Galliher, A.J.; Schiemann, W.P. β3 integrin and Src facilitate transforming growth factor-β mediated induction of epithelial-mesenchymal transition in mammary epithelial cells. Breast Cancer Res 2006, 8, R42:1–R42:16. [Google Scholar]
- Wendt, M.K.; Schiemann, W.P. Therapeutic targeting of the focal adhesion complex prevents oncogenic TGF-β signaling and metastasis. Breast Cancer Res 2009, 11, R68:1–R68:16. [Google Scholar]
- Sarkar, S.; Svoboda, M.; de Beaumont, R.; Freedman, A.S. The role of Aktand RAFTK in β1 integrin mediated survival of precursor B-acute lymphoblastic leukemia cells. Leuk. Lymphoma 2002, 43, 1663–1671. [Google Scholar]
- Sarkar, S. Tyrosine phosphorylation and translocation of LAT in platelets. FEBS Lett 1998, 441, 357–360. [Google Scholar]
- Sarkar, S.; Rooney, M.M.; Lord, S.T. Activation of integrin-β3-associated syk in platelets. Biochem. J 1999, 338, 677–680. [Google Scholar]
- Carraway, K.L., 3rd; Sweeney, C. Co-opted integrin signaling in ErbB2-induced mammary tumor progression. Cancer Cell 2006, 10, 93–95. [Google Scholar]
- Miller, P.G.; Al-Shahrour, F.; Hartwell, K.A.; Chu, L.P.; Jaras, M.; Puram, R.V.; Puissant, A.; Callahan, K.P.; Ashton, J.; McConkey, M.E.; et al. In vivo RNAi screening identifies a leukemia-specific dependence on integrin β 3 signaling. Cancer Cell 2013, 24, 45–58. [Google Scholar]
- Park, J.; Song, S.H.; Kim, T.Y.; Choi, M.C.; Jong, H.S.; Kim, T.Y.; Lee, J.W.; Kim, N.K.; Kim, W.H.; Bang, Y.J. Aberrant methylation of integrin α4 gene in human gastric cancer cells. Oncogene 2004, 23, 3474–3480. [Google Scholar]
- Ulazzi, L.; Sabbioni, S.; Miotto, E.; Veronese, A.; Angusti, A.; Gafa, R.; Manfredini, S.; Farinati, F.; Sasaki, T.; Lanza, G.; et al. Nidogen 1 and 2 gene promoters are aberrantly methylated in human gastrointestinal cancer. Mol. Cancer 2007. [Google Scholar]
- Nejjari, M.; Hafdi, Z.; Gouysse, G.; Fiorentino, M.; Beatrix, O.; Dumortier, J.; Pourreyron, C.; Barozzi, C.; D’Errico, A.; Grigioni, W.F.; et al. Expression, regulation, and function of αV integrins in hepatocellular carcinoma: An in vivo and in vitro study. Hepatology 2002, 36, 418–426. [Google Scholar]
- Mohamed, M.M.; Sloane, B.F. Cysteine cathepsins: Multifunctional enzymes in cancer. Nat. Rev. Cancer 2006, 6, 764–775. [Google Scholar]
- Gocheva, V.; Joyce, J.A. Cysteine cathepsins and the cutting edge of cancer invasion. Cell Cycle 2007, 6, 60–64. [Google Scholar]
- Laufs, S.; Schumacher, J.; Allgayer, H. Urokinase-receptor (u-PAR): An essential player in multiple games of cancer: A review on its role in tumor progression, invasion, metastasis, proliferation/dormancy, clinical outcome and minimal residual disease. Cell Cycle 2006, 5, 1760–1771. [Google Scholar]
- Egeblad, M.; Werb, Z. New functions for the matrix metalloproteinases in cancer progression. Nat. Rev. Cancer 2002, 2, 161–174. [Google Scholar]
- Lynch, C.C.; Matrisian, L.M. Matrix metalloproteinases in tumor-host cell communication. Differentiation 2002, 70, 561–573. [Google Scholar]
- Masterson, J.; O’Dea, S. Posttranslational truncation of E-cadherin and significance for tumour progression. Cells Tissues Organs 2007, 185, 175–179. [Google Scholar]
- Vihinen, P.; Kahari, V.M. Matrix metalloproteinases in cancer: Prognostic markers and therapeutic targets. Int. J. Cancer 2002, 99, 157–166. [Google Scholar]
- Miettinen, P.J.; Ebner, R.; Lopez, A.R.; Derynck, R. TGF-β induced transdifferentiation of mammary epithelial cells to mesenchymal cells: Involvement of type I receptors. J. Cell Biol 1994, 127, 2021–2036. [Google Scholar]
- Mani, S.A.; Guo, W.; Liao, M.J.; Eaton, E.N.; Ayyanan, A.; Zhou, A.Y.; Brooks, M.; Reinhard, F.; Zhang, C.C.; Shipitsin, M.; et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 2008, 133, 704–715. [Google Scholar]
- Battula, V.L.; Evans, K.W.; Hollier, B.G.; Shi, Y.; Marini, F.C.; Ayyanan, A.; Wang, R.Y.; Brisken, C.; Guerra, R.; Andreeff, M.; et al. Epithelial-mesenchymal transition-derived cells exhibit multilineage differentiation potential similar to mesenchymal stem cells. Stem Cells 2010, 28, 1435–1445. [Google Scholar]
- Taube, J.H.; Herschkowitz, J.I.; Komurov, K.; Zhou, A.Y.; Gupta, S.; Yang, J.; Hartwell, K.; Onder, T.T.; Gupta, P.B.; Evans, K.W.; et al. Core epithelial-to-mesenchymal transition interactome gene-expression signature is associated with claudin-low and metaplastic breast cancer subtypes. Proc. Natl. Acad. Sci. USA 2010, 107, 15449–15454. [Google Scholar]
- Kang, Y.; Massague, J. Epithelial-mesenchymal transitions: Twist in development and metastasis. Cell 2004, 118, 277–279. [Google Scholar]
- Watson, M.A.; Ylagan, L.R.; Trinkaus, K.M.; Gillanders, W.E.; Naughton, M.J.; Weilbaecher, K.N.; Fleming, T.P.; Aft, R.L. Isolation and molecular profiling of bone marrow micrometastases identifies TWIST1 as a marker of early tumor relapse in breast cancer patients. Clin. Cancer Res 2007, 13, 5001–5009. [Google Scholar]
- Cheng, G.Z.; Chan, J.; Wang, Q.; Zhang, W.; Sun, C.D.; Wang, L.H. Twist transcriptionally up-regulates AKT2 in breast cancer cells leading to increased migration, invasion, and resistance to paclitaxel. Cancer Res 2007, 67, 1979–1987. [Google Scholar]
- Cantrell, D.A. Phosphoinositide 3-kinase signalling pathways. J. Cell Sci 2001, 114, 1439–1445. [Google Scholar]
- Bachman, K.E.; Argani, P.; Samuels, Y.; Silliman, N.; Ptak, J.; Szabo, S.; Konishi, H.; Karakas, B.; Blair, B.G.; Lin, C.; et al. The PIK3CA gene is mutated with high frequency in human breast cancers. Cancer Biol. Ther 2004, 3, 772–775. [Google Scholar]
- Chau, N.M.; Ashcroft, M. Akt2: A role in breast cancer metastasis. Breast Cancer Res 2004, 6, 55–57. [Google Scholar]
- Liang, K.; Lu, Y.; Li, X.; Zeng, X.; Glazer, R.I.; Mills, G.B.; Fan, Z. Differential roles of phosphoinositide-dependent protein kinase-1 and akt1 expression and phosphorylation in breast cancer cell resistance to paclitaxel, doxorubicin, and gemcitabine. Mol. Pharmacol 2006, 70, 1045–1052. [Google Scholar]
- Wyman, S.K.; Parkin, R.K.; Mitchell, P.S.; Fritz, B.R.; O’Briant, K.; Godwin, A.K.; Urban, N.; Drescher, C.W.; Knudsen, B.S.; Tewari, M. Repertoire of microRNAs in epithelial ovarian cancer as determined by next generation sequencing of small RNA cDNA libraries. PLoS One 2009, 4, e5311. [Google Scholar]
- Wang, W.; Li, F.; Zhang, Y.; Tu, Y.; Yang, Q.; Gao, X. Reduced expression of miR-22 in gastric cancer is related to clinicopathologic characteristics or patient prognosis. Diagn. Pathol 2013. [Google Scholar] [CrossRef]
- Song, S.J.; Poliseno, L.; Song, M.S.; Ala, U.; Webster, K.; Ng, C.; Beringer, G.; Brikbak, N.J.; Yuan, X.; Cantley, L.C.; et al. MicroRNA-antagonism regulates breast cancer stemness and metastasis via TET-family-dependent chromatin remodeling. Cell 2013, 154, 311–324. [Google Scholar]
- Song, S.J.; Ito, K.; Ala, U.; Kats, L.; Webster, K.; Sun, S.M.; Jongen-Lavrencic, M.; Manova-Todorova, K.; Teruya-Feldstein, J.; Avigan, D.E.; et al. The oncogenic microRNA miR-22 targets the TET2 tumor suppressor to promote hematopoietic stem cell self-renewal and transformation. Cell Stem Cell 2013, 13, 87–101. [Google Scholar]
- Tavazoie, S.F.; Alarcon, C.; Oskarsson, T.; Padua, D.; Wang, Q.; Bos, P.D.; Gerald, W.L.; Massague, J. Endogenous human microRNAs that suppress breast cancer metastasis. Nature 2008, 451, 147–152. [Google Scholar]
- Federico, A.; Pallante, P.; Bianco, M.; Ferraro, A.; Esposito, F.; Monti, M.; Cozzolino, M.; Keller, S.; Fedele, M.; Leone, V.; et al. Chromobox protein homologue 7 protein, with decreased expression in human carcinomas, positively regulates E-cadherin expression by interacting with the histone deacetylase 2 protein. Cancer Res 2009, 69, 7079–7087. [Google Scholar]
- Gandellini, P.; Profumo, V.; Casamichele, A.; Fenderico, N.; Borrelli, S.; Petrovich, G.; Santilli, G.; Callari, M.; Colecchia, M.; Pozzi, S.; et al. MiR-205 regulates basement membrane deposition in human prostate: Implications for cancer development. Cell Death Differ 2012, 19, 1750–1760. [Google Scholar]
- Chaffer, C.L.; Weinberg, R.A. A perspective on cancer cell metastasis. Science 2011, 331, 1559–1564. [Google Scholar]
- Sanchez-Tillo, E.; Liu, Y.; de Barrios, O.; Siles, L.; Fanlo, L.; Cuatrecasas, M.; Darling, D.S.; Dean, D.C.; Castells, A.; Postigo, A. EMT-activating transcription factors in cancer: Beyond EMT and tumor invasiveness. Cell. Mol. Life Sci 2012, 69, 3429–3456. [Google Scholar]
- Lamouille, S.; Subramanyam, D.; Blelloch, R.; Derynck, R. Regulation of epithelial-mesenchymal and mesenchymal-epithelial transitions by microRNAs. Curr. Opin. Cell Biol 2013, 25, 200–207. [Google Scholar]
- Lee, J.; Choi, J.H.; Joo, C.K. TGF-β1 regulates cell fate during epithelial-mesenchymal transition by upregulating survivin. Cell Death Dis 2013. [Google Scholar] [CrossRef]
- Matsumura, N.; Huang, Z.; Mori, S.; Baba, T.; Fujii, S.; Konishi, I.; Iversen, E.S.; Berchuck, A.; Murphy, S.K. Epigenetic suppression of the TGF-β pathway revealed by transcriptome profiling in ovarian cancer. Genome Res 2011, 21, 74–82. [Google Scholar]
- Dong, Z.; Guo, W.; Guo, Y.; Kuang, G.; Yang, Z. Concordant promoter methylation of transforming growth factor-β receptor types I and II occurs early in esophageal squamous cell carcinoma. Am. J. Med. Sci 2012, 343, 375–381. [Google Scholar]
- Sarkar, S.; Faller, D.V. T-oligos inhibit growth and induce apoptosis in human ovarian cancer cells. Oligonucleotides 2011, 21, 47–53. [Google Scholar]
- Frew, A.J.; Lindemann, R.K.; Martin, B.P.; Clarke, C.J.; Sharkey, J.; Anthony, D.A.; Banks, K.M.; Haynes, N.M.; Gangatirkar, P.; Stanley, K.; et al. Combination therapy of established cancer using a histone deacetylase inhibitor and a TRAIL receptor agonist. Proc. Natl. Acad. Sci. USA 2008, 105, 11317–11322. [Google Scholar]
- Matei, D.E.; Nephew, K.P. Epigenetic therapies for chemoresensitization of epithelial ovarian cancer. Gynecol. Oncol 2010, 116, 195–201. [Google Scholar]
- Sarkar, S.; Abujamra, A.L.; Loew, J.E.; Forman, L.W.; Perrine, S.P.; Faller, D.V. Histone deacetylase inhibitors reverse CpG methylation by regulating DNMT1 through ERK signaling. Anticancer Res 2011, 31, 2723–2732. [Google Scholar]
- Zhou, Q.; Agoston, A.T.; Atadja, P.; Nelson, W.G.; Davidson, N.E. Inhibition of histone deacetylases promotes ubiquitin-dependent proteasomal degradation of DNA methyltransferase 1 in human breast cancer cells. Mol. Cancer Res 2008, 6, 873–883. [Google Scholar]
- Fujii, S.; Luo, R.Z.; Yuan, J.; Kadota, M.; Oshimura, M.; Dent, S.R.; Kondo, Y.; Issa, J.P.; Bast, R.C., Jr.; Yu, Y. Reactivation of the silenced and imprinted alleles of ARHI is associated with increased histone H3 acetylation and decreased histone H3 lysine 9 methylation. Hum. Mol. Genet 2003, 12, 1791–1800. [Google Scholar]
- Slamon, D.J.; Godolphin, W.; Jones, L.A.; Holt, J.A.; Wong, S.G.; Keith, D.E.; Levin, W.J.; Stuart, S.G.; Udove, J.; Ullrich, A.; et al. Studies of the HER-2/neu proto-oncogene in human breast and ovarian cancer. Science 1989, 244, 707–712. [Google Scholar]
- Zuo, T.; Liu, T.M.; Lan, X.; Weng, Y.I.; Shen, R.; Gu, F.; Huang, Y.W.; Liyanarachchi, S.; Deatherage, D.E.; Hsu, P.Y.; et al. Epigenetic silencing mediated through activated PI3K/AKT signaling in breast cancer. Cancer Res 2011, 71, 1752–1762. [Google Scholar]
- Esteve, P.O.; Chang, Y.; Samaranayake, M.; Upadhyay, A.K.; Horton, J.R.; Feehery, G.R.; Cheng, X.; Pradhan, S. A methylation and phosphorylation switch between an adjacent lysine and serine determines human DNMT1 stability. Nat. Struct. Mol. Biol 2011, 18, 42–48. [Google Scholar]
- Shen, Y.; Wang, P.; Li, Y.; Ye, F.; Wang, F.; Wan, X.; Cheng, X.; Lu, W.; Xie, X. MiR-375 is upregulated in acquired paclitaxel resistance in cervical cancer. Br. J. Cancer 2013, 109, 92–99. [Google Scholar]
- Sorrentino, A.; Liu, C.G.; Addario, A.; Peschle, C.; Scambia, G.; Ferlini, C. Role of microRNAs in drug-resistant ovarian cancer cells. Gynecol. Oncol 2008, 111, 478–486. [Google Scholar]
- Glover, A.B.; Leyland-Jones, B.R.; Chun, H.G.; Davies, B.; Hoth, D.F. Azacitidine: Ten years later. Cancer Treat. Rep 1987, 71, 737–746. [Google Scholar]
- Brock, M.V.; Hooker, C.M.; Ota-Machida, E.; Han, Y.; Guo, M.; Ames, S.; Glockner, S.; Piantadosi, S.; Gabrielson, E.; Pridham, G.; et al. DNA methylation markers and early recurrence in stage I lung cancer. N. Engl. J. Med 2008, 358, 1118–1128. [Google Scholar]
- Juergens, R.A.; Wrangle, J.; Vendetti, F.P.; Murphy, S.C.; Zhao, M.; Coleman, B.; Sebree, R.; Rodgers, K.; Hooker, C.M.; Franco, N.; et al. Combination epigenetic therapy has efficacy in patients with refractory advanced non-small cell lung cancer. Cancer Discov 2011, 1, 598–607. [Google Scholar]
- Sanchez-Gonzalez, B.; Yang, H.; Bueso-Ramos, C.; Hoshino, K.; Quintas-Cardama, A.; Richon, V.M.; Garcia-Manero, G. Antileukemia activity of the combination of an anthracycline with a histone deacetylase inhibitor. Blood 2006, 108, 1174–1182. [Google Scholar]
- Blum, K.A.; Liu, Z.; Lucas, D.M.; Chen, P.; Xie, Z.; Baiocchi, R.; Benson, D.M.; Devine, S.M.; Jones, J.; Andritsos, L.; et al. Phase I trial of low dose decitabine targeting DNA hypermethylation in patients with chronic lymphocytic leukaemia and non-Hodgkin lymphoma: Dose-limiting myelosuppression without evidence of DNA hypomethylation. Br. J. Haematol 2010, 150, 189–195. [Google Scholar]
- Blum, W.; Klisovic, R.B.; Hackanson, B.; Liu, Z.; Liu, S.; Devine, H.; Vukosavljevic, T.; Huynh, L.; Lozanski, G.; Kefauver, C.; et al. Phase I study of decitabine alone or in combination with valproic acid in acute myeloid leukemia. J. Clin. Oncol 2007, 25, 3884–3891. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Sarkar, S.; Horn, G.; Moulton, K.; Oza, A.; Byler, S.; Kokolus, S.; Longacre, M. Cancer Development, Progression, and Therapy: An Epigenetic Overview. Int. J. Mol. Sci. 2013, 14, 21087-21113. https://doi.org/10.3390/ijms141021087
Sarkar S, Horn G, Moulton K, Oza A, Byler S, Kokolus S, Longacre M. Cancer Development, Progression, and Therapy: An Epigenetic Overview. International Journal of Molecular Sciences. 2013; 14(10):21087-21113. https://doi.org/10.3390/ijms141021087
Chicago/Turabian StyleSarkar, Sibaji, Garrick Horn, Kimberly Moulton, Anuja Oza, Shannon Byler, Shannon Kokolus, and McKenna Longacre. 2013. "Cancer Development, Progression, and Therapy: An Epigenetic Overview" International Journal of Molecular Sciences 14, no. 10: 21087-21113. https://doi.org/10.3390/ijms141021087