MicroRNA-Regulated Pathways in Hematological Malignancies: How to Avoid Cells Playing Out of Tune



Abstract
:1. Introduction
2. Oncosuppressor miRNA Pathways
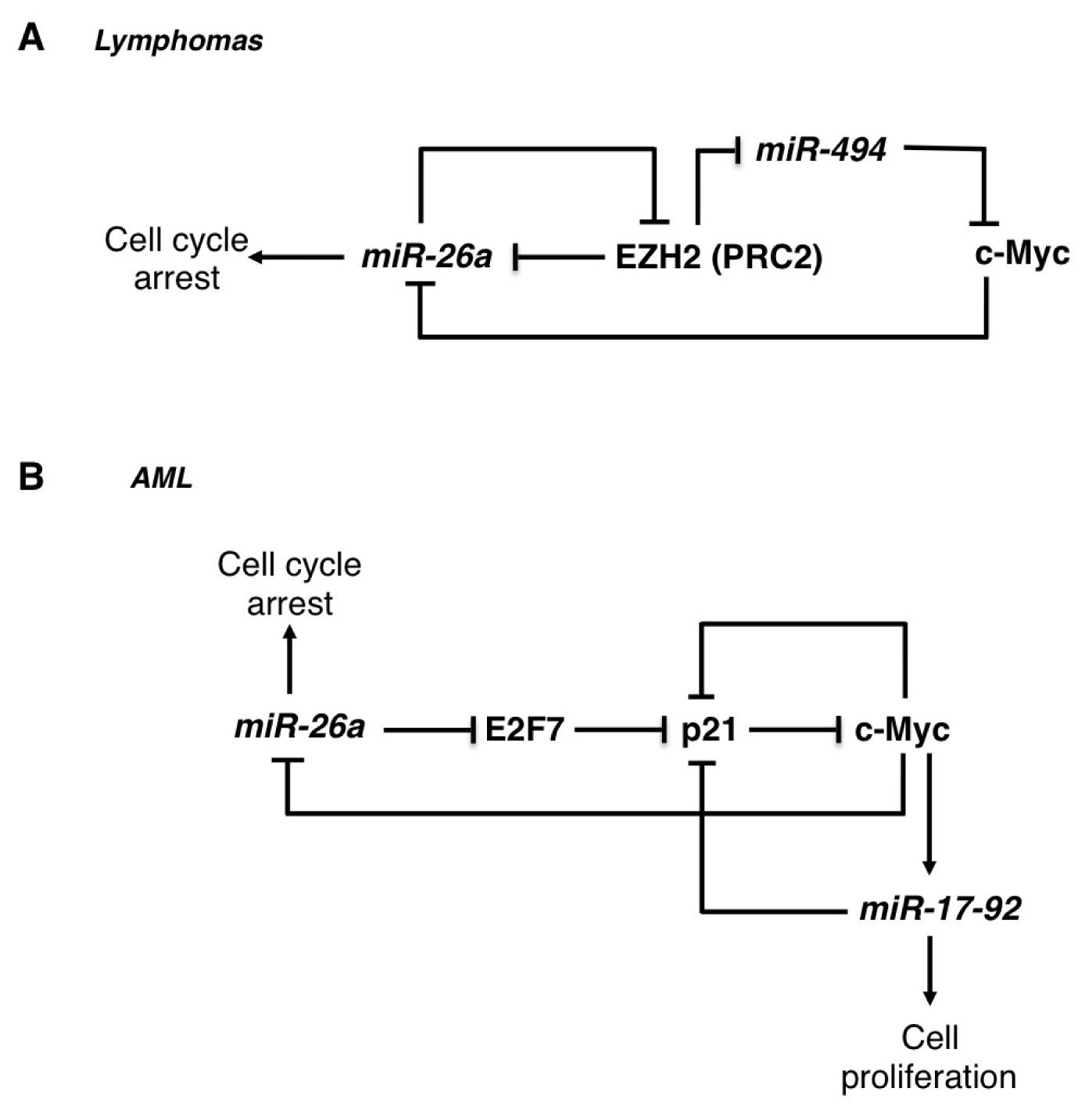
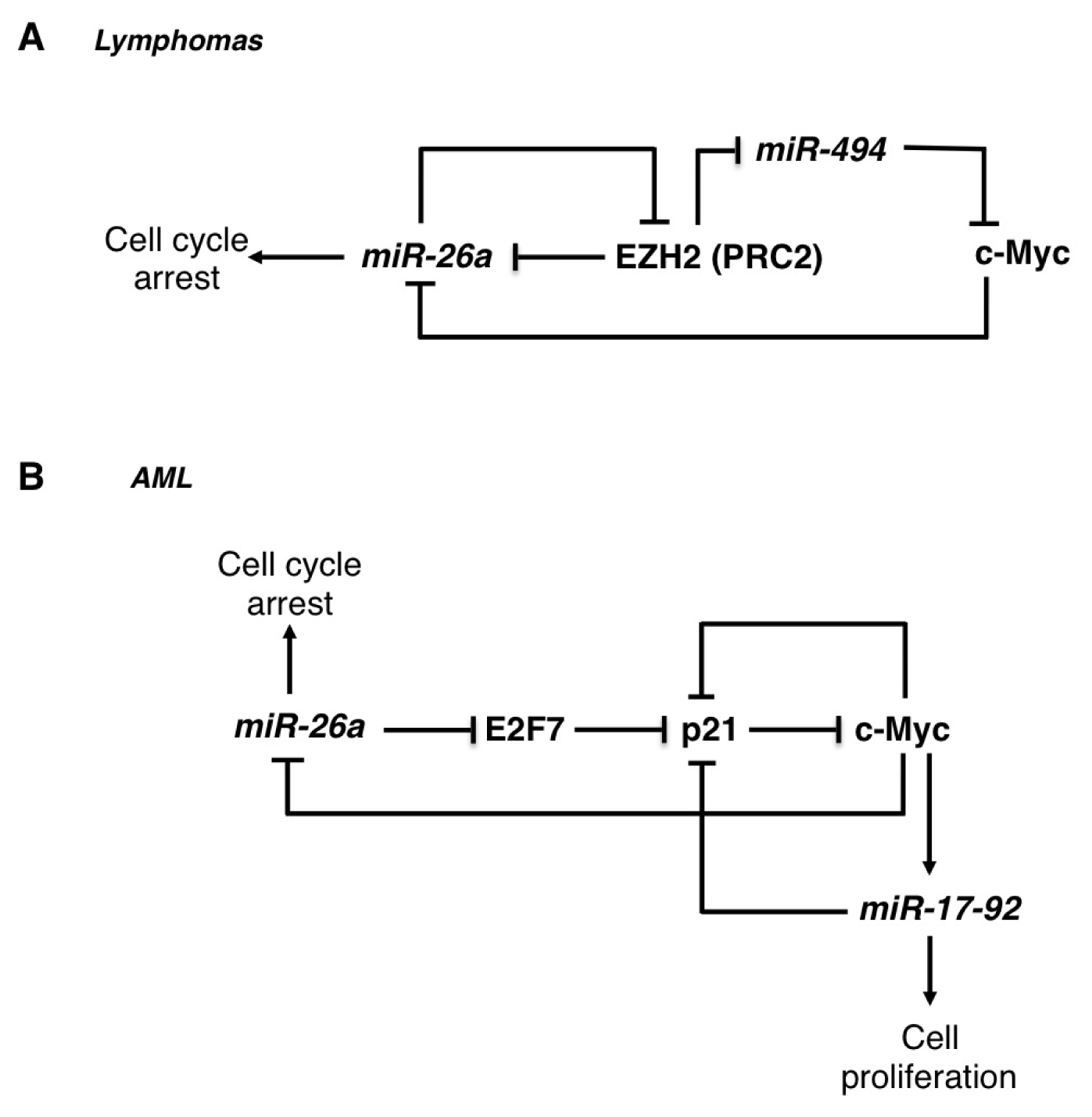
2.1. The miR-26 Family
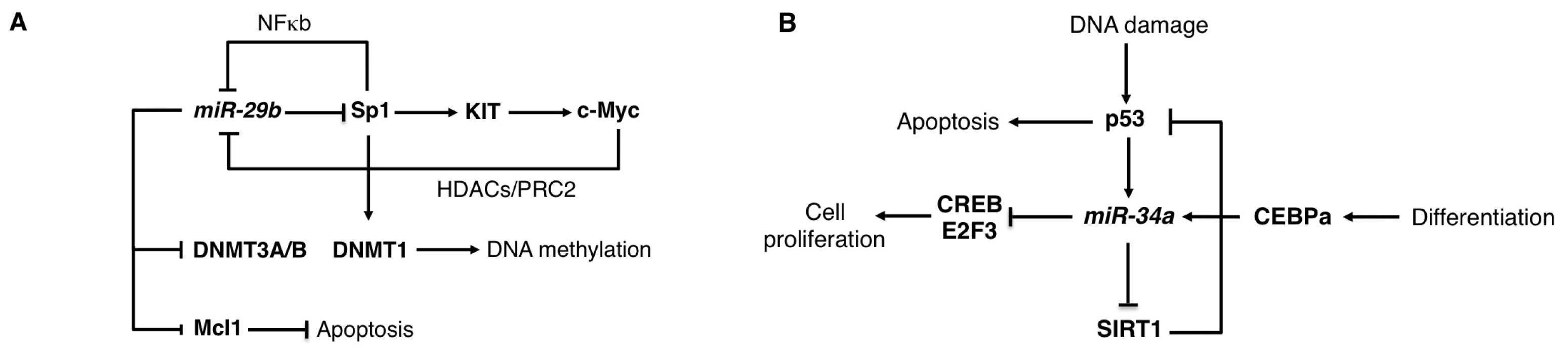
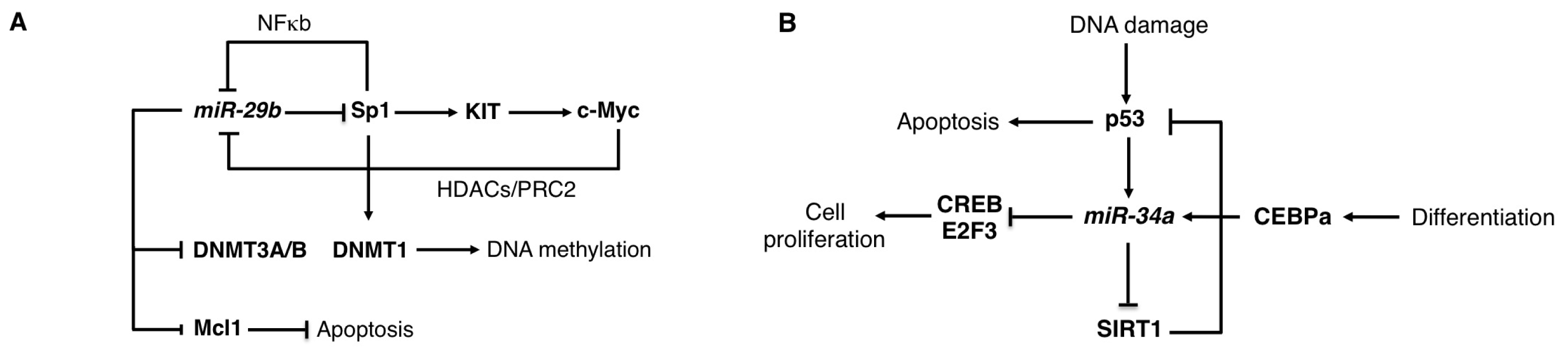
2.2. The miR-29 Family
2.3. The miR-34 Family
2.4. The miR-146 Family
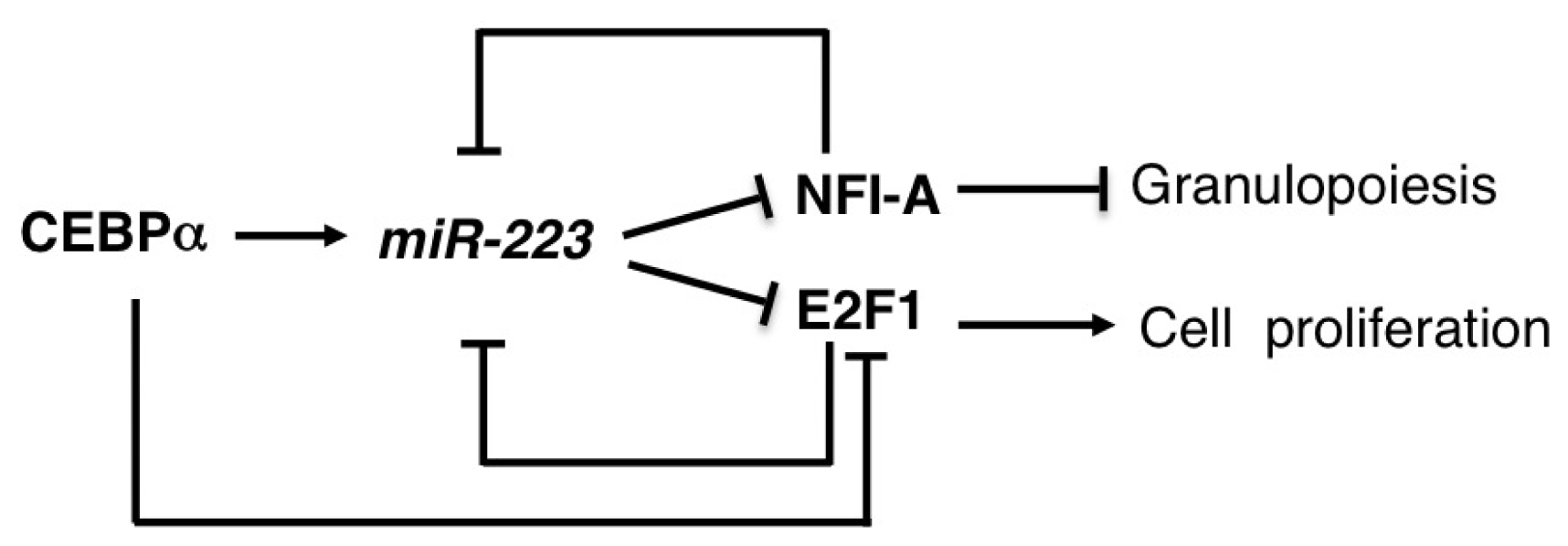
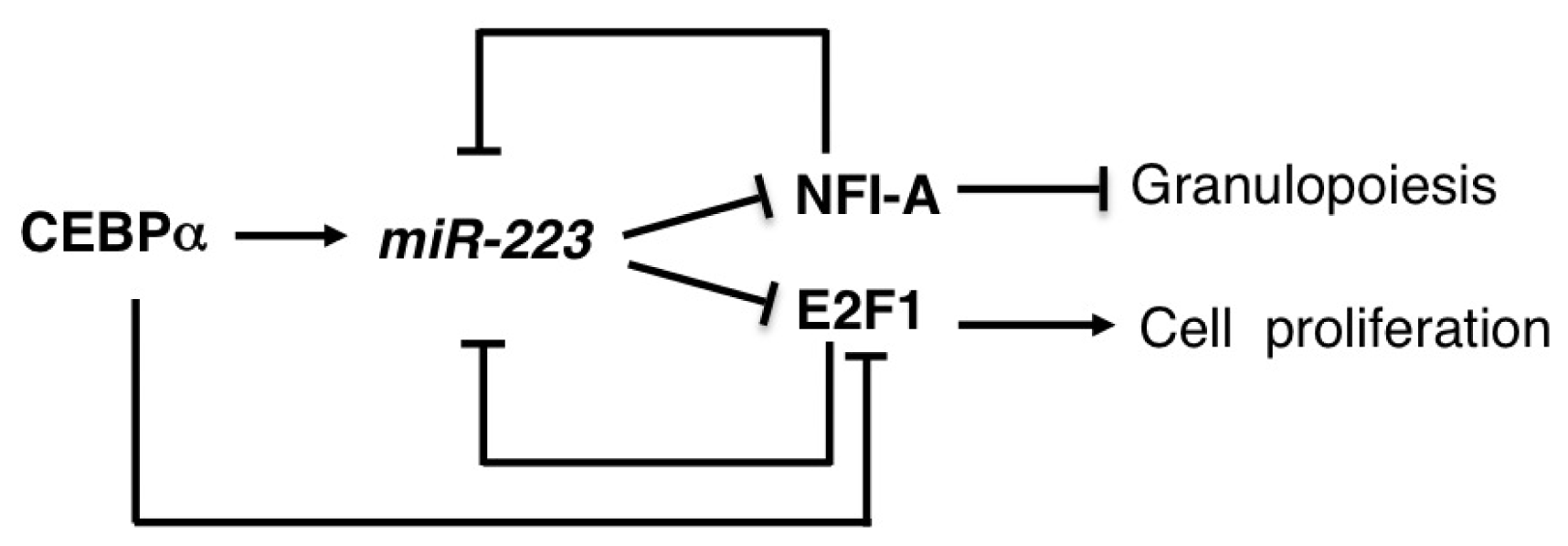
2.5. miR-223
2.6. miR-328
3. Oncogenic miRNA Pathways
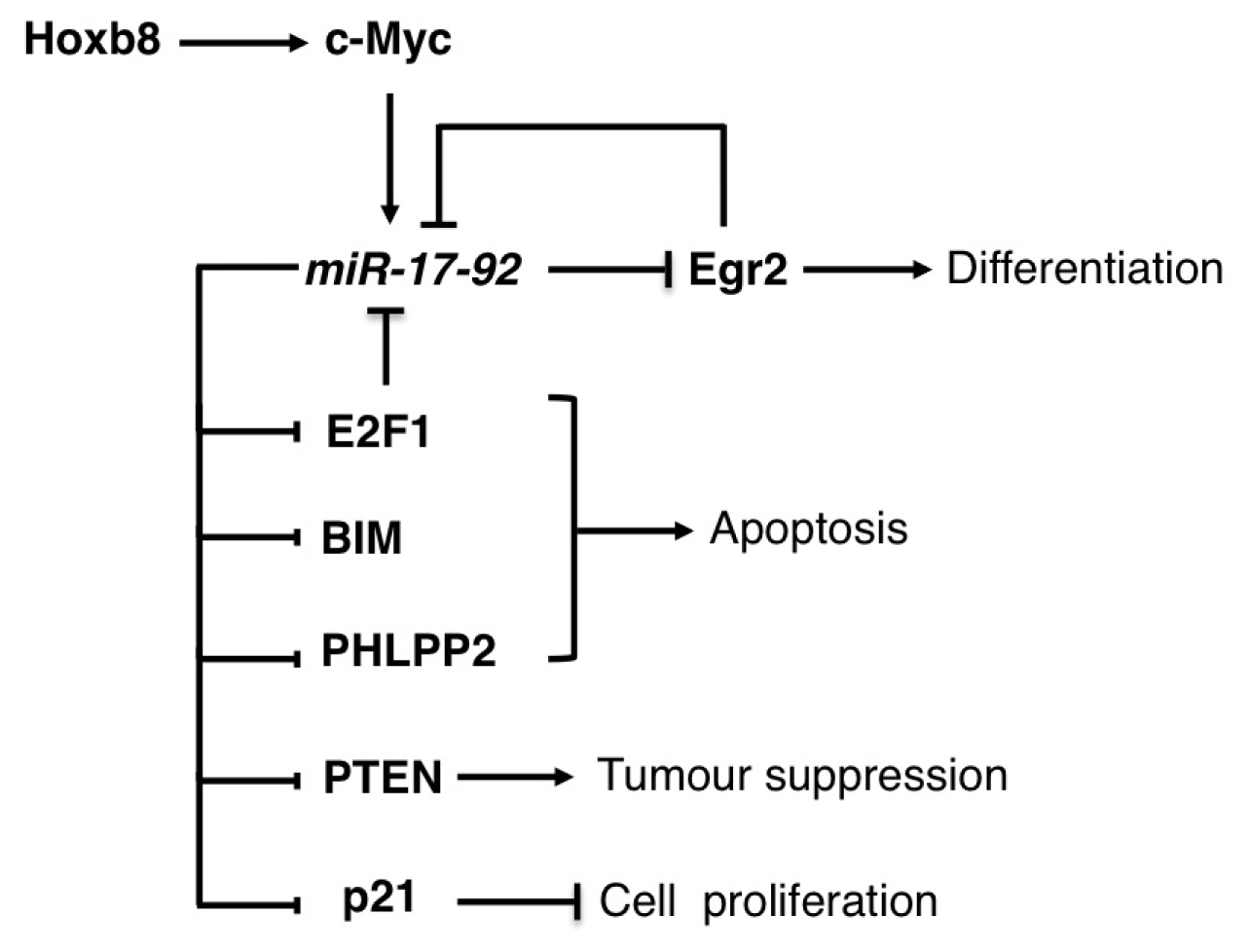
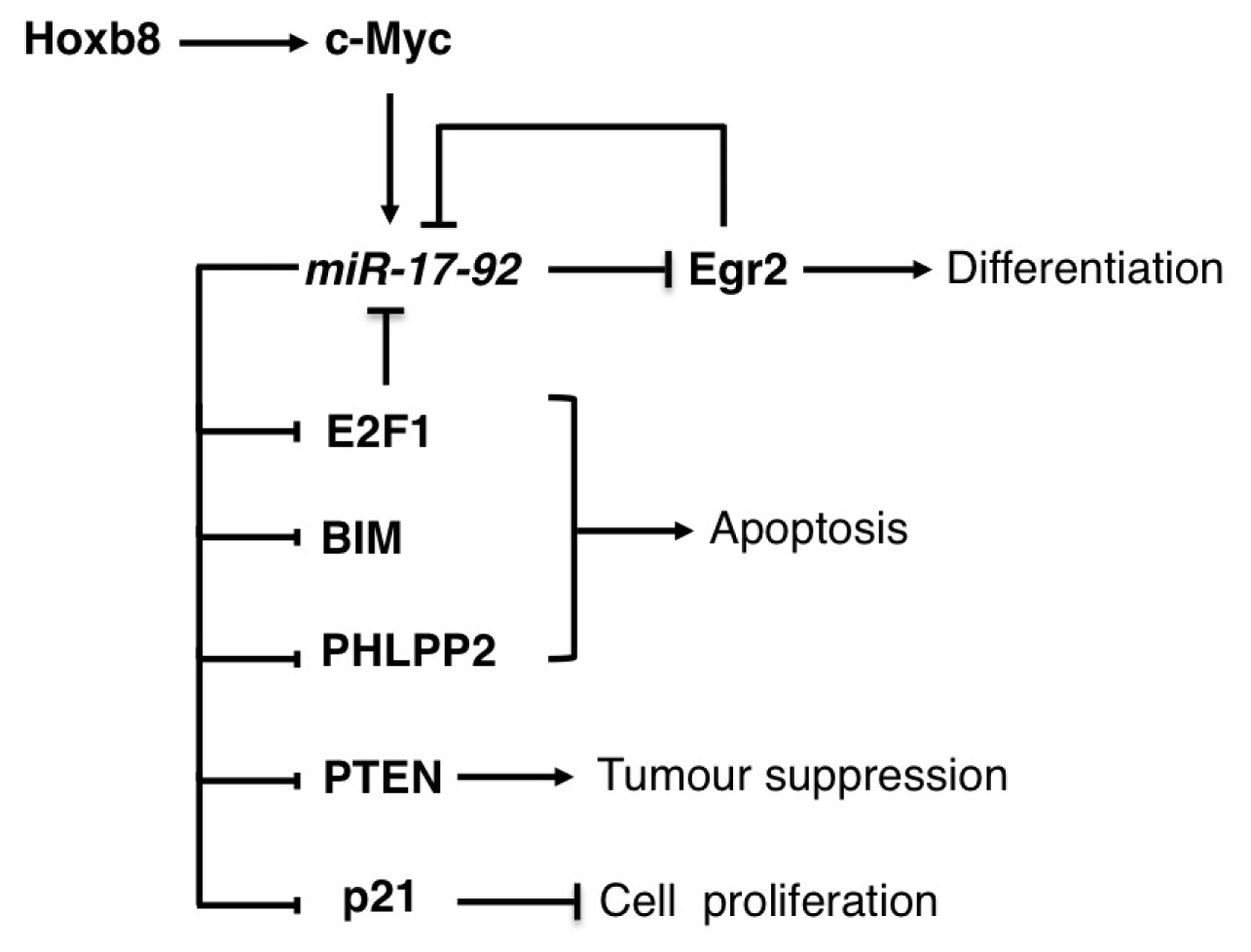
3.1. The miR-17-92 Family
3.2. The miR-125 Family
3.3. The miR-155
3.4. The miR-196b
4. miRNA Pathways and Drug Response Efficacy
5. Conclusions

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| MicroRNA | Malignancies | Function | Targets | References |
|---|---|---|---|---|
| miR-17-92 | Lymphomas | OG | E2Fs, PTEN, BIM, PHLP2 | [14–17] |
| T-ALL | OG | PTEN, BIM | [18] | |
| AML | OG | EGR2, BIM, P21 | [19–22] | |
| miR-26 | Lymphomas | OS | EZH2 | [23] |
| AML | OS | E2F7 | [24] | |
| T-ALL | OG | PTEN | [25] | |
| miR-29 | Lymphomas | OS | CDK6 | [26] |
| AML | OS | DNMT3A, DNMT3B, SP1, MCL1 | [27–29] | |
| B-CLL | OS/OG | HBP1, TCL1, PXDN | [26,30] | |
| miR-34 | AML | OS | CREB, E2F3 | [31,32] |
| CLL | OS | TCL1, SIRT1 | [33,34] | |
| miR-125 | AML and ALL | OG | CBFB, ABTB1, BAK1, PTPN18, PTPN7, PP1CA, PP2CA, LIN28 | [35–40] |
| miR-146 | MDS | OS | TRAF6 | [41,42] |
| miR-155 | Lymphomas and ALL | OG | SHIP, CEBPβ, HDAC4, BCL6 | [43–45] |
| AML | OG | BACH1, SLA, CUTL1, CSF1R, JARID2, CEBPβ, PU.1, ARNT1, HIF1A, PICALM | [46] | |
| miR-223 | AML | OS | E2F1, NFI-A | [47,48] |
| T-ALL | OG | FBXW7 | [49] | |
| miR-328 | CML | OS | PIM1 | [50] |
Acknowledgments
Conflicts of Interest
References
- Bartel, D.P. MicroRNAs: Target recognition and regulatory functions. Cell 2009, 136, 215–233. [Google Scholar]
- Kim, V.N. MicroRNA biogenesis: Coordinated cropping and dicing. Nat. Rev. Mol. Cell Biol 2005, 6, 376–385. [Google Scholar]
- Kim, V.N.; Han, J.; Siomi, M.C. Biogenesis of small RNAs in animals. Nat. Rev. Mol. Cell Biol 2009, 10, 126–139. [Google Scholar]
- Sun, K.; Lai, E.C. Adult-specific functions of animal microRNAs. Nat. Rev. Genet 2013, 14, 535–548. [Google Scholar]
- Tenen, D.G. Disruption of differentiation in human cancer: AML shows the way. Nat. Rev. Cancer 2003, 3, 89–101. [Google Scholar]
- Lujambio, A.; Lowe, S.W. The microcosmos of cancer. Nature 2012, 482, 347–355. [Google Scholar]
- Iwasaki, H.; Akashi, K. Hematopoietic developmental pathways: On cellular basis. Oncogene 2007, 26, 6687–6696. [Google Scholar]
- Doulatov, S.; Notta, F.; Laurenti, E.; Dick, J.E. Hematopoiesis: A human perspective. Cell Stem Cell 2012, 10, 120–136. [Google Scholar]
- Rosenbauer, F.; Tenen, D.G. Transcription factors in myeloid development: Balancing differentiation with transformation. Nat. Rev. Immunol 2007, 7, 105–117. [Google Scholar]
- Ebert, M.S.; Sharp, P.A. Roles for microRNAs in conferring robustness to biological processes. Cell 2012, 149, 515–524. [Google Scholar]
- Baltimore, D.; Boldin, M.P.; O’Connell, R.M.; Rao, D.S.; Taganov, K.D. MicroRNAs: New regulators of immune cell development and function. Nat. Immunol 2008, 9, 839–845. [Google Scholar]
- O’Connell, R.M.; Rao, D.S.; Chaudhuri, A.A.; Baltimore, D. Physiological and pathological roles for microRNAs in the immune system. Nat. Rev. Immunol 2010, 10, 111–122. [Google Scholar]
- Calin, G.A.; Sevignani, C.; Dumitru, C.D.; Hyslop, T.; Noch, E.; Yendamuri, S.; Shimizu, M.; Rattan, S.; Bullrich, F.; Negrini, M.; et al. Human microRNA genes are frequently located at fragile sites and genomic regions involved in cancers. Proc. Natl. Acad. Sci. USA 2004, 101, 2999–3004. [Google Scholar]
- Ota, A.; Tagawa, H.; Karnan, S.; Tsuzuki, S.; Karpas, A.; Kira, S.; Yoshida, Y.; Seto, M. Identification and characterization of a novel gene, C13orf25, as a target for 13q31–q32 amplification in malignant lymphoma. Cancer Res 2004, 64, 3087–3095. [Google Scholar]
- He, L.; Thomson, J.M.; Hemann, M.T.; Hernando-Monge, E.; Mu, D.; Goodson, S.; Powers, S.; Cordon-Cardo, C.; Lowe, S.W.; Hannon, G.J.; et al. A microRNA polycistron as a potential human oncogene. Nature 2005, 435, 828–833. [Google Scholar]
- Mu, P.; Han, Y.C.; Betel, D.; Yao, E.; Squatrito, M.; Ogrodowski, P.; de Stanchina, E.; D’Andrea, A.; Sander, C.; Ventura, A. Genetic dissection of the miR-17-92 cluster of microRNAs in Myc-induced B-cell lymphomas. Genes Dev 2009, 23, 2806–2811. [Google Scholar]
- Xiao, C.; Srinivasan, L.; Calado, D.P.; Patterson, H.C.; Zhang, B.; Wang, J.; Henderson, J.M.; Kutok, J.L.; Rajewsky, K. Lymphoproliferative disease and autoimmunity in mice with increased miR-17-92 expression in lymphocytes. Nat. Immunol 2008, 9, 405–414. [Google Scholar]
- Mavrakis, K.J.; Wolfe, A.L.; Oricchio, E.; Palomero, T.; de Keersmaecker, K.; McJunkin, K.; Zuber, J.; James, T.; Khan, A.A.; Leslie, C.S.; et al. Genome-wide RNA-mediated interference screen identifies miR-19 targets in Notch-induced T-cell acute lymphoblastic leukemia. Nat. Cell Biol 2010, 12, 372–379. [Google Scholar]
- Pospisil, V.; Vargova, K.; Kokavec, J.; Rybarova, J.; Savvulidi, F.; Jonasova, A.; Necas, E.; Zavadil, J.; Laslo, P.; Stopka, T. Epigenetic silencing of the oncogenic miR-17-92 cluster during PU.1-directed macrophage differentiation. EMBO J 2011, 30, 4450–4464. [Google Scholar]
- Fontana, L.; Pelosi, E.; Greco, P.; Racanicchi, S.; Testa, U.; Liuzzi, F.; Croce, C.M.; Brunetti, E.; Grignani, F.; Peschle, C. MicroRNAs 17-5p-20a-106a control monocytopoiesis through AML1 targeting and M-CSF receptor upregulation. Nat. Cell. Biol 2007, 9, 775–787. [Google Scholar]
- Salmanidis, M.; Brumatti, G.; Narayan, N.; Green, B.D.; van den Bergen, J.A.; Sandow, J.J.; Bert, A.G.; Silke, N.; Sladic, R.; Puthalakath, H.; et al. Hoxb8 regulates expression of microRNAs to control cell death and differentiation. Cell Death Differ 2013, 20, 1370–1380. [Google Scholar]
- Ivanovska, I.; Ball, A.S.; Diaz, R.L.; Magnus, J.F.; Kibukawa, M.; Schelter, J.M.; Kobayashi, S.V.; Lim, L.; Burchard, J.; Jackson, A.L.; et al. MicroRNAs in the miR-106b family regulate p21/CDKN1A and promote cell cycle progression. Mol. Cell. Biol 2008, 28, 2167–2174. [Google Scholar]
- Sander, S.; Bullinger, L.; Klapproth, K.; Fiedler, K.; Kestler, H.A.; Barth, T.F.; Möller, P.; Stilgenbauer, S.; Pollack, J.R.; Wirth, T. MYC stimulates EZH2 expression by repression of its negative regulator miR-26a. Blood 2008, 112, 4202–4212. [Google Scholar]
- Salvatori, B.; Iosue, I.; Mangiavacchi, A.; Loddo, G.; Padula, F.; Chiaretti, S.; Peragine, N.; Bozzoni, I.; Fazi, F.; Fatica, A. The microRNA-26a target E2F7 sustains cell proliferation and inhibits monocytic differentiation of acute myeloid leukemia cells. Cell Death Dis 2012, 3, e413. [Google Scholar]
- Mavrakis, K.J.; van der Meulen, J.; Wolfe, A.L.; Liu, X.; Mets, E.; Taghon, T.; Khan, A.A.; Setty, M.; Rondou, P.; Vandenberghe, P.; et al. A cooperative microRNA-tumour suppressor gene network in acute T-cell lymphoblastic leukemia (T-ALL). Nat. Genet 2011, 43, 673–678. [Google Scholar]
- Zhao, J.J.; Lin, J.; Lwin, T.; Yang, H.; Guo, J.; Kong, W.; Dessureault, S.; Moscinski, L.C.; Rezania, D.; Dalton, W.S.; et al. microRNA expression profile and identification of miR-29 as a prognostic marker and pathogenetic factor by targeting CDK6 in mantle cell lymphoma. Blood 2010, 115, 2630–2639. [Google Scholar]
- Garzon, R.; Liu, S.; Fabbri, M.; Liu, Z.; Heaphy, C.E.; Callegari, E.; Schwind, S.; Pang, J.; Yu, J.; Muthusamy, N.; et al. MicroRNA-29b induces global DNA hypomethylation and tumour suppressor gene reexpression in acute myeloid leukemia by targeting directly DNMT3A and 3B and indirectly DNMT1. Blood 2009, 113, 6411–6418. [Google Scholar]
- Garzon, R.; Heaphy, C.E.; Havelange, V.; Fabbri, M.; Volinia, S.; Tsao, T.; Zanesi, N.; Kornblau, S.M.; Marcucci, G.; Calin, G.A.; et al. MicroRNA 29b functions in acute myeloid leukemia. Blood 2009, 114, 5331–5341. [Google Scholar]
- Zhang, Y.K.; Wang, H.; Leng, Y.; Li, Z.L.; Yang, Y.F.; Xiao, F.J.; Li, Q.F.; Chen, X.Q.; Wang, L.S. Overexpression of microRNA-29b induces apoptosis of multiple myeloma cells through down regulating Mcl-1. Biochem. Biophys. Res. Commun 2011, 414, 233–239. [Google Scholar]
- Liu, S.; Wu, L.C.; Pang, J.; Santhanam, R.; Schwind, S.; Wu, Y.Z.; Hickey, C.J.; Yu, J.; Becker, H.; Maharry, K.; et al. Sp1/NFkappaB/HDAC/miR-29b regulatory network in KIT-driven myeloid leukemia. Cancer Cell 2010, 17, 333–347. [Google Scholar]
- Pigazzi, M.; Manara, E.; Baron, E.; Basso, G. miR-34b targets cyclic AMP-responsive element binding protein in acute myeloid leukemia. Cancer Res 2009, 69, 2471–2478. [Google Scholar]
- Pulikkan, J.A.; Peramangalam, P.S.; Dengler, V.; Ho, P.A.; Preudhomme, C.; Meshinchi, S.; Christopeit, M.; Nibourel, O.; Müller-Tidow, C.; Bohlander, S.K.; et al. C/EBPα regulated microRNA-34a targets E2F3 during granulopoiesis and is down-regulated in AML with CEBPA mutations. Blood 2010, 116, 5638–5649. [Google Scholar]
- Cardinaud, B.; Moreilhon, C.; Marcet, B.; Robbe-Sermesant, K.; LeBrigand, K.; Mari, B.; Eclache, V.; Cymbalista, F.; Raynaud, S.; Barbry, P. miR-34b/miR-34c: A regulator of TCL1 expression in 11q-chronic lymphocytic leukemia? Leukemia 2009, 23, 2174–2177. [Google Scholar]
- Audrito, V.; Vaisitti, T.; Rossi, D.; Gottardi, D.; D’Arena, G.; Laurenti, L.; Gaidano, G.; Malavasi, F.; Deaglio, S. Nicotinamide blocks proliferation and induces apoptosis ofchronic lymphocytic leukemia cells through activation of the p53/miR-34a/SIRT1 tumour suppressor network. Cancer Res 2011, 71, 4473–4483. [Google Scholar]
- Surdziel, E.; Cabanski, M.; Dallmann, I.; Lyszkiewicz, M.; Krueger, A.; Ganser, A.; Scherr, M.; Eder, M. Enforced expression of miR-125b affects myelopoiesis by targeting multiple signaling pathways. Blood 2011, 117, 4338–4348. [Google Scholar]
- Bousquet, M.; Harris, M.H.; Zhou, B.; Lodish, H.F. MicroRNA miR-125b causes leukemia. Proc. Natl. Acad. Sci. USA 2010, 107, 21558–21563. [Google Scholar]
- O’Connell, R.M.; Chaudhuri, A.A.; Rao, D.S.; Gibson, W.S.; Balazs, A.B.; Baltimore, D. MicroRNAs enriched in hematopoietic stem cells differentially regulate long-term hematopoietic output. Proc. Natl. Acad. Sci. USA 2010, 107, 14235–14240. [Google Scholar]
- Chaudhuri, A.A.; So, A.Y.; Mehta, A.; Minisandram, A.; Sinha, N.; Jonsson, V.D.; Rao, D.S.; O’Connell, R.M.; Baltimore, D. Oncomir miR-125b regulates hematopoiesis by targeting the gene Lin28A. Proc. Natl. Acad. Sci. USA 2012, 109, 4233–4238. [Google Scholar]
- Guo, S.; Bai, H.; Megyola, C.M.; Halene, S.; Krause, D.S.; Scadden, D.T.; Lu, J. Complex oncogene dependence in microRNA-125a-induced myeloproliferative neoplasms. Proc. Natl. Acad. Sci. USA 2012, 109, 16636–16641. [Google Scholar]
- Zhong, X.; Li, N.; Liang, S.; Huang, Q.; Coukos, G.; Zhang, L. Identification of microRNAs regulating reprogramming factor LIN28 in embryonic stem cells and cancer cells. J. Biol. Chem 2010, 285, 41961–41971. [Google Scholar]
- Boldin, M.P.; Taganov, K.D.; Rao, D.S.; Yang, L.; Zhao, J.L.; Kalwani, M.; Garcia-Flores, Y.; Luong, M.; Devrekanli, A.; Xu, J.; et al. miR-146a is a significant brake on autoimmunity, myeloproliferation, and cancer in mice. J. Exp. Med 2011, 208, 1189–1201. [Google Scholar]
- Zhao, J.L.; Rao, D.S.; Boldin, M.P.; Taganov, K.D.; O’Connell, R.M.; Baltimore, D. NF-kappaB dysregulation in microRNA-146a-deficient mice drives the development of myeloid malignancies. Proc. Natl. Acad. Sci. USA 2011, 108, 9184–9189. [Google Scholar]
- Kluiver, J.; Poppema, S.; de Jong, D.; Blokzijl, T.; Harms, G.; Jacobs, S.; Kroesen, B.J.; van den Berg, A. BIC and miR-155 are highly expressed in Hodgkin, primary mediastinal and diffuse large B cell lymphomas. J. Pathol 2005, 207, 243–249. [Google Scholar]
- Sandhu, S.K.; Volinia, S.; Costinean, S.; Galasso, M.; Neinast, R.; Santhanam, R.; Parthun, M.R.; Perrotti, D.; Marcucci, G.; Garzon, R.; et al. miR-155 targets histone deacetylase 4 (HDAC4) and impairs transcriptional activity of B-cell lymphoma 6 (BCL6) in the Eμ-miR-155 transgenic mouse model. Proc. Natl. Acad. Sci. USA 2012, 109, 20047–20052. [Google Scholar]
- O’Connell, R.M.; Chaudhuri, A.A.; Rao, D.S.; Baltimore, D. Inositol phosphatase SHIP1 is a primary target of miR-155. Proc. Natl. Acad. Sci. USA 2009, 106, 7113–7118. [Google Scholar]
- Marcucci, G.; Maharry, K.S.; Metzeler, K.H.; Volinia, S.; Wu, Y.Z.; Mrózek, K.; Nicolet, D.; Kohlschmidt, J.; Whitman, S.P.; Mendler, J.H; et al. Clinical Role of microRNAs in cytogenetically normal acute myeloid leukemia: miR-155 upregulation independently identifies high-risk patients. J. Clin. Oncol 2013, 31, 2086–2093. [Google Scholar]
- Fazi, F.; Rosa, A.; Fatica, A.; Gelmetti, V.; de Marchis, M.L.; Nervi, C.; Bozzoni, I. A minicircuitry comprised of microRNA-223 and transcription factors NFI-A and C/EBPalpha regulates human granulopoiesis. Cell 2005, 123, 819–831. [Google Scholar]
- Pulikkan, J.A.; Dengler, V.; Peramangalam, P.S.; Peer Zada, A.A.; Müller-Tidow, C.; Bohlander, S.K.; Tenen, D.G.; Behre, G. Cell-cycle regulator E2F1 and microRNA-223 comprise an autoregulatory negative feedback loop in acute myeloid leukemia. Blood 2010, 115, 1768–1778. [Google Scholar]
- Mansour, M.R.; Sanda, T.; Lawton, L.N.; Li, X.; Kreslavsky, T.; Novina, C.D.; Brand, M.; Gutierrez, A.; Kelliher, M.A.; Jamieson, C.H.; et al. The TAL1 complex targets the FBXW7 tumour suppressor by activating miR-223 in human T cell acute lymphoblastic leukemia. J. Exp. Med 2013, 210, 1545–1557. [Google Scholar]
- Eiring, A.M.; Harb, J.G.; Neviani, P.; Garton, C.; Oaks, J.J.; Spizzo, R.; Liu, S.; Schwind, S.; Santhanam, R.; Hickey, C.J.; et al. miR-328 functions as an RNA decoy to modulate hnRNP E2 regulation of mRNA translation in leukemic blasts. Cell 2010, 140, 652–665. [Google Scholar]
- Hoffman, B.; Amanullah, A.; Shafarenko, M.; Liebermann, D.A. The proto-oncogene c-myc in hematopoietic development and leukemogenesis. Oncogene 2002, 21, 3414–3421. [Google Scholar]
- Chang, T.C.; Yu, D.; Lee, Y.S.; Wentzel, E.A.; Arking, D.E.; West, K.M.; Dang, C.V.; Thomas-Tikhonenko, A.; Mendell, J.T. Widespread microRNA repression by Myc contributes to tumourigenesis. Nat. Genet 2008, 40, 43–50. [Google Scholar]
- Harris, A.W.; Pinkert, C.A.; Crawford, M.; Langdon, W.Y.; Brinster, R.L.; Adams, J.M. The Eμ-Myc transgenic mouse: A model for high-incidence spontaneous lymphoma and leukemia of early B cells. J. Exp. Med 1988, 167, 353–371. [Google Scholar]
- Luo, H.; O’Neal, J.; Kreisel, F.; Le Beau, M.M.; Tomasson, M.H. c-Myc rapidly induces acute myeloid leukemia in mice without evidence of lymphoma-associated antiapoptotic mutations. Blood 2005, 106, 2542–2561. [Google Scholar]
- Salvatori, B.; Iosue, I.; Djodji Damas, N.; Mangiavacchi, A.; Chiaretti, S.; Messina, M.; Padula, F.; Guarini, A.; Bozzoni, I.; Fazi, F.; et al. Critical role of c-Myc in acute myeloid leukemia involving direct regulation of miR-26a and histone methyltransferase EZH2. Genes Cancer 2011, 2, 585–592. [Google Scholar]
- Zhao, X.; Lwin, T.; Zhang, X.; Huang, A.; Wang, J.; Marquez, V.E.; Chen-Kiang, S.; Dalton, W.S.; Sotomayor, E.; Tao, J. Disruption of the MYC-miRNA-EZH2 loop to suppress aggressive B-cell lymphoma survival and clonogenicity. Leukemia 2013. [Google Scholar] [CrossRef]
- Cao, R.; Zhang, Y. The functions of E(Z)/EZH2-mediated methylation of lysine 27 in histone H3. Curr. Opin. Genet. Dev 2004, 14, 155–164. [Google Scholar]
- Zhu, Y.; Lu, Y.; Zhang, Q.; Liu, J.J.; Li, T.J.; Yang, J.R.; Zeng, C.; Zhuang, S.M. MicroRNA-26a/b and their host genes cooperate to inhibit the G1/S transition by activating the pRb protein. Nucleic Acids Res 2012, 40, 4615–4625. [Google Scholar]
- Zheng, Y.S.; Zhang, H.; Zhang, X.J.; Feng, D.D.; Luo, X.Q.; Zeng, C.W.; Lin, K.Y.; Zhou, H.; Qu, L.H.; Zhang, P.; et al. miR-100 regulates cell differentiation and survival by targeting RBSP3, a phosphatase-like tumour suppressor in acute myeloid leukemia. Oncogene 2012, 31, 80–92. [Google Scholar]
- Kota, J.; Chivukula, R.R.; O’Donnell, K.A.; Wentzel, E.A.; Montgomery, C.L.; Hwang, H.W.; Vivekanandan, P.; Torbenson, M.; Clark, K.R.; Mendell, J.R.; et al. Therapeutic microRNA delivery suppresses tumourigenesis in a murine liver cancer model. Cell 2009, 137, 1005–1017. [Google Scholar]
- Kim, H.; Huang, W.; Jiang, X.; Pennicooke, B.; Park, P.J.; Johnson, M.D. Integrative genome analysis reveals an oncomir/oncogene cluster regulating glioblastoma survivorship. Proc. Natl. Acad. Sci. USA 2010, 107, 2183–2188. [Google Scholar]
- Wang, X.S.; Gong, J.N.; Yu, J.; Wang, F.; Zhang, X.H.; Yin, X.L.; Tan, Z.Q.; Luo, Z.M.; Yang, G.H.; Shen, C.; et al. MicroRNA-29a and microRNA-142-3p are regulators of myeloid differentiation and acute myeloid leukemia. Blood 2012, 119, 4992–5004. [Google Scholar]
- Han, Y.C.; Park, C.Y.; Bhagat, G.; Zhang, J.; Wang, Y.; Fan, J.B.; Liu, M.; Zou, Y.; Weissman, I.L.; Gu, H. MicroRNA-29a induces aberrant self-renewal capacity in hematopoietic progenitors, biased myeloid development, and acute myeloid leukemia. J. Exp. Med 2010, 207, 475–489. [Google Scholar]
- Sampath, D.; Liu, C.; Vasan, K.; Sulda, M.; Puduvalli, V.K.; Wierda, W.G.; Keating, M.J. Histone deacetylases mediate the silencing of miR-15a, miR-16, and miR-29b in chronic lymphocytic leukemia. Blood 2012, 119, 1162–1172. [Google Scholar]
- Pekarsky, Y.; Santanam, U.; Cimmino, A.; Palamarchuk, A.; Efanov, A.; Maximov, V.; Volinia, S.; Alder, H.; Liu, C.G.; Rassenti, L; et al. Tcl1 expression in chronic lymphocytic leukemia is regulated by miR-29 and miR-181. Cancer Res 2006, 66, 11590–11593. [Google Scholar]
- Santanam, U.; Zanesi, N.; Efanov, A.; Costinean, S.; Palamarchuk, A.; Hagan, J.P.; Volinia, S.; Alder, H.; Rassenti, L.; Kipps, T.; et al. Chronic lymphocytic leukemia modeled in mouse by targeted miR-29 expression. Proc. Natl. Acad. Sci. USA 2010, 107, 12210–12215. [Google Scholar]
- Ciafrè, S.A.; Galardi, S. MicroRNAs and RNA-binding proteins: A complex network of interactions and reciprocal regulations in cancer. RNA Biol 2013, 10, 935–943. [Google Scholar]
- Zhang, X.; Zhao, X.; Fiskus, W.; Lin, J.; Lwin, T.; Rao, R.; Zhang, Y.; Chan, J.C.; Fu, K.; Marquez, V.E.; et al. Coordinated silencing of MYC-mediated miR-29 by HDAC3 and EZH2 as a therapeutic target of histone modification in aggressive B-Cell lymphomas. Cancer Cell 2012, 22, 506–523. [Google Scholar]
- Chim, C.S.; Wong, K.Y.; Qi, Y.; Loong, F.; Lam, W.L.; Wong, L.G.; Jin, D.Y.; Costello, J.F.; Liang, R. Epigenetic inactivation of the miR-34a in hematological malignancies. Carcinogenesis 2010, 31, 745–750. [Google Scholar]
- Hermeking, H. The miR-34 family in cancer and apoptosis. Cell Death Differ 2010, 17, 193–199. [Google Scholar]
- Dufour, A.; Palermo, G.; Zellmeier, E.; Mellert, G.; Duchateau-Nguyen, G.; Schneider, S.; Benthaus, T.; Kakadia, P.M.; Spiekermann, K.; Hiddemann, W.; et al. Inactivation of TP53 correlates with disease progression and low miR-34a expression in previously treated chronic lymphocytic leukemia patients. Blood 2013, 121, 3650–3657. [Google Scholar]
- Zenz, T.; Mohr, J.; Eldering, E.; Kater, A.P.; Bühler, A.; Kienle, D.; Winkler, D.; Dürig, J.; van Oers, M.H.; Mertens, D.; et al. miR-34a as part of the resistance network in chronic lymphocytic leukemia. Blood 2009, 113, 3801–3808. [Google Scholar]
- Pramanik, D.; Campbell, N.R.; Karikari, C.; Chivukula, R.; Kent, O.A.; Mendell, T.; Maitra, A. Restitution of tumour suppressor microRNAs using a systemic nanovector inhibits pancreatic cancer growth in mice. Mol. Cancer Ther 2011, 10, 1470–1480. [Google Scholar]
- Liu, C.; Kelnar, K.; Liu, B.; Chen, X.; Calhoun-Davis, T.; Li, H.; Patrawala, L.; Yan, H.; Jeter, C.; Honorio, S.; et al. The microRNA miR-34a inhibits prostate cancer stem cells and metastasis by directly repressing CD44. Nat. Med 2011, 17, 211–215. [Google Scholar]
- Trang, P.; Wiggins, J.F.; Daige, C.L.; Cho, C.; Omotola, M.; Brown, D.; Weidhaas, J.B.; Bader, A.G.; Slack, F.J. Systemic delivery of tumour suppressor microRNA mimics using a neutral lipid emulsion inhibits lung tumours in mice. Mol. Ther 2011, 19, 1116–1122. [Google Scholar]
- Ghani, S.; Riemke, P.; Schönheit, J.; Lenze, D.; Stumm, J.; Hoogenkamp, M.; Lagendijk, A.; Heinz, S.; Bonifer, C.; Bakkers, J.; et al. Macrophage development from HSCs requires PU.1-coordinated microRNA expression. Blood 2011, 118, 2275–2284. [Google Scholar]
- Curtale, G.; Citarella, F.; Carissimi, C.; Goldoni, M.; Carucci, N.; Fulci, V.; Franceschini, D.; Meloni, F.; Barnaba, V.; Macino, G. An emerging player in the adaptive immune response: microRNA-146a is a modulator of IL-2 expression and activation-induced cell death in T lymphocytes. Blood 2010, 115, 265–273. [Google Scholar]
- Garcia, A.I.; Buisson, M.; Bertrand, P.; Rimokh, R.; Rouleau, E.; Lopez, B.S.; Lidereau, R.; Mikaélian, I.; Mazoyer, S. Down-regulation of BRCA1 expression by miR-146a and miR-146b-5p in triple negative sporadic breast cancers. EMBO Mol. Med 2011, 3, 279–290. [Google Scholar]
- So, A.Y.; Zhao, J.L.; Baltimore, D. The Yin and Yang of microRNAs: Leukemia and immunity. Immunol. Rev 2013, 253, 129–145. [Google Scholar]
- Starnes, L.M.; Sorrentino, A.; Pelosi, E.; Ballarino, M.; Morsilli, O.; Biffoni, M.; Santoro, S.; Felli, N.; Castelli, G.; de Marchis, M.L.; et al. NFI-A directs the fate of hematopoietic progenitors to the erythroid or granulocytic lineage and controls beta-globin and G-CSF receptor expression. Blood 2009, 114, 1753–1763. [Google Scholar]
- Zardo, G.; Ciolfi, A.; Vian, L.; Starnes, L.M.; Billi, M.; Racanicchi, S.; Maresca, C.; Fazi, F.; Travaglini, L.; Noguera, N.; et al. Polycombs and microRNA-223 regulate human granulopoiesis by transcriptional control of target gene expression. Blood 2007, 119, 4034–4046. [Google Scholar]
- Fazi, F.; Racanicchi, S.; Zardo, G.; Starnes, L.M.; Mancini, M.; Travaglini, L.; Diverio, D.; Ammatuna, E.; Cimino, G.; Lo-Coco, F.; et al. Epigenetic silencing of the myelopoiesis regulator microRNA-223 by the AML1/ETO oncoprotein. Cancer Cell 2007, 12, 457–466. [Google Scholar]
- Concepcion, C.P.; Bonetti, C.; Ventura, A. The microRNA-17-92 family of microRNA clusters in development and disease. Cancer J 2012, 18, 262–267. [Google Scholar]
- O’Donnell, K.A.; Wentzel, E.A.; Zeller, K.I.; Dang, C.V.; Mendell, J.T. c-Myc-regulated microRNAs modulate E2F1 expression. Nature 2005, 435, 839–843. [Google Scholar]
- Woods, K.; Thomson, J.M.; Hammond, S.M. Direct regulation of an oncogenic micro-RNA cluster by E2F transcription factors. J. Biol. Chem 2007, 282, 2130–2134. [Google Scholar]
- Sylvestre, Y.; de Guire, V.; Querido, E.; Mukhopadhyay, U.K.; Bourdeau, V.; Major, F.; Ferbeyre, G.; Chartrand, P. An E2F/miR-20a autoregulatory feedback loop. J. Biol. Chem 2007, 282, 2135–2143. [Google Scholar]
- Ventura, A.; Young, A.G.; Winslow, M.M.; Lintault, L.; Meissner, A.; Erkeland, S.J.; Newman, J.; Bronson, R.T.; Crowley, D.; Stone, J.R.; et al. Targeted deletion reveals essential and overlapping functions of the miR-17 through 92 family of miRNA clusters. Cell 2008, 132, 875–886. [Google Scholar]
- Rao, E.; Jiang, C.; Ji, M.; Huang, X.; Iqbal, J.; Lenz, G.; Wright, G.; Staudt, L.M.; Zhao, Y.M.; McKeithan, T.W.; et al. The miRNA-17-92 cluster mediates chemoresistance and enhances tumour growth in mantle cell lymphoma via PI3K/AKT pathway activation. Leukemia 2012, 26, 1064–1072. [Google Scholar]
- Li, Z.; Lu, J.; Sun, M.; Mi, S.; Zhang, H.; Luo, R.T.; Chen, P.; Wang, Y.; Yan, M.; Qian, Z.; et al. Distinct microRNA expression profiles in acute myeloid leukemia with common translocations. Proc. Natl. Acad. Sci. USA 2008, 105, 15535–15540. [Google Scholar]
- Wong, P.; Iwasaki, M.; Somervaille, T.C.; Ficara, F.; Carico, C.; Arnold, C.; Chen, C.Z.; Cleary, M.L. The miR-17-92 microRNA polycistron regulates MLL leukemia stem cell potential by modulating p21 expression. Cancer Res 2010, 70, 3833–3842. [Google Scholar]
- Fontana, L.; Fiori, M.E.; Albini, S.; Cifaldi, L.; Giovinazzi, S.; Forloni, M.; Boldrini, R.; Donfrancesco, A.; Federici, V.; Giacomini, P.; et al. Antagomir-17-5p abolishes the growth of therapy-resistant neuroblastoma through p21 and BIM. PLoS One 2008, 3, e2236. [Google Scholar]
- Shaham, L.; Binder, V.; Gefen, N.; Borkhardt, A.; Izraeli, S. miR-125 in normal and malignant hematopoiesis. Leukemia 2012, 26, 2011–2018. [Google Scholar]
- Lin, K.Y.; Zhang, X.; Feng, D.D.; Zhang, H.; Zeng, C.W.; Han, B.W.; Zhou, A.D.; Qu, L.H.; Xu, L.; Chen, Y. miR-125b, a target of CDX2, regulates cell differentiation through repression of the core binding factor in hematopoietic malignancies. J. Biol. Chem 2011, 286, 38253–38263. [Google Scholar]
- Androulidaki, A.; Iliopoulos, D; Arranz, A; Doxaki, C.; Schworer, S.; Zacharioudaki, V.; Margioris, A.N.; Tsichlis, P.N.; Tsatsanis, C. The kinase Akt1 controls macrophage response to lipopolysaccharide by regulating microRNAs. Immunity 2009, 31, 220–231. [Google Scholar]
- O’Connell, R.M.; Taganov, K.D.; Boldin, M.P.; Cheng, G.; Baltimore, D. MicroRNA-155 is induced during the macrophage inflammatory response. Proc. Natl. Acad. Sci. USA 2007, 104, 1604–1609. [Google Scholar]
- Yin, Q.; Wang, X.; McBride, J.; Fewell, C.; Flemington, E. B-cell receptor activation induces BIC/miR-155 expression through a conserved AP-1 element. J. Biol. Chem 2008, 283, 2654–2662. [Google Scholar]
- Ruggiero, T.; Trabucchi, M.; de Santa, F.; Zupo, S.; Harfe, B.D.; McManus, M.T.; Rosenfeld, M.G.; Briata, P.; Gherzi, R. LPS induces KH-type splicing regulatory protein-dependent processing of microRNA-155 precursors in macrophages. FASEB J 2009, 23, 2898–2908. [Google Scholar]
- Eis, P.S.; Tam, W.; Sun, L.; Chadburn, A.; Li, Z.; Gomez, M.F.; Lund, E.; Dahlberg, J.E. Accumulation of miR-155 and BIC RNA in human B cell lymphomas. Proc. Natl. Acad. Sci. USA 2005, 102, 3627–3632. [Google Scholar]
- Costinean, S.; Zanesi, N.; Pekarsky, Y.; Tili, E.; Volinia, S.; Heerema, N.; Croce, C.M. Pre-B cell proliferation and lymphoblastic leukemia/high-grade lymphoma in E(mu)-miR155 transgenic mice. Proc. Natl. Acad. Sci. USA 2006, 103, 7024–7029. [Google Scholar]
- O’Connell, R.M.; Rao, D.S.; Chaudhuri, A.A.; Boldin, M.P.; Taganov, K.D.; Nicoll, J.; Paquette, R.L.; Baltimore, D. Sustained expression of microRNA-155 in hematopoietic stem cells causes a myeloproliferative disorder. J. Exp. Med 2008, 205, 585–594. [Google Scholar]
- Faraoni, I.; Laterza, S.; Ardiri, D.; Ciardi, C.; Fazi, F.; Lo-Coco, F. miR-424 and miR-155 deregulated expression in cytogenetically normal acute myeloid leukaemia: Correlation with NPM1 and FLT3 mutation status. J. Hematol. Oncol 2012, 5. [Google Scholar] [CrossRef]
- Croce, C.M. MicroRNA dysregulation in acute myeloid leukemia. J. Clin. Oncol 2013, 31, 2065–2066. [Google Scholar]
- Hu, Y.L.; Fong, S.; Largman, C.; Shen, W.F. HOXA9 regulates miR-155 in hematopoietic cells. Nucleic Acids Res 2010, 38, 5472–5478. [Google Scholar]
- Babar, I.A.; Cheng, C.J.; Booth, C.J.; Liang, X.; Weidhaas, J.B.; Saltzman, W.M.; Slack, F.J. Nanoparticle-based therapy in an in vivo microRNA-155 (miR-155)-dependent mouse model of lymphoma. Proc. Natl. Acad. Sci. USA 2012, 109, E1695–E1704. [Google Scholar]
- Costinean, S.; Sandhu, S.K.; Pedersen, I.M.; Tili, E.; Trotta, R.; Perrotti, D.; Ciarlariello, D.; Neviani, P.; Harb, J.; Kauffman, L.R.; et al. Src homology 2 domain-containing inositol-5-phosphatase and CCAAT enhancer-binding protein beta are targeted by miR-155 in B cells of Emicro-miR-155 transgenic mice. Blood 2009, 114, 1374–1382. [Google Scholar]
- Zhang, Y.; Roccaro, A.M.; Rombaoa, C.; Flores, L.; Obad, S.; Fernandes, S.M.; Sacco, A.; Liu, Y.; Ngo, H.; Quang, P.; et al. LNA-mediated anti-miR-155 silencing in low-grade B-cell lymphomas. Blood 2012, 120, 1678–1686. [Google Scholar]
- Ernst, P.; Mabon, M.; Davidson, A.J.; Zon, L.I.; Korsmeyer, S.J. An Mll-dependent Hox program drives hematopoietic progenitor expansion. Curr. Biol 2004, 14, 2063–2069. [Google Scholar]
- Milne, T.A.; Briggs, S.D.; Brock, H.W.; Martin, M.E.; Gibbs, D.; Allis, C.D.; Hess, J.L. MLL targets SET domain methyltransferase activity to Hox gene promoters. Mol. Cell 2002, 10, 1107–1117. [Google Scholar]
- Erfurth, F.E.; Popovic, R.; Grembecka, J.; Cierpicki, T.; Theisler, C.; Xia, Z.B.; Stuart, T.; Diaz, M.O.; Bushweller, J.H.; Zeleznik-Le, N.J. MLL protects CpG clusters from methylation within the Hoxa9 gene, maintaining transcript expression. Proc. Natl. Acad. Sci. USA 2008, 105, 7517–7522. [Google Scholar]
- Armstrong, S.A.; Staunton, J.E.; Silverman, L.B.; Pieters, R.; den Boer, M.L.; Minden, M.D.; Sallan, S.E.; Lander, E.S.; Golub, T.R.; Korsmeyer, S.J. MLL translocations specify a distinct gene expression profile that distinguishes a unique leukemia. Nat. Genet 2002, 30, 41–47. [Google Scholar]
- Milne, T.A.; Martin, M.E.; Brock, H.W.; Slany, R.K.; Hess, J.L. Leukemogenic MLL fusion proteins bind across a broad region of the Hoxa9 locus, promoting transcription and multiple histone modifications. Cancer Res 2005, 65, 11367–11374. [Google Scholar]
- Popovic, R.; Riesbeck, L.E.; Velu, C.S.; Chaubey, A.; Zhang, J.; Achille, N.J.; Erfurth, F.E.; Eaton, K.; Lu, J.; Grimes, H.L.; et al. Regulation of miR-196b by MLL and its overexpression by MLL fusions contributes to immortalization. Blood 2009, 113, 3314–3322. [Google Scholar]
- Li, Z.; Huang, H.; Chen, P.; He, M.; Li, Y.; Arnovitz, S.; Jiang, X.; He, C.; Hyjek, E.; Zhang, J.; et al. miR-196b directly targets both Hoxa9/MEIS1 oncogenes and FAS tumour suppressor in MLL-rearranged leukaemia. Nat. Commun 2012, 3. [Google Scholar] [CrossRef]
- Schotte, D.; Lange-Turenhout, E.A.; Stumpel, D.J.; Stam, R.W.; Buijs-Gladdines, J.G.; Meijerink, J.P.; Pieters, R.; den Boer, M.L. Expression of miR-196b is not exclusively MLL-driven but is especially linked to activation of Hoxa genes in pediatric acute lymphoblastic leukemia. Haematologica 2010, 95, 1675–1682. [Google Scholar]
- Bai, H.; Cao, Z.; Deng, C.; Zhou, L.; Wang, C. miR-181a sensitizes resistant leukaemia HL-60/Ara-C cells to Ara-C by inducing apoptosis. J. Cancer Res. Clin. Oncol 2012, 138, 595–602. [Google Scholar]
- Gocek, E.; Wang, X.; Liu, X.; Liu, C.G.; Studzinski, G.P. MicroRNA-32 upregulation by 1,25-dihydroxyvitamin D3 in human myeloid leukemia cells leads to Bim targeting and inhibition of Ara-C-induced apoptosis. Cancer Res 2011, 71, 6230–6239. [Google Scholar]
- Christman, J.K. 5-Azacytidine and 5-aza-2′-deoxycytidine as inhibitors of DNA methylation: Mechanistic studies and their implications for cancer therapy. Oncogene 2002, 21, 5483–5495. [Google Scholar]
- Blum, W.; Garzon, R.; Klisovic, R.B.; Schwind, S.; Walker, A.; Geyer, S.; Liu, S.; Havelange, V.; Becker, H.; Schaaf, L.; et al. Clinical response and miR-29b predictive significance in older AML patients treated with a 10-day schedule of decitabine. Proc. Natl. Acad. Sci. USA 2010, 107, 7473–7478. [Google Scholar]
- Mims, A.; Walker, A.R.; Huang, X.; Sun, J.; Wang, H.; Santhanam, R.; Dorrance, A.M.; Walker, C.; Hoellerbauer, P.; Tarighat, S.S.; et al. Increased anti-leukemic activity of decitabine via AR-42-induced upregulation of miR-29b: A novel epigenetic-targeting approach in acute myeloid leukemia. Leukemia 2013, 27, 871–878. [Google Scholar]
- Huang, X.; Schwind, S.; Yu, B.; Santhanam, R.; Wang, H.; Hoellerbauer, P.; Mims, A.; Klisovic, R.; Walker, A.R.; Chan, K.K.; et al. Targeted delivery of microRNA-29b by transferrin-conjugated anionic lipopolyplex nanoparticles: A novel therapeutic strategy in acute myeloid leukemia. Clin. Cancer Res 2013, 19, 2355–2367. [Google Scholar]
- Amodio, N.; Leotta, M.; Bellizzi, D.; di Martino, M.T.; D’Aquila, P.; Lionetti, M.; Fabiani, F.; Leone, E.; Gullà, A.M.; Passarino, G.; et al. DNA-demethylating and anti-tumour activity of synthetic miR-29b mimics in multiple myeloma. Oncotarget 2012, 3, 1246–1258. [Google Scholar]
- Bueno, M.J.; Pérez de Castro, I.; Gómez de Cedrón, M.; Santos, J.; Calin, G.A.; Cigudosa, J.C.; Croce, C.M.; Fernández-Piqueras, J.; Malumbres, M. Genetic and epigenetic silencing of microRNA-203 enhances ABL1 and BCR-ABL1 oncogene expression. Cancer Cell 2008, 13, 496–506. [Google Scholar]
- Faber, J.; Gregory, R.I.; Armstrong, S.A. Linking miRNA regulation to BCR-ABL expression: The next dimension. Cancer Cell 2008, 13, 467–469. [Google Scholar]
- Shibuta, T.; Honda, E.; Shiotsu, H.; Tanaka, Y.; Vellasamy, S.; Shiratsuchi, M.; Umemura, T. Imatinib induces demethylation of miR-203 gene: An epigenetic mechanism of anti-tumor effect of imatinib. Leuk. Res 2013, 37, 1278–1286. [Google Scholar]
- Kasinski, A.L.; Slack, F.J. Epigenetics and genetics. MicroRNAs en route to the clinic: Progress in validating and targeting microRNAs for cancer therapy. Nat. Rev. Cancer 2011, 11, 849–864. [Google Scholar]
- Batista, P.J.; Chang, H.Y. Long noncoding RNAs: Cellular address codes in development and disease. Cell 2013, 152, 1298–1307. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Fatica, A.; Fazi, F. MicroRNA-Regulated Pathways in Hematological Malignancies: How to Avoid Cells Playing Out of Tune. Int. J. Mol. Sci. 2013, 14, 20930-20953. https://doi.org/10.3390/ijms141020930
Fatica A, Fazi F. MicroRNA-Regulated Pathways in Hematological Malignancies: How to Avoid Cells Playing Out of Tune. International Journal of Molecular Sciences. 2013; 14(10):20930-20953. https://doi.org/10.3390/ijms141020930
Chicago/Turabian StyleFatica, Alessandro, and Francesco Fazi. 2013. "MicroRNA-Regulated Pathways in Hematological Malignancies: How to Avoid Cells Playing Out of Tune" International Journal of Molecular Sciences 14, no. 10: 20930-20953. https://doi.org/10.3390/ijms141020930
APA StyleFatica, A., & Fazi, F. (2013). MicroRNA-Regulated Pathways in Hematological Malignancies: How to Avoid Cells Playing Out of Tune. International Journal of Molecular Sciences, 14(10), 20930-20953. https://doi.org/10.3390/ijms141020930