Protein Glutathionylation in Cardiovascular Diseases


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
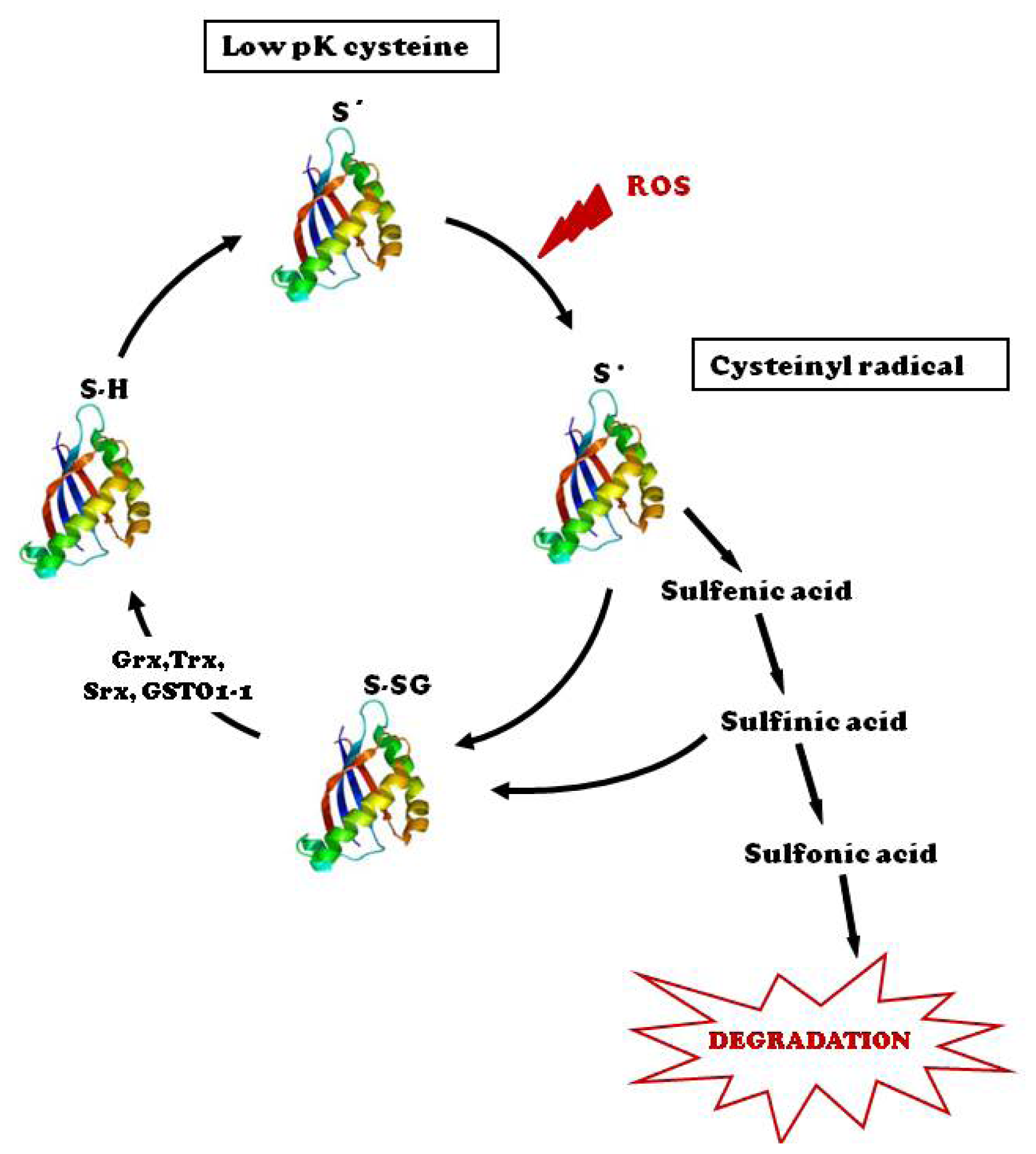
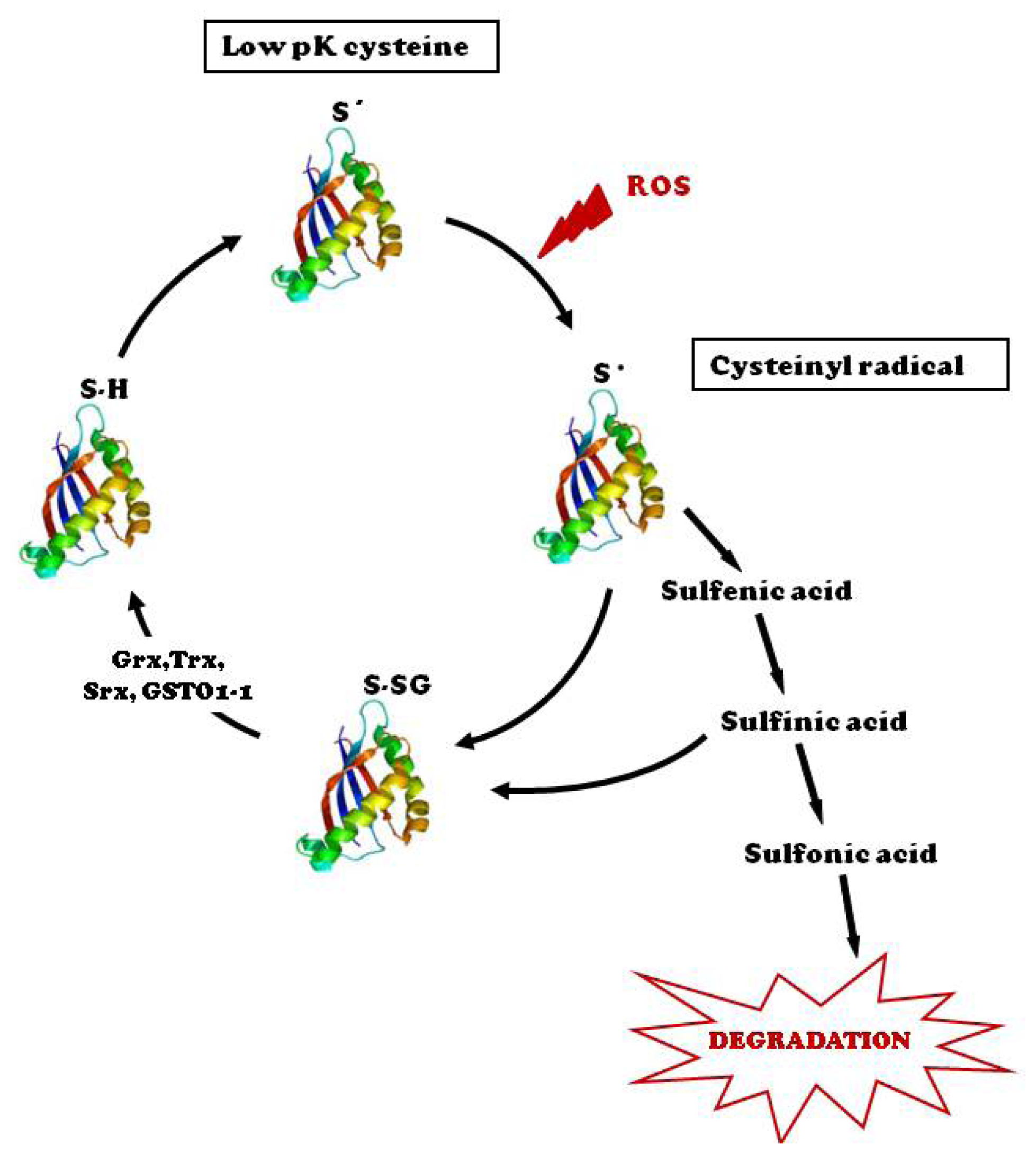
:1. Introduction
2. Protein-S-Glutathionylation Status in Cardiovascular Diseases
2.1. Myocardial Infarction
2.2. Cardiac Hypertrophy
2.3. Atherosclerosis
3. Tools and Strategies to Monitor Protein S-Glutathionylation
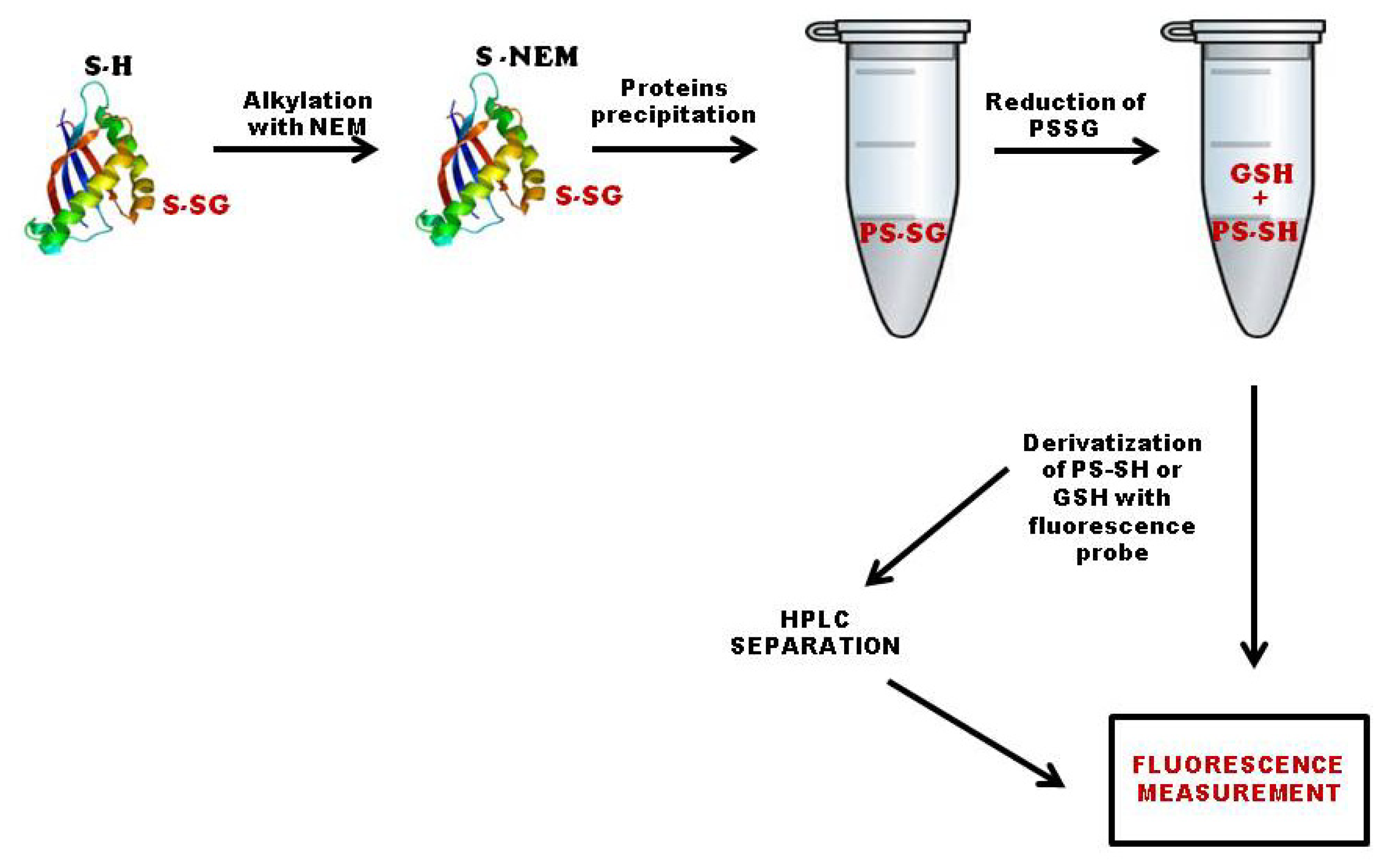
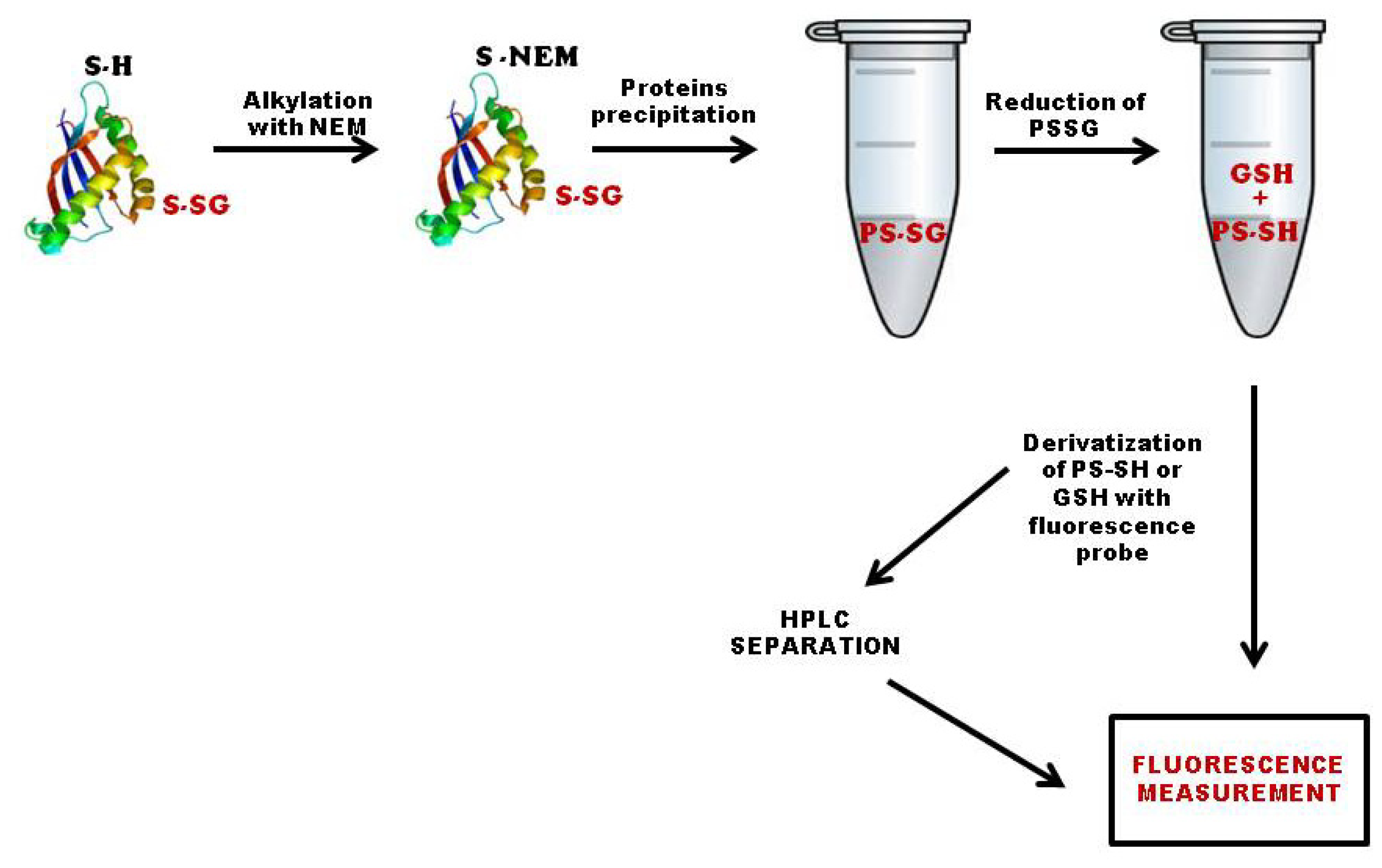
3.1. Quantification of Total S-Glutathionylated Proteins
3.2. Methods Based on Labeling of Glutathione
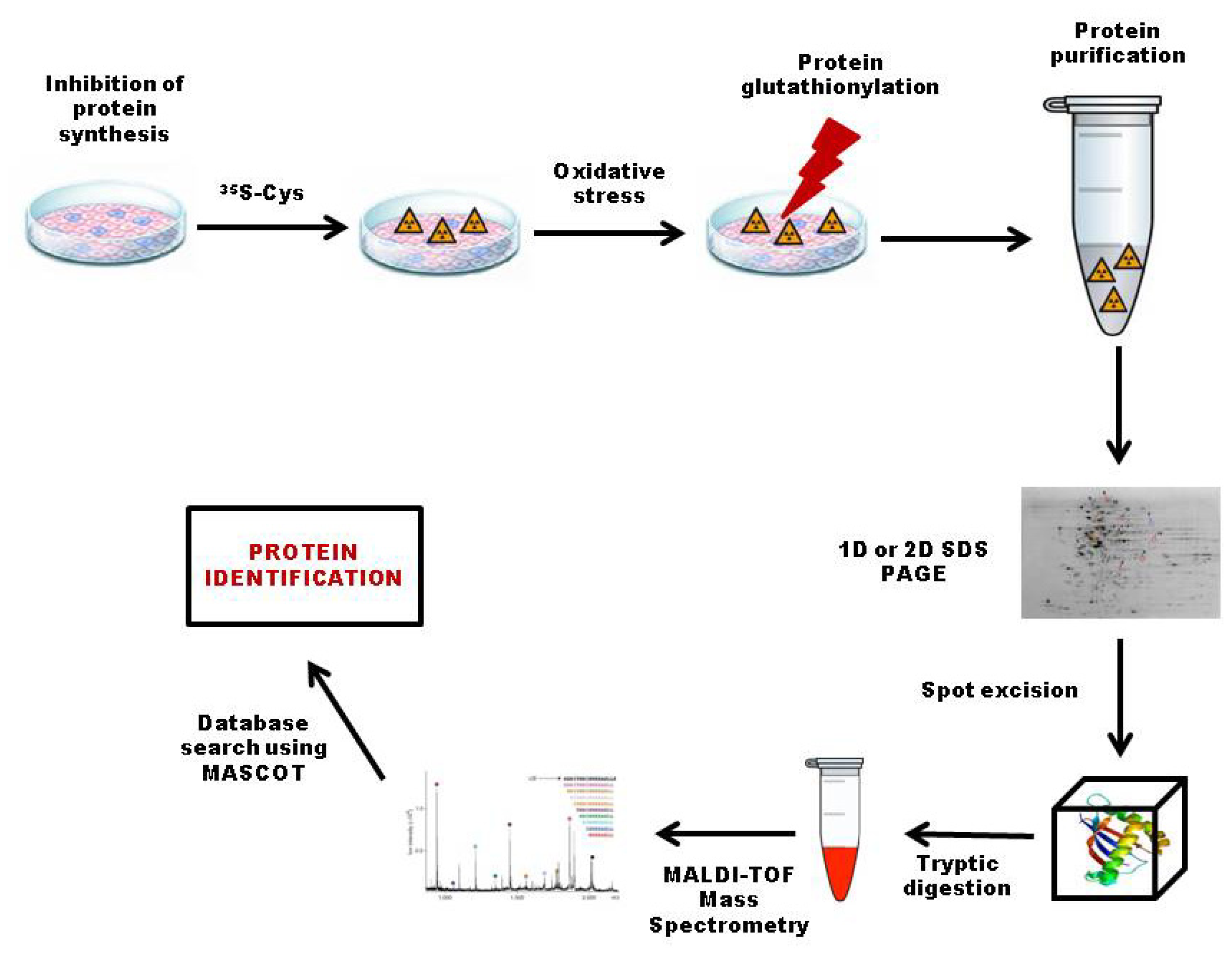
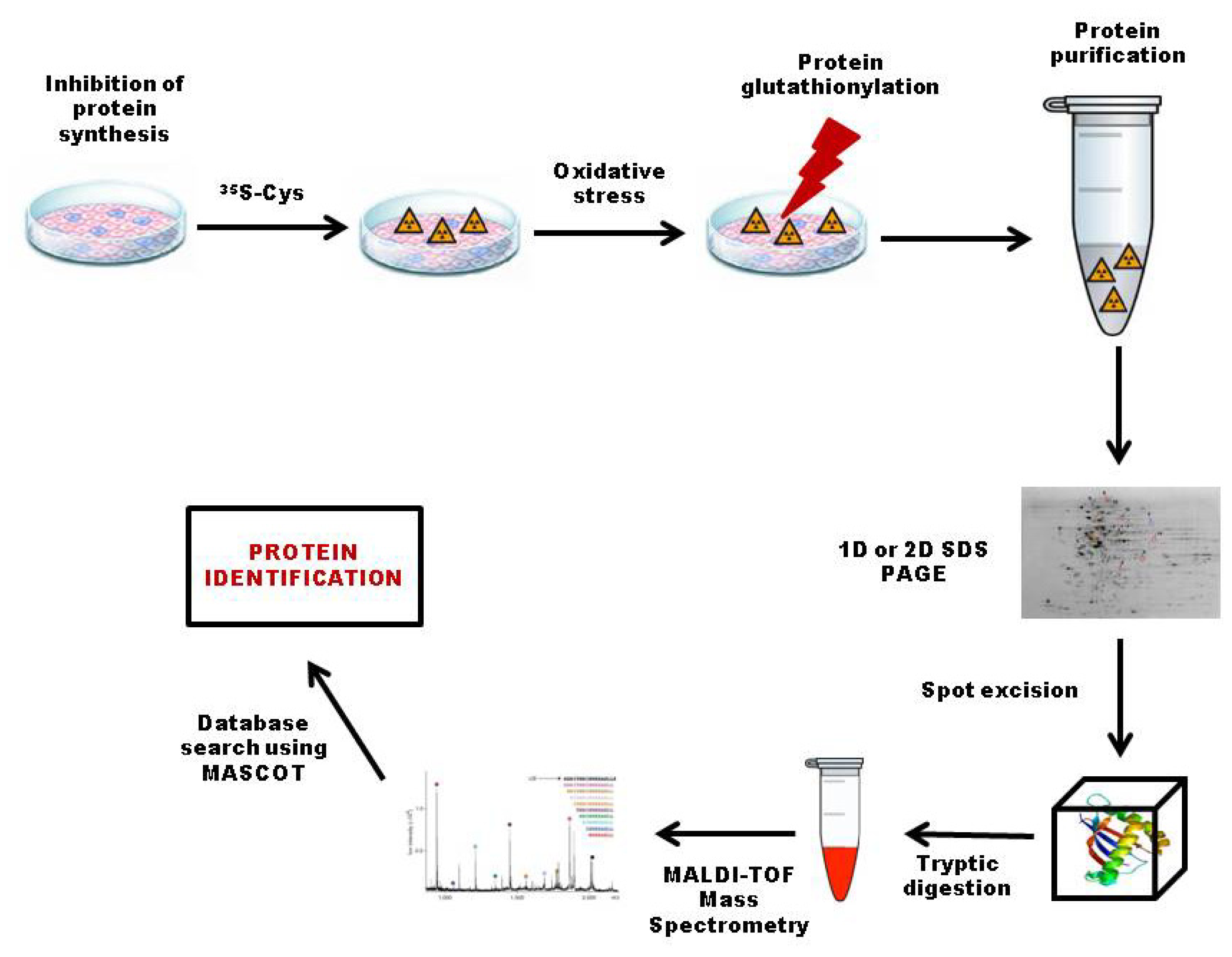
3.2.1. 35S Radiolabeling
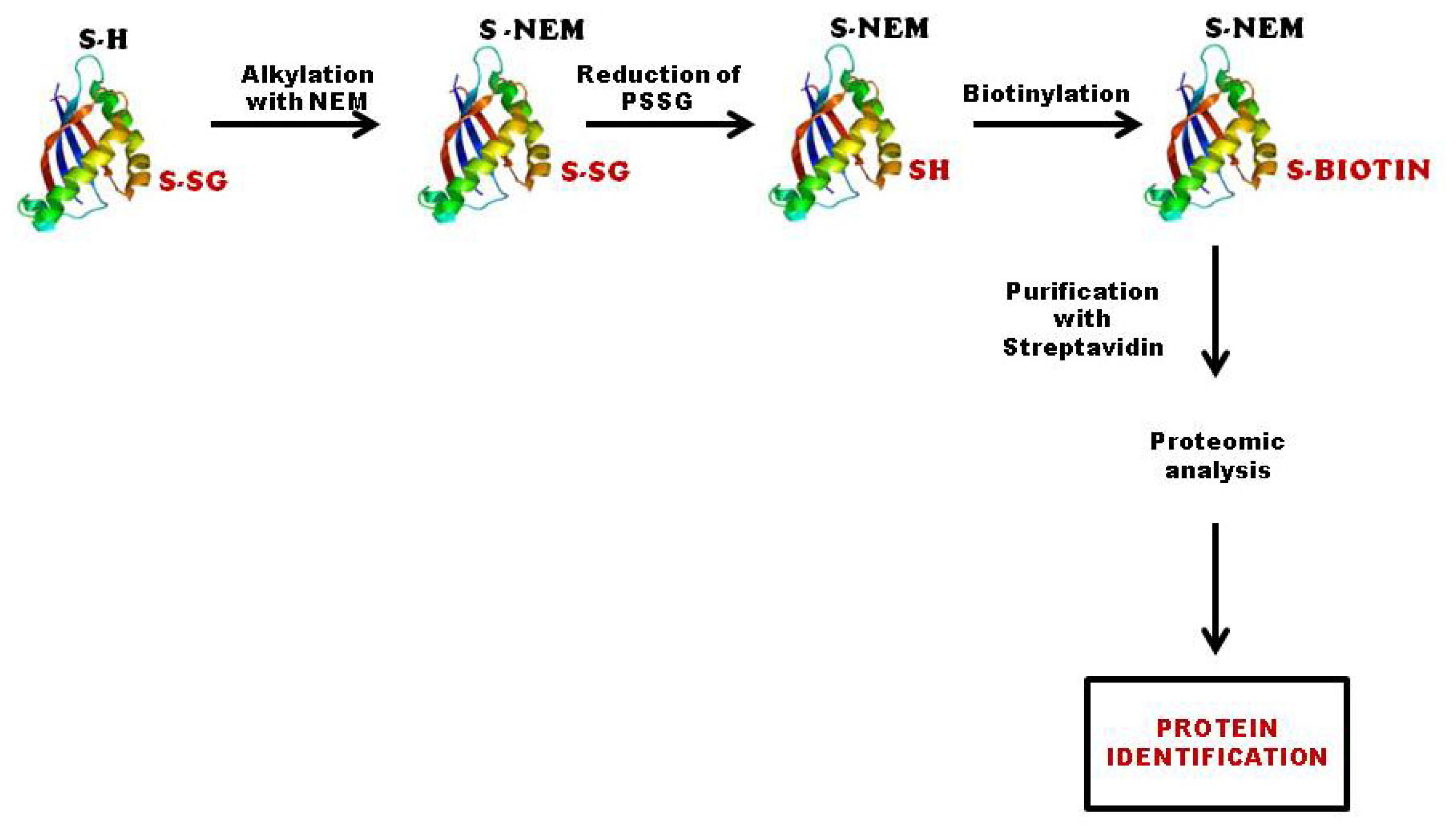
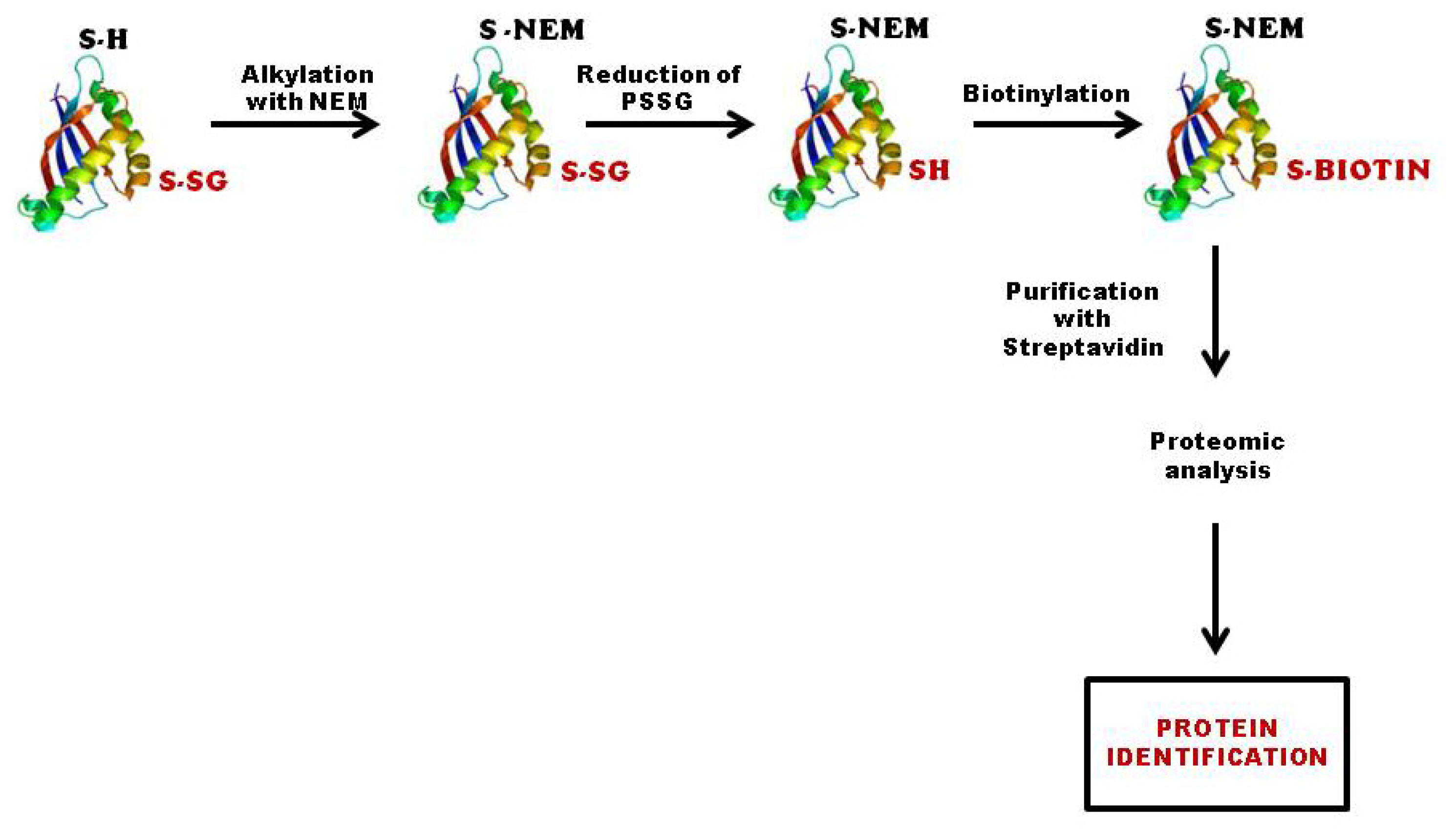
3.2.2. Biotinylated Glutathione
3.3. Methods Utilizing Anti-Glutathione Antibodies
3.4. Top-Down Proteomics Approach
3.5. Other Methods
4. Physiological Effects of Protein Glutathionylation
4.1. Myocardial Contraction
4.1.1. Ryanodine Receptor
4.1.2. Sarco/Endoplasmic Reticulum Ca2+ ATPase (SERCA)
4.1.3. The Endothelial Nitric Oxide Synthase (eNOS)
4.1.4. Na+-K+ ATPase
4.1.5. Contractile Proteins
4.1.5.1. Myosin
4.1.5.2. α-Actin
4.1.5.3. Myosin Binding Protein C (MyBP-C)
4.1.5.4. Troponin
4.2. Myocardial Metabolism
4.2.1. Complex I (CI)
4.2.2. Complex II (CII)
4.2.3. ATP synthase Complex (CV)
4.2.4. Sirtuin-1
4.2.5. Aldose Reductase
4.2.6. Hemoglobin
4.3. Myocardial Proliferation and Hypertrophy
p21Ras
4.4. Myocardial Inflammation
4.4.1. NFkB
4.4.2. Interferon Regulator Protein 3
5. Concluding Remarks
6. Future Directions
Conflicts of Interest
References
- Elahi, M.M.; Kong, Y.X.; Matata, B.M. Oxidative stress as a mediator of cardiovascular disease. Oxid. Med. Cell Longev 2009, 2, 259–269. [Google Scholar]
- Mieyal, J.J.; Gallogly, M.M.; Qanungo, S.; Sabens, E.A.; Shelton, M.D. Molecular mechanisms and clinical implications of reversible protein S-glutathionylation. Antioxid. Redox Signal 2008, 10, 1941–1988. [Google Scholar]
- Pimentel, D.; Haeussler, D.J.; Matsui, R.; Burgoyne, J.R.; Cohen, R.A.; Bachschmid, M.M. Regulation of cell physiology and pathology by protein S-glutathionylation: Lessons learned from the cardiovascular system. Antioxid. Redox Signal 2012, 16, 524–42. [Google Scholar]
- Kumar, V.; Calamaras, T.D.; Haeussler, D.; Colucci, W.S.; Cohen, R.A.; McComb, M.E.; Pimentel, D.; Bachschmid, M.M. Cardiovascular redox and ox stress proteomics. Antioxid. Redox Signal 2012, 17, 1528–1559. [Google Scholar]
- Kim, G.H.; Ryan, J.J.; Archer, S.L. The role of redox signaling in epigenetics and cardiovascular disease. Antioxid. Redox Signal 2013, 18, 1920–1936. [Google Scholar]
- Eaton, P.; Wright, N.; Hearse, D.J.; Shattock, M.J. Glyceraldehyde phosphate dehydrogenase oxidation during cardiac ischemia and reperfusion. J. Mol. Cell. Cardiol 2002, 34, 1549–1560. [Google Scholar]
- Chen, F.C.; Ogut, O. Decline of contractility during ischemia-reperfusion injury: Actin glutathionylation and its effect on allosteric interaction with tropomyosin. Am. J. Physiol. Cell. Physiol 2006, 290, C719–C727. [Google Scholar]
- Wang, J.; Boja, E.S.; Tan, W.; Tekle, E.; Fales, H.M.; English, S.; Mieyal, J.J.; Chock, P.B. Reversible glutathionylation regulates actin polymerization in A431 cells. J. Biol. Chem 2001, 276, 47763–47766. [Google Scholar]
- Chen, Y.R.; Chen, C.L.; Pfeiffer, D.R.; Zweier, J.L. Mitochondrial complex II in the post-ischemic heart: Oxidative injury and the role of protein S-glutathionylation. J. Biol. Chem 2007, 282, 32640–32654. [Google Scholar]
- Malik, G.; Nagy, N.; Ho, Y.S.; Maulik, N.; Das, D.K. Role of glutaredoxin-1 in cardioprotection: An insight with Glrx1 transgenic and knockout animals. J. Mol. Cell Cardiol 2008, 44, 261–269. [Google Scholar]
- Nagy, N.; Malik, G.; Tosaki, A.; Ho, Y.S.; Maulik, N.; Das, D.K. Overexpression of glutaredoxin-2 reduces myocardial cell death by preventing both apoptosis and necrosis. J. Mol. Cell Cardiol 2008, 44, 252–260. [Google Scholar]
- Mukherjee, S.; Gangopadhyay, H.; Das, D.K. Broccoli: A unique vegetable that protects mammalian hearts through the redox cycling of the thioredoxin superfamily. J. Agric. Food Chem 2008, 56, 609–617. [Google Scholar]
- Taylor, E.R.; Hurrell, F.; Shannon, R.J.; Lin, T.K.; Hirst, J.; Murphy, M.P. Reversible glutathionylation of Complex I increases mitochondrial superoxide formation. J. Biol. Chem 2003, 278, 19603–19610. [Google Scholar]
- Heineke, J.; Molkentin, J.D. Regulation of cardiac hypertrophy by intracellular signalling pathways. Nat. Rev. Mol. Cell Biol 2006, 7, 589–600. [Google Scholar]
- Pimentel, D.R; Adachi, T.; Ido, Y.; Heibeck, T.; Jiang, B.; Lee, Y.; Melendez, J.A.; Cohen, R.A.; Colucci, W.S. Strain-stimulated hypertrophy in cardiac myocytes is mediated by reactive oxygen species-dependent Ras S-glutathiolation. J. Mol. Cell Cardiol 2006, 41, 613–22. [Google Scholar]
- Pantano, C.; Reynaert, N.L.; Van Der Vliet, A.; Janssen-Heininger, Y.M.W. Redox-sensitive kinases of the nuclear factor-kB signaling pathway. Antioxid. Redox Signal 2006, 8, 1791–1806. [Google Scholar]
- Wang, Y.; Qiao, M.; Mieyal, J.J.; Asmis, L.M.; Asmis, R. Molecular mechanism of glutathione-mediated protection from oxidized low-density lipoprotein-induced cell injury in human macrophages: Role of glutathione reductase and glutaredoxin. Free Radic. Biol. Med 2006, 41, 775–785. [Google Scholar]
- Okuda, M.; Inoue, N.; Azumi, H.; Seno, T.; Sumi, Y.; Hirata, K.; Kawashima, S.; Hayashi, Y.; Itoh, H.; Yodoi, J.; Yokoyama, M. Expression of glutaredoxin in human coronary arteries: Its potential role in antioxidant protection against atherosclerosis. Arterioscler. Thromb. Vasc. Biol 2001, 21, 1483–1487. [Google Scholar]
- Kher, N.; Marsh, J.D. Pathobiology of atherosclerosis—A brief review. Semin. Thromb. Hemost 2004, 30, 665–672. [Google Scholar]
- Nonaka, K.; Kume, N.; Urata, Y.; Seto, S.; Kohno, T.; Honda, S.; Ikeda, S.; Muroya, T.; Ikeda, Y.; Ihara, Y.; Kita, T.; Kondo, T. Serum levels of S-glutathionylated proteins as a risk marker for arteriosclerosis obliterans. Circ. J 2007, 71, 100–105. [Google Scholar]
- Adachi, T.; Weisbrod, R.M.; Pimentel, D.R.; Ying, J.; Sharov, V.S.; Schoneich, C.; Cohen, R.A. S-Glutathiolation by peroxynitrite activates SERCA during arterial relaxation by nitric oxide. Nat. Med 2004, 10, 1200–1207. [Google Scholar]
- Adachi, T.; Pimentel, D.R.; Heibeck, T.; Hou, X.; Lee, Y.J.T.; Jiang, B.; Cohen, R.A. S-glutathiolation of Ras mediates redox-sensitive signaling by Angiotensin ii in vascular smooth muscle cells. J. Biol. Chem 2004, 279, 29857–29862. [Google Scholar]
- Landmesser, U.; Cai, H.; Dikalov, S.; McCann, L.; Hwang, J.; Jo, H.; Holland, S.M.; Harrison, D.G. Role of p47(phox) in vascular oxidative stress and hypertension caused by angiotensin II. Hypertension 2002, 40, 511–515. [Google Scholar]
- Wang, H.D.; Xu, S.; Johns, D.G.; Du, Y.; Quinn, M.T.; Cayatte, A.J.; Cohen, R.A. Role of NADPH oxidase in the vascular hypertrophic and oxidative stress response to angiotensin II in mice. Circ. Res 2001, 88, 947–953. [Google Scholar]
- Clavreul, N.; Adachi, T.; Pimental, D.R.; Ido, Y.; Schoneich, C.; Cohen, R.A. S-glutathiolation by peroxynitrite of p21ras at cysteine-118 mediates its direct activation and downstream signaling in endothelial cells. FASEB J 2006, 20, 518–520. [Google Scholar]
- Wang, J.; Pan, S.; Berk, B.C. Glutaredoxin mediates Akt and eNOS activation by flow in a glutathione reductase dependent manner. Arterioscler. Thromb. Vasc.Biol 2007, 27, 1283–1288. [Google Scholar]
- Mullonkal, C.J.; Toledo-Pereyra, L.H. Akt in ischemia and reperfusion. J. Invest. Surg 2007, 20, 195–203. [Google Scholar]
- Dixon, W.G.; Symmons, D.P. What effects might anti-TNFalpha treatment be expected to have on cardiovascular morbidity and mortality in rheumatoid arthritis? A review of the role of TNFalpha in cardiovascular pathophysiology. Ann. Rheum. Dis 2007, 66, 1132–1136. [Google Scholar]
- Pan, S.; Berk, B.C. Glutathiolation regulates tumor necrosis factor-alpha-induced caspase-3 cleavage and apoptosis: Key role for glutaredoxin in the death pathway. Circ. Res 2007, 100, 213–219. [Google Scholar]
- Thomas, J.A.; Zhao, W.; Hendrich, S.; Haddock, P. Analysis of cells and tissues for S-thiolation of specific proteins. Methods Enzymol 1995, 251, 423–429. [Google Scholar]
- Dalle-Donne, I.; Milzani, A.; Gagliano, N.; Colombo, R.; Giustarini, D.; Rossi, R. Molecular mechanisms and potential clinical significance of S-glutathionylation. Antioxid. Redox Signal 2008, 10, 445–473. [Google Scholar]
- Lu, J.; Holmgren, A. The thioredoxin antioxidant system. Free Radic. Biol. Med 2013, in press. [Google Scholar]
- Findlay, V.J.; Townsend, D.M.; Morris, T.E.; Fraser, J.P.; He, L.; Tew, K.D. A novel role for human sulfiredoxin in the reversal of glutathionylation. Cancer Res 2006, 66, 6800–6806. [Google Scholar]
- Menon, D.M; Board,, P.G. A role for glutathione transferase omega 1 (GSTO1–1) in the glutathionylation cycle. J. Biol. Chem 2013, 288, 25769–25779. [Google Scholar]
- Tew, K.D.; Townsend, D.M. Redox platforms in cancer drug discovery and development. Curr. Opin. Chem. Biol 2011, 15, 156–161. [Google Scholar]
- Ansong, C.; Wu, S.; Meng, D.; Liu, X.; Brewer, H.M.; Deatherage Kaiser, B.L.; Nakayasu, E.S.; Cort, J.R.; Pevzner, P.; Smith, R.D.; et al. Top-down proteomics reveals a unique protein S-thiolation switch in Salmonella Typhimurium in response to infection-like conditions. Proc. Natl. Acad. Sci. USA 2013, 25, 10153–10158. [Google Scholar]
- Townsend, D.M.; Manevich, Y.; He, L.; Hutchens, S.; Pazoles, C.J.; Tew, K.D. Novel role for glutathione S-transferase pi. Regulator of protein S-Glutathionylation following oxidative and nitrosative stress. J. Biol. Chem 2009, 284, 436–345. [Google Scholar]
- Pastore, A.; Martinelli, D.; Piemonte, F.; Tozzi, G.; Boenzi, S.; Di Giovamberardino, G.; Petrillo, S.; Bertini, E.; Dionisi-Vici, C. Glutathione metabolism in cobalamin deficiency type C (cblC). J. Inherit. Metab. Dis 2013, in press. [Google Scholar]
- Petrillo, S.; Piemonte, F.; Pastore, A.; Tozzi, G.; Aiello, C.; Pujol, A.; Cappa, M.; Bertini, E. Glutathione imbalance in patients with X-linked adrenoleukodystrophy. Mol. Genet. Metab 2013, 109, 366–370. [Google Scholar]
- Pastore, A.; Petrillo, S.; Tozzi, G.; Carrozzo, R.; Martinelli, D.; Dionisi-Vici, C.; Di Giovamberardino, G.; Ceravolo, F.; Klein, M.B.; Miller, G.; et al. Glutathione: A redox signature in monitoring EPI-743 therapy in children with mitochondrial encephalomyopathies. Mol. Genet. Metab 2013, 109, 208–214. [Google Scholar]
- Menon, D.; Board, P.G. A fluorometric method to quantify protein glutathionylation using glutathione derivatization with 2,3-naphthalenedicarboxaldehyde. Anal. Biochem 2013, 433, 132–136. [Google Scholar]
- Lind, C.; Gerdes, R.; Schuppe-Koistinen, I.; Cotgreave, I.A. Studies on the mechanism of oxidative modification of human glyceraldehyde-3-phosphate dehydrogenase by glutathione: Catalysis by glutaredoxin. Biochem. Biophys. Res. Commun 1998, 247, 481–486. [Google Scholar]
- Ward, N.E.; Stewart, J.R.; Ioannides, C.G.; O’Brian, C.A. Oxidant-induced S-glutathiolation inactivates protein kinase C-alpha (PKC-alpha): A potential mechanism of PKC isozyme regulation. Biochemistry 2000, 39, 10319–10329. [Google Scholar]
- Manevich, Y.; Feinstein, S.I.; Fisher, A.B. Activation of the antioxidant enzyme 1-CYS peroxiredoxin requires glutathionylation mediated by heterodimerization with pi GST. Proc. Natl. Acad. Sci. USA 2004, 101, 3780–3785. [Google Scholar]
- Hidalgo, C.; Sanchez, G.; Barrientos, G.; Aracena-Parks, P. A transverse tubule NADPH oxidase activity stimulates calcium release from isolated triads via ryanodine receptor type 1 S-glutathionylation. J. Biol. Chem 2006, 281, 26473–26482. [Google Scholar]
- Rokutan, K.; Thomas, J.A.; Johnston, R.B., Jr. Phagocytosis and stimulation of the respiratory burst by phorbol diester initiate S-thiolation of specific proteins in macrophages. J. Immunol 1991, 147, 260–264. [Google Scholar]
- Ravichandran, V.; Seres, T.; Moriguchi, T.; Thomas, J.A.; Johnston, R.B., Jr. S-thiolation of glyceraldehyde-3-phosphate dehydrogenase induced by the phagocytosis-associated respiratory burst in blood monocytes. J. Biol. Chem 1994, 269, 25010–25015. [Google Scholar]
- Seres, T.; Ravichandran, V.; Moriguchi, T.; Rokutan, K.; Thomas, J.A.; Johnston, R.B., Jr. Protein S-thiolation and dethiolation during the respiratory burst in human monocytes: A reversible post-translational modification with potential for buffer ring the effects of oxidant stress. J. Immunol 1996, 156, 1973–1980. [Google Scholar]
- Fratelli, M.; Demol, H.; Puype, M.; Casagrande, S.; Eberini, I.; Salmona, M.; Bonetto, V.; Mengozzi, M.; Duffieux, F.; Miclet, E.; et al. Identification by redox proteomics of glutathionylated proteins in oxidatively stressed human T lymphocytes. Proc. Natl. Acad. Sci. USA 2002, 99, 3505–3510. [Google Scholar]
- Brennan, J.P.; Miller, J.I.; Fuller, W.; Wait, R.; Begum, S.; Dunn, M.J.; Eaton, P. The utility of N,N-biotinyl glutathione disulfide in the study of protein S-glutathiolation. Mol. Cell. Proteomics 2006, 5, 215–225. [Google Scholar]
- Reynaert, N.L.; Van der Vliet, A.; Guala, A.S.; McGovern, T.; Hristova, M.; Pantano, C.; Heintz, N.H.; Heim, J.; Ho, Y.S.; Matthews, D.E.; et al. Dynamic redox control of NF-kappaB through glutaredoxin regulated S-glutathionylation of inhibitory kappaB kinase beta. Proc. Natl. Acad. Sci. USA 2006, 103, 13086–13091. [Google Scholar]
- Dalle-Donne, I.; Giustarini, D.; Rossi, R.; Colombo, R.; Milzani, A. Reversible S-glutathionylation of Cys374 regulates actin filament formation by inducing structural changes in the actin molecule. Free Radic. Biol. Med 2003, 34, 23–32. [Google Scholar]
- Wang, J.; Tekle, E.; Oubrahim, H.; Mieyal, J.J.; Stadtman, E.R.; Chock, P.B. Stable and controllable RNA interference: Investigating the physiological function of glutathionylated actin. Proc. Natl. Acad. Sci. USA 2003, 100, 5103–5106. [Google Scholar]
- Pastore, A.; Tozzi, G.; Gaeta, L.M.; Bertini, E.; Serafini, V.; Di Cesare, S.; Bonetto, V.; Casoni, F.; Carrozzo, R.; Federici, G.; et al. Actin glutathionylation increases in fibroblasts of patients with Friedreich’s ataxia: A potential role in the pathogenesis of the disease. J. Biol. Chem 2003, 43, 42588–42595. [Google Scholar]
- Fiaschi, T.; Cozzi, G.; Raugei, G.; Formigli, L.; Ramponi, G.; Chiarugi, P. Redox regulation of beta-actin during integrin-mediated cell adhesion. J. Biol. Chem 2006, 281, 22983–22991. [Google Scholar]
- Sparaco, M.; Gaeta, L.M.; Tozzi, G.; Bertini, E.; Pastore, A.; Simonati, A.; Santorelli, F.M.; Piemonte, F. Protein glutathionylation in human central nervous system: Potential role in redox regulation of neuronal defense against free radicals. J. Neurosci. Res 2006, 83, 256–263. [Google Scholar]
- Passarelli, C.; Di Venere, A.; Piroddi, N.; Pastore, A.; Scellini, B.; Tesi, C.; Petrini, S.; Sale, P.; Bertini, E.; Poggesi, C.; et al. Susceptibility of isolated myofibrils to in vitro glutathionylation: potential relevance to muscle functions. Cytoskeleton 2010, 67, 81–89. [Google Scholar]
- Passarelli, C.; Petrini, S.; Pastore, A.; Bonetto, V.; Sale, P.; Gaeta, L.M.; Tozzi, G.; Bertini, E.; Canepari, M.; Rossi, R.; et al. Myosin as a potential redox-sensor: An in vitro study. J. Muscle Res. Cell. Motil 2008, 29, 119–126. [Google Scholar]
- Landino, L.M.; Robinson, S.H.; Skreslet, T.E.; Cabral, D.M. Redox modulation of tau and microtubule-associated protein-2 by the glutathione/glutaredoxin reductase system. Biochem. Biophys. Res. Commun 2004, 323, 112–117. [Google Scholar]
- Fernandes, A.P.; Fladvad, M.; Berndt, C.; Andresen, C.; Lillig, C.H.; Neubauer, P.; Sunnerhagen, M.; Holmgren, A.; Vlamis-Gardikas, A. A novel monothiol glutaredoxin (Grx4) from Escherichia coli can serve as a substrate for thioredoxin reductase. J. Biol. Chem 2005, 280, 24544–24552. [Google Scholar]
- Hoppe, G.; Chai, Y.C.; Crabb, J.W.; Sears, J. Protein S-glutathionylation in retinal pigment epithelium converts heat shock protein 70 to an active chaperone. Exp. Eye Res 2004, 78, 1085–1092. [Google Scholar]
- West, M.B.; Hill, B.G.; Xuan, Y.T.; Bhatnagar, A. Protein glutathiolation by nitric oxide: An intracellular mechanism regulating redox protein modification. FASEB J 2006, 20, 1715–1717. [Google Scholar]
- Aracena, P.; Tang, W.; Hamilton, S.L.; Hidalgo, C. Effects of S-glutathionylation and S-nitrosylation on calmodulin binding to triads and FKBP12 binding to type 1 calcium release channels. Antioxid. Redox Signal 2005, 7, 870–881. [Google Scholar]
- Aracena-Parks, P.; Goonasekera, S.A.; Gilman, C.; Dirksen, R.T.; Hidalgo, C.; Hamilton, S.L. Identification of cysteines involved in S-nitrosylation, S-glutathionylation, and oxidation to disulfides in RyR1. J. Biol. Chem 2006, 281, 40354–40368. [Google Scholar]
- Craghill, J.; Cronshaw, A.D.; Harding, J.J. The identification of a reaction site of glutathione mixed-disulphide formation on cS-crystallin in human lens. Biochem. J 2004, 379, 595–600. [Google Scholar]
- Newman, S.F.; Sultana, R.; Perluigi, M.; Coccia, R.; Cai, J.; Pierce, W.M.; Klein, J.B.; Turner, D.M.; Butterfield, D.A. An increase in S-glutathionylated proteins in the Alzheimer’s disease inferior parietal lobule, a proteomics approach. J. Neurosci. Res 2007, 85, 1506–1514. [Google Scholar]
- Townsend, D.M.; Findlay, V.L.; Fazilev, F.; Ogle, M.; Fraser, J.; Saavedra, J.E.; Ji, X.; Keefer, L.K.; Tew, K.D. A glutathione S-transferase pi-activated prodrug causes kinase activation concurrent with Sglutathionylation of proteins. Mol. Pharmacol 2006, 69, 501–508. [Google Scholar]
- Lampela, O.; Juffer, A.H.; Rauk, A. Conformational analysis of glutathione in aqueous solution with molecular dynamics. J. Phys. Chem. A 2003, 107, 9208–9220. [Google Scholar]
- Smith, L.M.; Kelleher, N.L. Consortium for Top Down Proteomics. Proteoform: A single term describing protein complexity. Nat. Methods 2013, 10, 186–187. [Google Scholar]
- Niture, S.K.; Velu, C.S.; Bailey, N.I.; Srivenugopal, K.S. S-thiolation mimicry: Quantitative and kinetic analysis of redox status of protein cysteines by glutathione-affinity chromatography. Arch. Biochem. Biophys 2005, 444, 174–184. [Google Scholar]
- Velu, C.S.; Niture, S.K.; Doneanu, C.E.; Pattabiraman, N.; Srivenugopal, K.S. Human p53 is inhibited by glutathionylation of cysteines present in the proximal DNA-binding domain during oxidative stress. Biochemistry 2007, 46, 7765–7780. [Google Scholar]
- Naito, C.; Kajita, M.; Niwa, T. Determination of glutathionyl hemoglobin in hemodialysis patients using electrospray ionization liquid chromatography-mass spectrometry. J. Chromatogr. B 1999, 731, 121–124. [Google Scholar]
- Pastore, A.; Mozzi, A.F.; Tozzi, G.; Gaeta, L.M.; Federici, G.; Bertini, E.; Lo Russo, A.; Mannucci, L.; Piemonte, F. Determination of glutathionyl-hemoglobin in human erythrocytes by cation-exchange high-performance liquid chromatography. Anal. Biochem 2003, 312, 85–90. [Google Scholar]
- Sun, C.; Shi, Z.Z.; Zhou, X.; Chen, L.; Zhao, X.M. Prediction of S-glutathionylation sites based on protein sequences. PLoS One 2013, 8, e55512. [Google Scholar]
- Sanchez, G.; Pedrozo, Z.; Domenech, R.J.; Hidalgo, C.; Donoso, P. Tachycardia increases NADPH oxidase activity and RyR2 S-glutathionylation in ventricular muscle. J. Mol. Cell Cardiol 2005, 39, 982–991. [Google Scholar]
- Sanchez, G.; Escobar, M.; Pedrozo, Z.; Macho, P.; Domenech, R.; Hartel, S.; Hidalgo, C.; Donoso, P. Exercise and tachycardia increase NADPH oxidase and ryanodine receptor-2 activity: Possible role in cardioprotection. Cardiovasc. Res 2008, 77, 380–386. [Google Scholar]
- Heumuller, S.; Wind, S.; Barbosa-Sicard, E.; Schmidt, H.H.; Busse, R.; Schroder, K.; Brandes, R.P. Apocynin is not an inhibitor of vascular NADPH oxidases but an antioxidant. Hypertension 2008, 51, 211–217. [Google Scholar]
- Donoso, P.; Sanchez, G.; Bull, R.; Hidalgo, C. Modulation of cardiac ryanodine receptor activity by ROS and RNS. Front. Biosci 2011, 16, 553–567. [Google Scholar]
- Vangheluwe, P.; Raeymaekers, L.; Dode, L.; Wuytack, F. Modulating sarco(endo)plasmic reticulum Ca2+ ATPase 2 (SERCA2) activity: Cell biological implications. Cell Calcium 2005, 38, 291–302. [Google Scholar]
- Vangheluwe, P.; Louch, W.E.; Ver Heyen, M.; Sipido, K.; Raeymaekers, L.; Wuytack, F. Ca2+ transport ATPase isoforms SERCA2a and SERCA2b are targeted to the same sites in the murine heart. Cell Calcium 2003, 34, 457–464. [Google Scholar]
- Cohen, R.A.; Adachi, T. Nitric-oxide-induced vasodilatation: Regulation by physiologic S-glutathiolation and pathologic oxidation of the sarcoplasmic endoplasmic reticulum calcium ATPase. Trends Cardiovasc. Med 2006, 16, 109–114. [Google Scholar]
- Lock, J.T.; Sinkins, W.G.; Schilling, W.P. Effect of protein S-glutathionylation on Ca2+ homeostasis in cultured aortic endothelial cells. Am. J. Physiol. Heart Circ. Physiol 2011, 300, 493–506. [Google Scholar]
- Lancel, S.; Zhang, J.; Evangelista, A.; Trucillo, M.P.; Tong, X.; Siwik, D.A.; Cohen, R.A.; Colucci, W.S. Nitroxyl activates SERCA in cardiac myocytes via glutathiolation of cysteine 674. Circ. Res 2009, 104, 720–723. [Google Scholar]
- Chen, C.A.; Wang, T.Y.; Varadharaj, S.; Reyes, L.A.; Hemann, C.; Talukder, M.A.; Chen, Y.R.; Druhan, L.J.; Zweier, J.L. S-glutathionylation uncouples eNOS and regulates its cellular and vascular function. Nature 2010, 468, 1115–1118. [Google Scholar]
- Zweier, J.L.; Chen, C.A.; Druhan, L.J. S-glutathionylation reshapes our understanding of endothelial nitric oxide synthase uncoupling and nitric oxide/reactive oxygen species-mediated signaling. Antioxid. Redox Signal 2011, 14, 1769–1775. [Google Scholar]
- Kaplan, J.H. Biochemistry of Na, K-ATPase. Annu. Rev.Biochem 2002, 71, 511–535. [Google Scholar]
- Galougahi, K.K.; Liu, C.C.; Garcia, A.; Fry, N.A.; Hamilton, E.J.; Rasmussen, H.H.; Figtree, G.A. Protein kinase-dependent oxidative regulation of the cardiac Na+-K+ pump: Evidence from in vivo and in vitro modulation of cell signaling. J. Physiol 2013, 591, 2999–3015. [Google Scholar]
- Jorgensen, P.L.; Hakansson, K.O.; Karlish, S.J. Structure and mechanism of Na, K-ATPase: Functional sites and their interactions. Annu. Rev. Physiol 2003, 65, 817–849. [Google Scholar]
- Pieske, B.; Houser, S.R. [Na+], handling in the failing human heart. Cardiovasc. Res 2003, 57, 874–886. [Google Scholar]
- Pogwizd, S.M.; Sipido, K.R.; Verdonck, F.; Bers, D.M. Intracellular Na in animal models of hypertrophy and heart failure: Contractile function and arrhythmogenesis. Cardiovasc. Res 2003, 57, 887–896. [Google Scholar]
- Rasmussen, H.H.; Hamilton, E.J.; Liu, C.C.; Figtree, G.A. Reversible oxidative modification: Implications for cardiovascular physiology and pathophysiology. Trends Cardiovasc. Med 2010, 20, 85–90. [Google Scholar]
- Shimon, M.B.; Goldshleger, R.; Karlish, S.J. Specific Cu2+-catalyzed oxidative cleavage of Na, K-ATPase at the extracellular surface. J. Biol. Chem 1998, 273, 34190–34195. [Google Scholar]
- Figtree, G.A.; Rasmussen, H.H.; Liu, C.C. Oxidative regulation of the Na+ -K+ pump in cardiac physiology and pathology: Clarifying the published evidence. Circ. Res 2013, 112, 1. [Google Scholar]
- Liu, C.C.; Garcia, A.; Mahmmoud, Y.A.; Hamilton, E.J.; Galougahi, K.K.; Fry, N.A.; Figtree, G.A.; Cornelius, F.; Clarke, R.J.; Rasmussen, H.H. Susceptibility of β1 Na+-K+ pump subunit to glutathionylation and oxidative inhibition depends on conformational state of pump. J. Biol. Chem 2012, 287, 12353–12364. [Google Scholar]
- Morth, J.P.; Pedersen, B.P.; Toustrup-Jensen, M.S.; Sørensen, T.L.; Petersen, J.; Andersen, J.P.; Vilsen, B.; Nissen, P. Crystal structure of the sodium-potassium pump. Nature 2007, 450, 1043–1049. [Google Scholar]
- Shinoda, T.; Ogawa, H.; Cornelius, F.; Toyoshima, C. Crystal structure of the sodium-potassium pump at 2.4 A resolution. Nature 2009, 459, 446–450. [Google Scholar]
- Morth, J.P.; Pedersen, B.P.; Buch-Pedersen, M.J.; Andersen, J.P.; Vilsen, B.; Palmgren, M.G.; Nissen, P. A structural overview of the plasma membrane Na+, K+-ATPase and H+ATPase ion pumps. Nat. Rev. Mol. Cell Biol 2011, 12, 60–70. [Google Scholar]
- Dempski, R.E.; Hartung, K.; Friedrich, T.; Bamberg, E. Fluorometric measurements of intermolecular distances between the alpha- and beta-subunits of the Na+/K+-ATPase. J. Biol. Chem 2006, 281, 36338–36346. [Google Scholar]
- Liu, C.C.; Karimi Galougahi, K.; Weisbrod, R.M.; Hansen, T.; Ravaie, R.; Nunez, A.; Liu, Y.B.; Fry, N.; Garcia, A.; Hamilton, E.J.; et al. Oxidative inhibition of the vascular Na(+)-K(+) pump via NADPH oxidase-dependent β(1)-subunit glutathionylation: Implications for angiotensin II-induced vascular dysfunction. Free Radic. Biol. Med 2013, 65, 563–572. [Google Scholar]
- Johansson, M.; Lundberg, M. Glutathionylation of beta-actin via a cysteinyl sulfenic acid intermediary. Biochemistry 2007, 8, 26–32. [Google Scholar]
- Dalle-Donne, I.; Rossi, R.; Giustarini, D.; Colombo, R.; Milzani, A. Actin S-glutathionylation: Evidence against a thiol-disulphide exchange mechanism. Free Rad. Biol. Med 2003, 35, 1185–1193. [Google Scholar]
- Pizarro, G.O.; Ogut, O. Impact of actin glutathionylation on the actomyosin-S1 ATPase. Biochemistry 2009, 48, 7533–7538. [Google Scholar]
- Lovelock, J.D.; Monasky, M.M.; Jeong, E.M.; Lardin, H.A.; Liu, H.; Patel, B.G.; Taglieri, D.M.; Gu, L.; Kumar, P.; Pokhrel, N.; et al. Ranolazine improves cardiac diastolic dysfunction through modulation of myofilament calcium sensitivity. Circ. Res 2012, 110, 841–850. [Google Scholar]
- Harris, S.P.; Lyons, R.G.; Bezold, K.L. In the thick of it: HCM-causing mutations in myosin binding proteins of the thick filament. Circ. Res 2011, 108, 751–764. [Google Scholar]
- Mollica, J.P.; Dutka, T.L.; Merry, T.L.; Lamboley, C.R.; McConell, G.K.; McKenna, M.J.; Murphy, R.M.; Lamb, G.D. S-glutathionylation of troponin I (fast) increases contractile apparatus Ca2+ sensitivity in fast-twitch muscle fibres of rats and humans. J. Physiol 2012, 590, 1443–1463. [Google Scholar]
- Chong, P.C.; Hodges, R.S. Proximity of sulfhydryl groups to the sites of interaction between components of the troponin complex from rabbit skeletal muscle. J. Biol. Chem 1982, 257, 2549–2555. [Google Scholar]
- Allen, D.G.; Lamb, G.D.; Westerblad, H. Skeletal muscle fatigue: Cellular mechanisms. Physiol. Rev 2008, 88, 287–332. [Google Scholar]
- Dutka, T.L.; Mollica, J.P.; Posterino, G.S.; Lamb, G.D. Modulation of contractile apparatus Ca2+ sensitivity and disruption of excitation-contraction coupling by S-nitrosoglutathione in rat muscle fibres. J. Physiol 2011, 589, 2181–2196. [Google Scholar]
- Stull, J.T.; Kamm, K.E.; Vandenboom, R. Myosin light chain kinase and the role of myosin light chain phosphorylation in skeletal muscle. Arch. Biochem. Biophys 2011, 510, 120–128. [Google Scholar]
- Andrade, F.H.; Reid, M.B.; Allen, D.G.; Westerblad, H. Effect of hydrogen peroxide and dithiothreitol on contractile function of single skeletal muscle fibres from the mouse. J. Physiol 1998, 509, 565–575. [Google Scholar]
- Lamb, G.D.; Westerblad, H. Acute effects of reactive oxygen and nitrogen species on the contractile function of skeletal muscle. J. Physiol 2011, 589, 2119–2127. [Google Scholar]
- Beer, S.M.; Taylor, E.R.; Brown, S.E.; Dahm, C.C.; Costa, N.J.; Runswick, M.J.; Murphy, M.P. Glutaredoxin 2 catalyzes the reversible oxidation and glutathionylation of mitochondrial membrane thiol proteins: Implications for mitochondrial redox regulation and antioxidant defense. J. Biol. Chem 2004, 279, 47939–47951. [Google Scholar]
- Hurd, T.R.; Requejo, R.; Filipovska, A.; Brown, S.; Prime, T.A.; Robinson, A.J.; Fearnley, I.M.; Murphy, M.P. Complex I within oxidatively stressed bovine heart mitochondria is glutathionylated on Cys-531 and Cys-704 of the 75-kDa subunit. J. Biol. Chem 2008, 283, 24801–24815. [Google Scholar]
- Kang, P.T.; Zhang, L.; Chen, C.L.; Chen, J.; Green, K.B.; Chen, Y.R. Protein thiyl radical mediates S-glutathionylation of complex I. Free Radic. Biol. Med 2012, 53, 962–973. [Google Scholar]
- Passarelli, C.; Tozzi, G.; Pastore, A.; Bertini, E.; Piemonte, F. GSSG-mediated Complex I defect in isolated cardiac mitochondria. Int. J. Mol. Med 2010, 26, 95–99. [Google Scholar]
- Papa, S.; De Rasmo, D.; Scacco, S.; Signorile, A.; Technikova-Dobrova, Z.; Palmisano, G.; Sardanelli, A.M.; Papa, F.; Panelli, D.; Scaringi, R.; et al. Mammalian Complex I: A regulable and vulnerable pacemaker in mitochondrial respiratory function. Biochim. Biophys. Acta 2008, 1777, 719–728. [Google Scholar]
- Zhang, L.; Kang, P.T.; Chen, C.L.; Green, K.B.; Chen, Y.R. Oxidative modifications of mitochondria complex II. Methods Mol. Biol 2013, 1005, 143–56. [Google Scholar]
- Wang, S.B.; Foster, D.B.; Rucker, J.; O’Rourke, B.; Kass, D.A.; Van Eyk, J.E. Redox regulation of mitochondrial ATP synthase: Implications for cardiac resynchronization therapy. Circ. Res 2011, 109, 750–757. [Google Scholar]
- Lee, I.H.; Cao, L.; Mostoslavsky, R.; Lombard, D.B.; Liu, J; Bruns, N.E.; Tsokos, M.; Alt, F.W.; Finkel, T. A role for the NAD-dependent deacetylase Sirt1 in the regulation of autophagy. Proc. Natl. Acad. Sci. USA 2008, 105, 3374–3379. [Google Scholar]
- Zee, R.S.; Yoo, C.B.; Pimentel, D.R.; Perlman, D.H.; Burgoyne, J.R.; Hou, X.; McComb, M.E.; Costello, C.E.; Cohen, R.A.; Bachschmid, M.M. Redox regulation of sirtuin-1 by Sglutathiolation. Antioxid. Redox Signal 2010, 13, 1023–1032. [Google Scholar]
- Caito, S.; Rajendrasozhan, S.; Cook, S.; Chung, S.; Yao, H.; Friedman, A.E.; Brookes, P.S.; Rahman, I. SIRT1 is a redox sensitive deacetylase that is post-translationally modified by oxidants and carbonyl stress. FASEB J 2010, 24, 3145–3159. [Google Scholar]
- Srivastava, S.K.; Ramana, K.V.; Bhatnagar, A. Role of aldose reductase and oxidative damage in diabetes and the consequent potential for therapeutic options. Endocr. Rev 2005, 26, 380–392. [Google Scholar]
- Srivastava, S.K.; Yadav, U.C.; Reddy, A.B.; Saxena, A.; Tammali, R.; Shoeb, M.; Ansari, N.H.; Bhatnagar, A.; Petrash, M.J.; Srivastava, S.; et al. Aldose reductase inhibition suppresses oxidative stress induced inflammatory disorders. Chem. Biol. Interact 2011, 191, 330–338. [Google Scholar]
- Ruef, J.; Liu, S.Q.; Bode, C.; Tocchi, M.; Srivastava, S.; Runge, M.S.; Bhatnagar, A. Involvement of aldose reductase in vascular smooth muscle cell growth and lesion formation after arterial injury. Arterioscler. Thromb. Vasc. Biol 2000, 20, 1745–1752. [Google Scholar]
- Iwata, T.; Sato, S.; Jimenez, J.; McGowan, M.; Moroni, M.; Dey, A.; Ibaraki, N.; Reddy, V.N.; Carper, D. Osmotic response element is required for the induction of aldose reductase by tumor necrosis factor-alpha. J. Biol. Chem 1999, 274, 7993–8001. [Google Scholar]
- Ramana, K.V.; Bhatnagar, A.; Srivastava, S.K. Inhibition of aldose reductase attenuates TNF-alpha-induced expression of adhesion molecules in endothelial cells. FASEB J 2004, 18, 1209–1218. [Google Scholar]
- Cappiello, M.; Voltarelli, M.; Cecconi, I.; Vilardo, P.G.; Dal Monte, M.; Marini, I.; Del Corso, A.; Wilson, D.K.; Quiocho, F.A.; Petrash, J.M.; et al. Specifically targeted modification of human aldose reductase by physiological disulfides. J. Biol. Chem 1996, 271, 33539–33544. [Google Scholar]
- Bohren, K.M.; Gabbay, K.H. Cys298 is responsible for reversible thiol-induced variation in aldose reductase activity. Adv. Exp. Med. Biol 1993, 328, 267–277. [Google Scholar]
- Liu, S.Q.; Bhatnagar, A.; Ansari, N.H.; Srivastava, S.K. Identification of the reactive cysteine residue in human placenta aldose reductase. Biochim. Biophys. Acta 1993, 1164, 268–272. [Google Scholar]
- Cappiello, M.; Amodeo, P.; Mendez, B.L.; Scaloni, A.; Vilardo, P.G.; Cecconi, I.; Dal Monte, M.; Banditelli, S.; Talamo, F.; Micheli, V.; et al. Modulation of aldose reductase activity through S-thiolation by physiological thiols. Chem. Biol. Interact 2001, 130–132, 597–608. [Google Scholar]
- Ramana, K.V.; Tammali, R.; Reddy, A.B.; Bhatnagar, A.; Srivastava, S.K. Aldose reductase-regulated tumor necrosis factor-alpha production is essential for high glucoseinduced vascular smooth muscle cell growth. Endocrinology 2007, 148, 4371–4384. [Google Scholar]
- Kaiserova, K.; Srivastava, S.; Hoetker, J.D.; Awe, S.O.; Tang, X.L.; Cai, J.; Bhatnagar, A. Redox activation of aldose reductase in the ischemic heart. J. Biol. Chem 2006, 281, 15110–15120. [Google Scholar]
- Kaiserova, K.; Tang, X.L.; Srivastava, S.; Bhatnagar, A. Role of nitric oxide in regulating aldose reductase activation in the ischemic heart. J. Biol. Chem 2008, 283, 9101–9112. [Google Scholar]
- Garel, M.C.; Domenget, C.; Caburi-Martin, J.; Prehu, C.; Galacteros, F.; Beuzard, Y. Covalent binding of glutathione to hemoglobin. I. Inhibition of hemoglobin S polymerization. J. Biol. Chem 1986, 261, 14704–14709. [Google Scholar]
- Craescu, C.T.; Poyart, C.; Schaeffer, C.; Garel, M.C.; Kister, J.; Beuzard, Y. Covalent binding of glutathione to hemoglobin. II. Functional consequences and structural changes reflected in NMR spectra. J. Biol. Chem 1986, 261, 14710–14716. [Google Scholar]
- Mawatari, S.; Murakami, K. Different types of glutathionylation of hemoglobin can exist in intact erythrocytes. Arch. Biochem. Biophys 2004, 421, 108–114. [Google Scholar]
- Regazzoni, L.; Panusa, A.; Yeum, K.J.; Carini, M; Aldini, G. Hemoglobin glutathionylation can occur through cysteine sulfenic acid intermediate: Electrospray ionization LTQ-Orbitrap hybrid mass spectrometry studies. J. Chromatogr. B 2009, 877, 3456–3461. [Google Scholar]
- Metere, A.; Iorio, E.; Scorza, G.; Camerini, S.; Casella, M.; Crescenzi, M.; Minetti, M.; Pietraforte, D. Carbon monoxide signaling in human red blood cells: evidence for pentose phosphate pathway activation and protein deglutathionylation. Antioxid. Redox Signal 2013, in press. [Google Scholar]
- Head, C.A.; Brugnara, C.; Martinez-Ruiz, R.; Kacmarek, R.M.; Bridges, K.R.; Kuter, D.; Bloch, K.D.; Zapol, W.M. Low concentrations of nitric oxide increase oxygen affinity of sickle erythrocytes in vitro and in vivo. J. Clin. Invest. 1997, 100, 1193–1198. [Google Scholar]
- Jia, L.; Bonaventura, C.; Bonaventura, J.; Stamler, J.S. S-nitrosohaemoglobin: A dynamic activity of blood involved in vascular control. Nature 1996, 380, 221–226. [Google Scholar]
- Singel, D.J.; Stamler, J.S. Chemical physiology of blood flow regulation by red blood cells: The role of nitric oxide and S-nitrosohemoglobin. Annu. Rev. Physiol 2005, 67, 99–145. [Google Scholar]
- Niwa, T. Protein glutathionylation and oxidative stress. J. Chromatogr. B 2007, 855, 59–65. [Google Scholar]
- Sethuraman, M.; Clavreul, N.; Huang, H.; McComb, M.E.; Costello, C.E.; Cohen, R.A. Quantification of oxidative posttranslational modifications of cysteine thiols of p21ras associated with redox modulation of activity using isotopecoded affinity tags and mass spectrometry. Free Radic. Biol. Med 2007, 42, 823–829. [Google Scholar]
- Zhao, C.; Sethuraman, M.; Clavreul, N.; Kaur, P.; Cohen, R.A.; O’Connor, P.B. Detailed map of oxidative post-translational modifications of human p21ras using Fourier transform mass spectrometry. Anal. Chem 2006, 78, 5134–5142. [Google Scholar]
- Kuster, G.M.; Pimentel, D.R.; Adachi, T.; Ido, Y.; Brenner, D.A.; Cohen, R.A.; Liao, R.; Siwik, D.A.; Colucci, W.S. Alpha-adrenergic receptor-stimulated hypertrophy in adult rat ventricular myocytes is mediated via thioredoxin-1-sensitive oxidative modification of thiols on Ras. Circulation 2005, 111, 1192–1198. [Google Scholar]
- Takayama, N.; Kai, H.; Kudo, H.; Yasuoka, S.; Mori, T.; Anegawa, T.; Koga, M.; Kajimoto, H.; Hirooka, Y.; Imaizumi, T. Simvastatin prevents large blood pressure variability induced aggravation of cardiac hypertrophy in hypertensive rats by inhibiting RhoA/Ras-ERK pathways. Hypertens. Res 2011, 34, 341–347. [Google Scholar]
- Shelton, M.D.; Mieyal, J.J. Regulation by reversible Sglutathionylation: Molecular targets implicated in inflammatory diseases. Mol. Cells 2008, 25, 332–346. [Google Scholar]
- Pineda-Molina, E.; Klatt, P.; Vazquez, J.; Marina, A.; Garcia de Lacoba, M.; Perez-Sala, D.; Lamas, S. Glutathionylation of the p50 subunit of NF-kappaB: A mechanism for redoxinduced inhibition of DNA binding. Biochemistry 2001, 40, 14134–14142. [Google Scholar]
- Qanungo, S.; Starke, D.W.; Pai, H.V.; Mieyal, J.J.; Nieminen, A.L. Glutathione supplementation potentiates hypoxic apoptosis by S-glutathionylation of p65-NFkappaB. J. Biol. Chem 2007, 282, 18427–18436. [Google Scholar]
- Baeuerle, P. AIkB-NF-kB structures: At the interface of inflammation control. Cell 1998, 95, 729–731. [Google Scholar]
- Kabe, Y.; Ando, K.; Hirao, S.; Yoshida, M.; Handa, H. Redox regulation of NFkappaB activation: Distinct redox regulation between the cytoplasm and the nucleus. Antioxid. Redox Signal 2005, 7, 395–403. [Google Scholar]
- Braun, T.; Carvalho, G.; Fabre, C.; Grosjean, J.; Fenaux, P.; Kroemer, G. Targeting NF-kappaB in hematologic malignancies. Cell Death. Differ 2006, 13, 748–758. [Google Scholar]
- Oakley, F.D.; Smith, R.L.; Engelhardt, J.F. Lipid rafts and caveolin-1 coordinate interleukin-1beta (IL-1beta)-dependent activation of NFkappaB by controlling endocytosis of Nox2 and IL-1beta receptor 1 from the plasma membrane. J. Biol. Chem 2009, 284, 33255–33564. [Google Scholar]
- Oakley, F.D.; Abbott, D.; Li, Q.; Engelhardt, J.F. Signaling components of redox active endosomes: The redoxosomes. Antioxid. Redox Signal 2009, 11, 1313–1333. [Google Scholar]
- Li, Q.; Spencer, N.Y.; Oakley, F.D.; Buettner, G.R.; Engelhardt, J.F. Endosomal Nox2 facilitates redox dependent induction of NF-kappa B by TNF-alpha. Antioxid. Redox Signal 2009, 11, 1249–1263. [Google Scholar]
- Prinarakis, E.; Chantzoura, E.; Thanos, D.; Spyrou, G. S-glutathionylation of IRF3 regulates IRF3-CBP interaction and activation of the IFN beta pathway. EMBO J 2008, 27, 865–875. [Google Scholar]
- Madhani, M.; Hall, A.R.; Cuello, F.; Charles, R.L.; Burgoyne, J.R.; Fuller, W.; Hobbs, A.J.; Shattock, M.J.; Eaton, P. Phospholemman Ser69 phosphorylation contributes to sildenafilinduced cardioprotection against reperfusion injury. Am. J. Physiol. Heart Circ. Physiol 2010, 299, H827–H836. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Pastore, A.; Piemonte, F. Protein Glutathionylation in Cardiovascular Diseases. Int. J. Mol. Sci. 2013, 14, 20845-20876. https://doi.org/10.3390/ijms141020845
Pastore A, Piemonte F. Protein Glutathionylation in Cardiovascular Diseases. International Journal of Molecular Sciences. 2013; 14(10):20845-20876. https://doi.org/10.3390/ijms141020845
Chicago/Turabian StylePastore, Anna, and Fiorella Piemonte. 2013. "Protein Glutathionylation in Cardiovascular Diseases" International Journal of Molecular Sciences 14, no. 10: 20845-20876. https://doi.org/10.3390/ijms141020845
APA StylePastore, A., & Piemonte, F. (2013). Protein Glutathionylation in Cardiovascular Diseases. International Journal of Molecular Sciences, 14(10), 20845-20876. https://doi.org/10.3390/ijms141020845