Supramolecular Interactions of Terpyridine-Derived Cores of Metallomesogen Precursors


Abstract
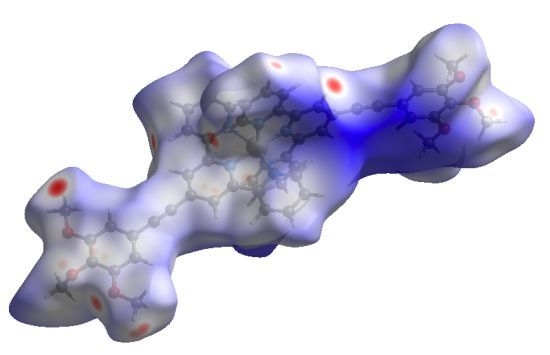
:
1. Introduction
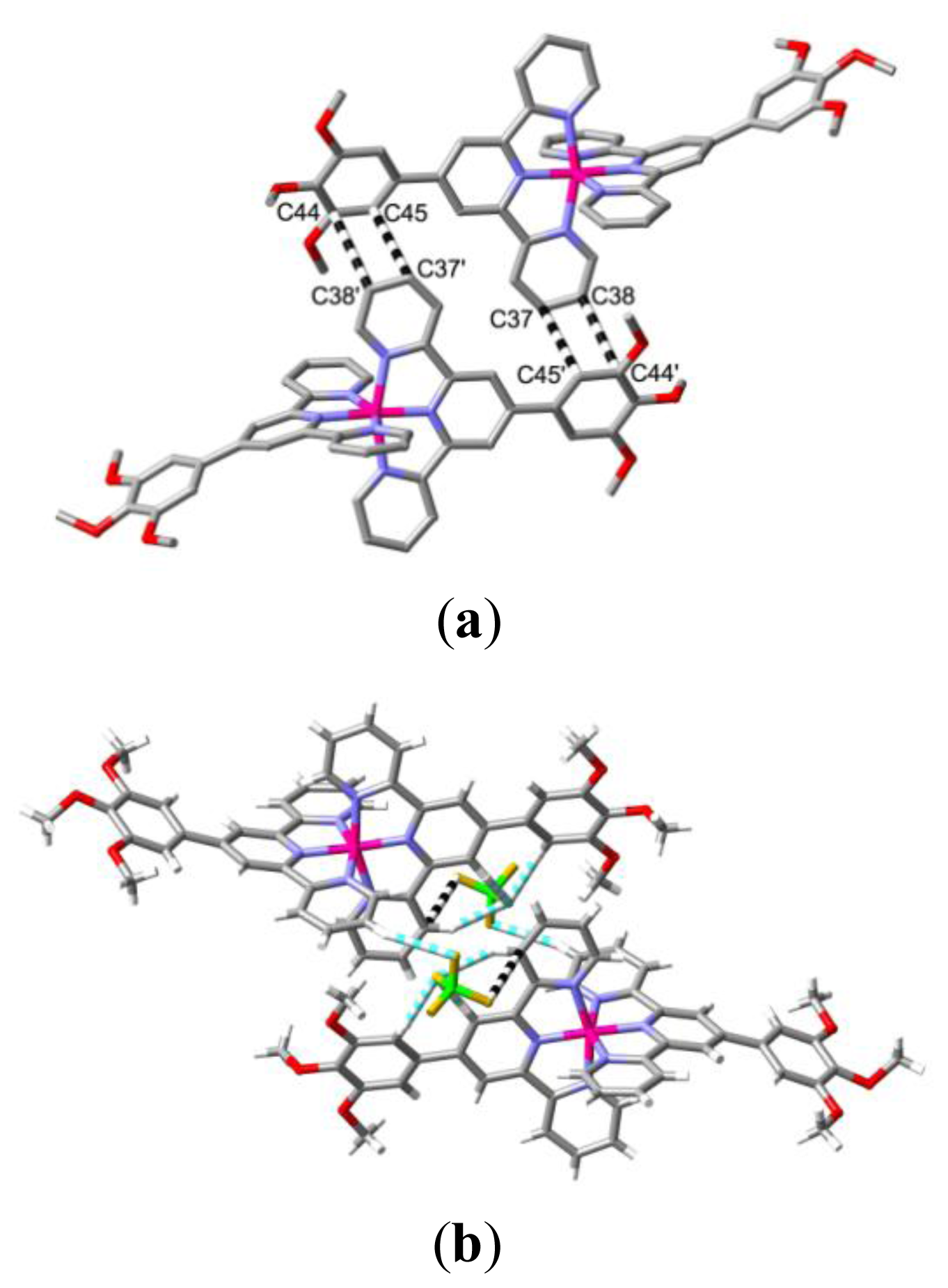
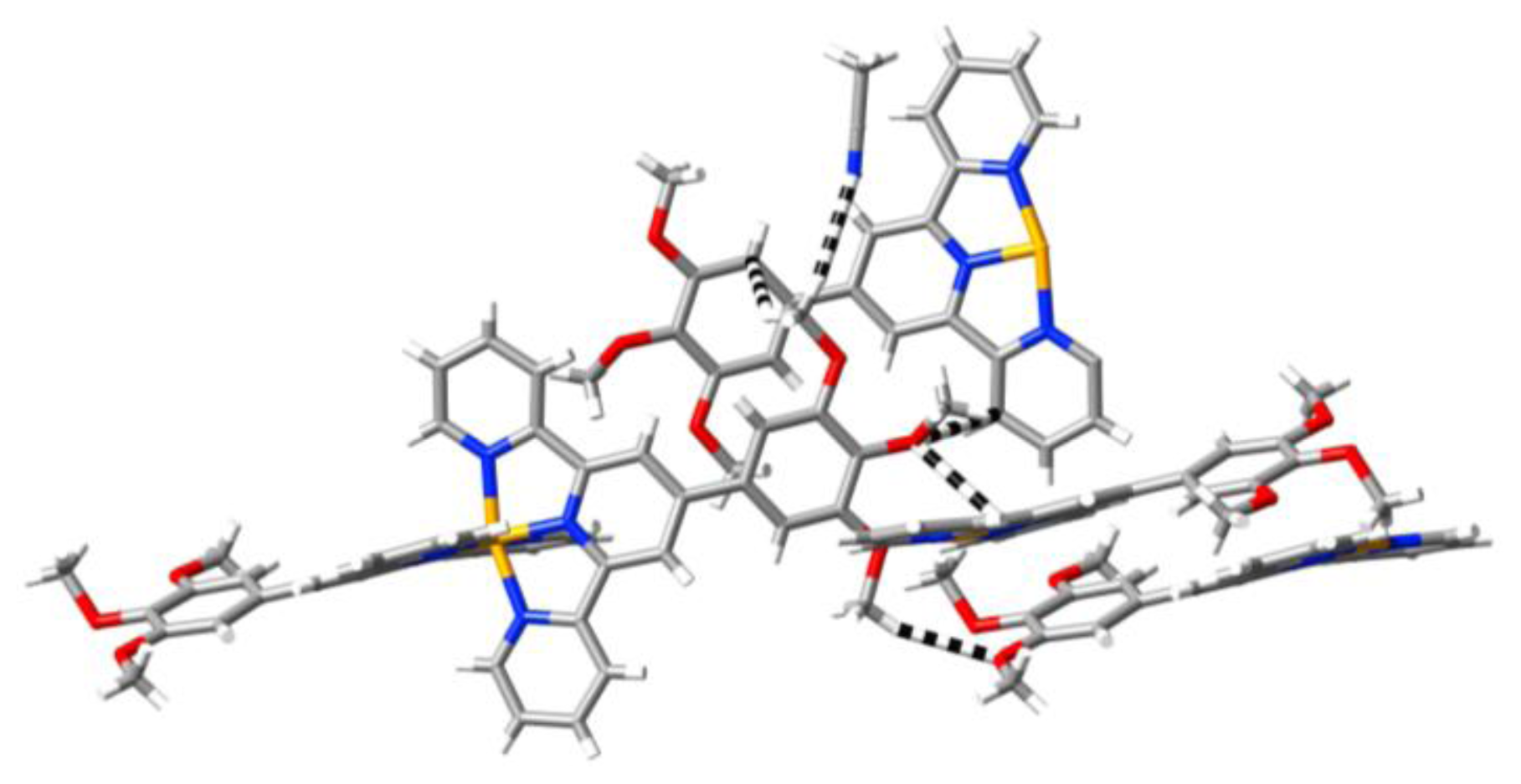
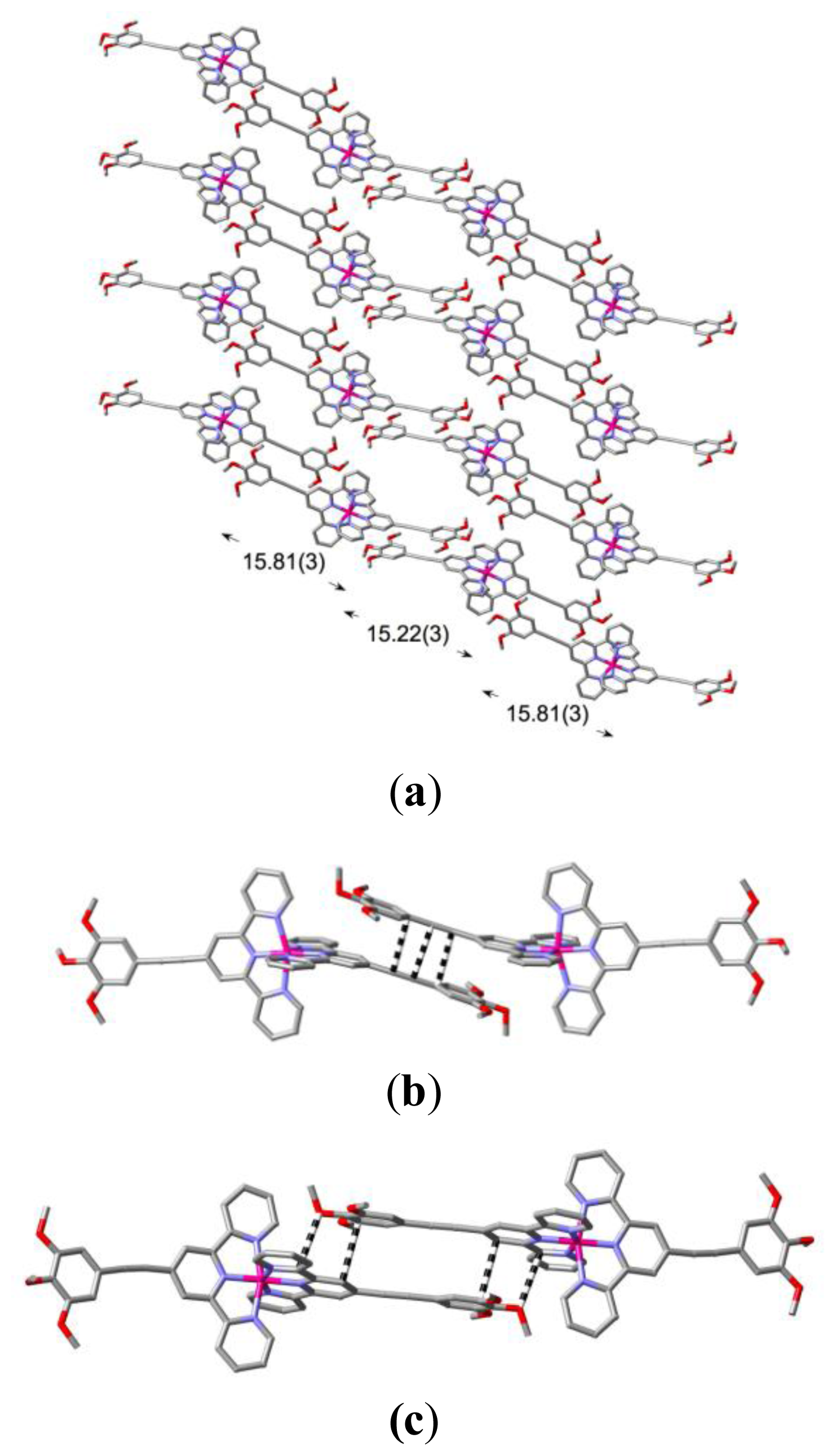
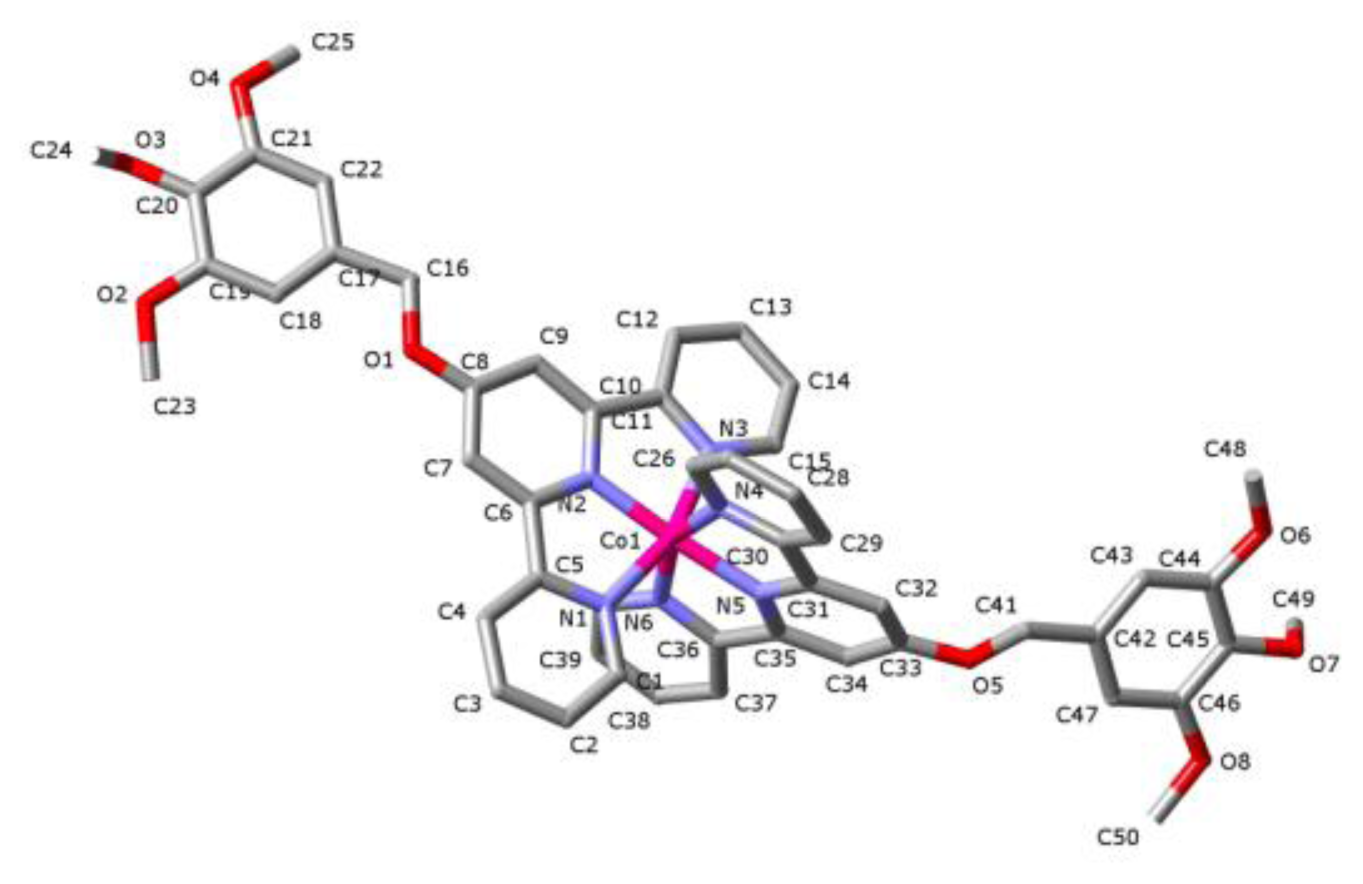
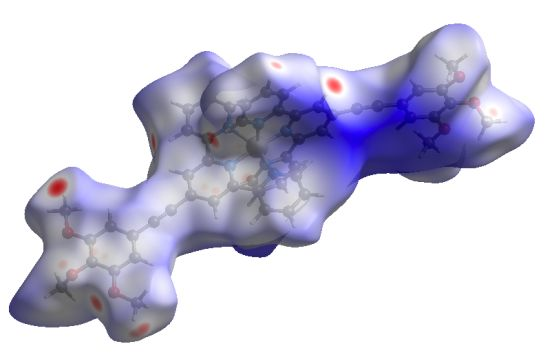
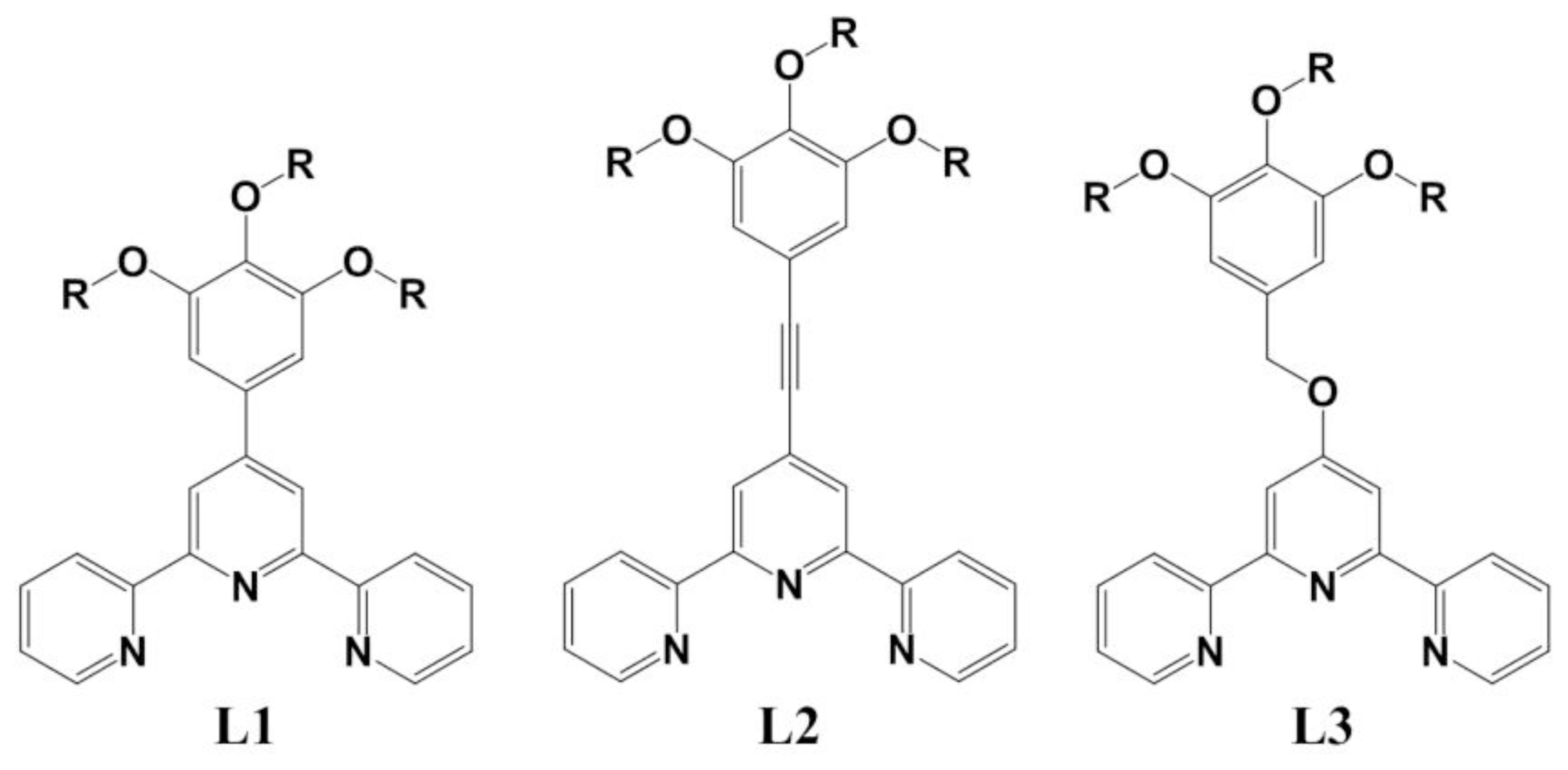
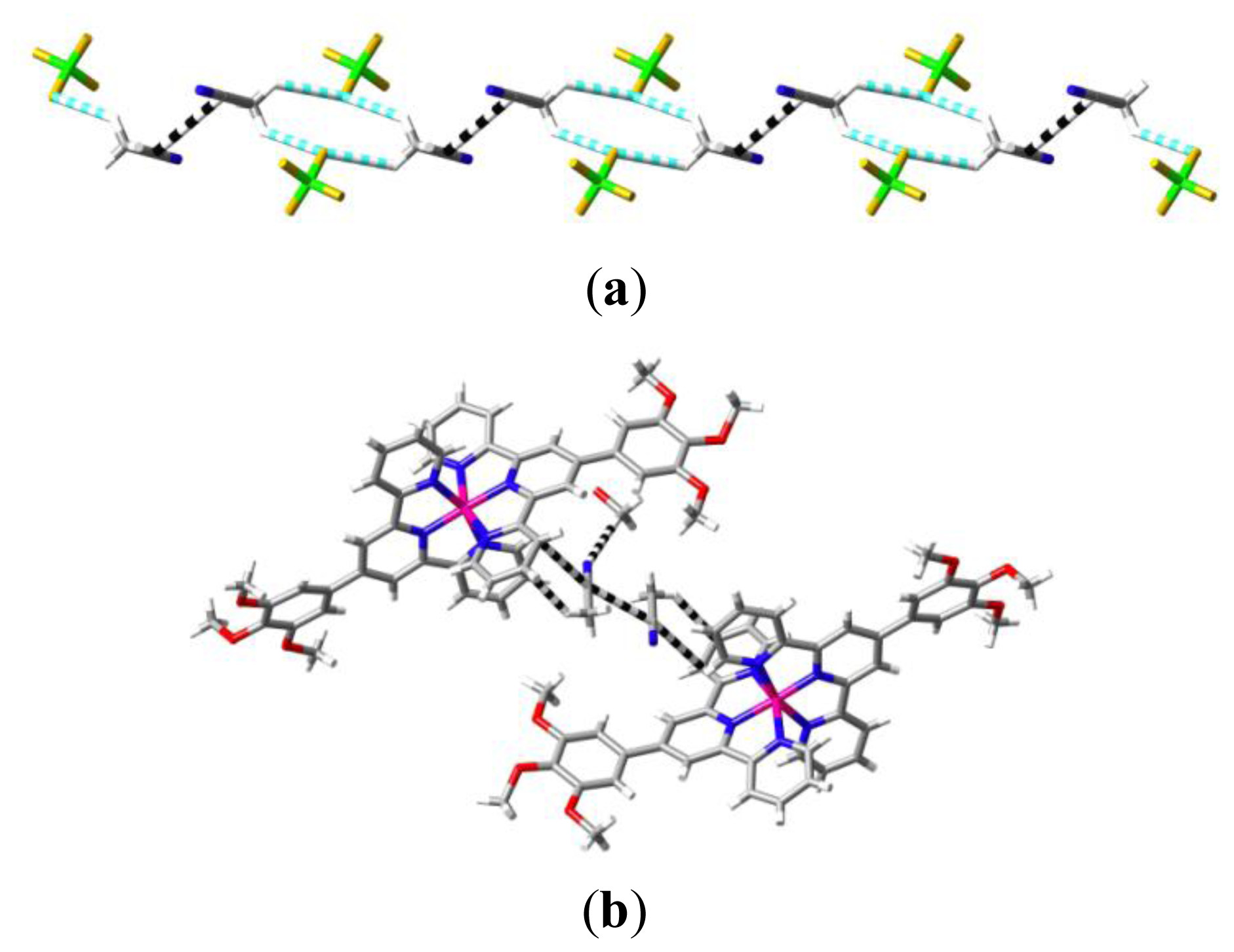
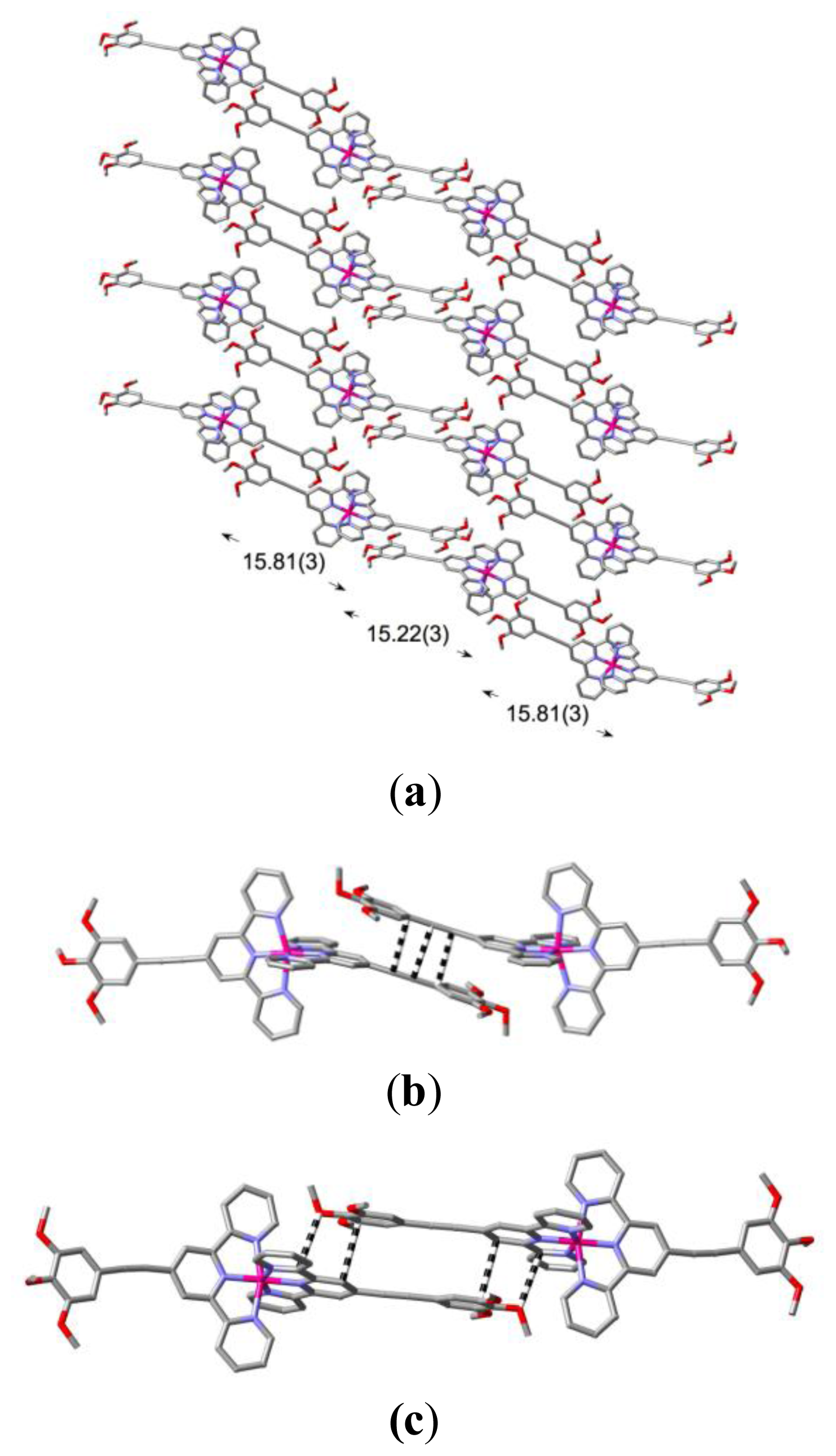
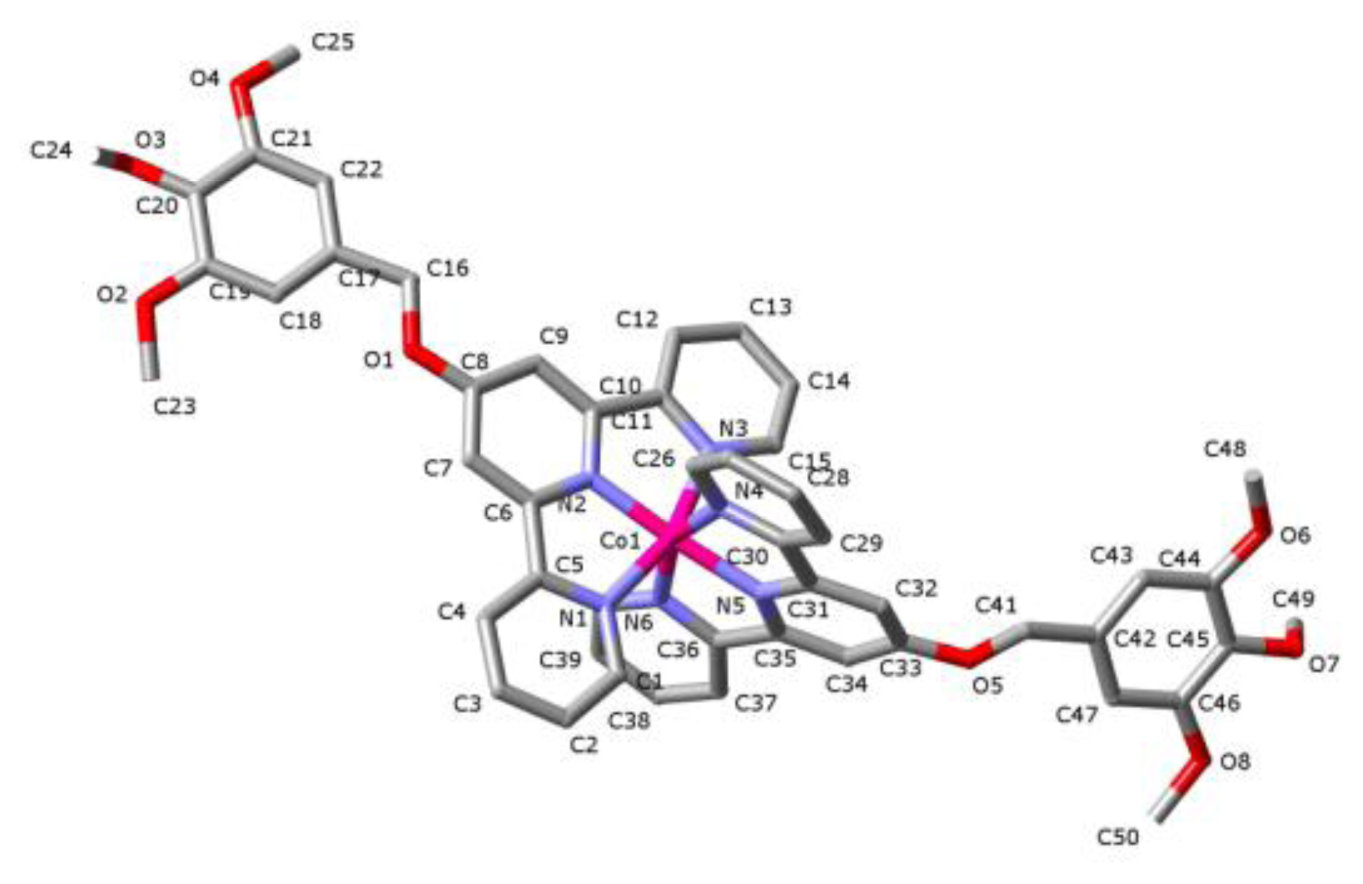
2. Results and Discussion
3. Experimental
3.1. Materials, Software and Instrumentation
3.2. Synthesis
3.3. Crystallography
4. Conclusions
Supplementary Information
ijms-14-20729-s001.pdf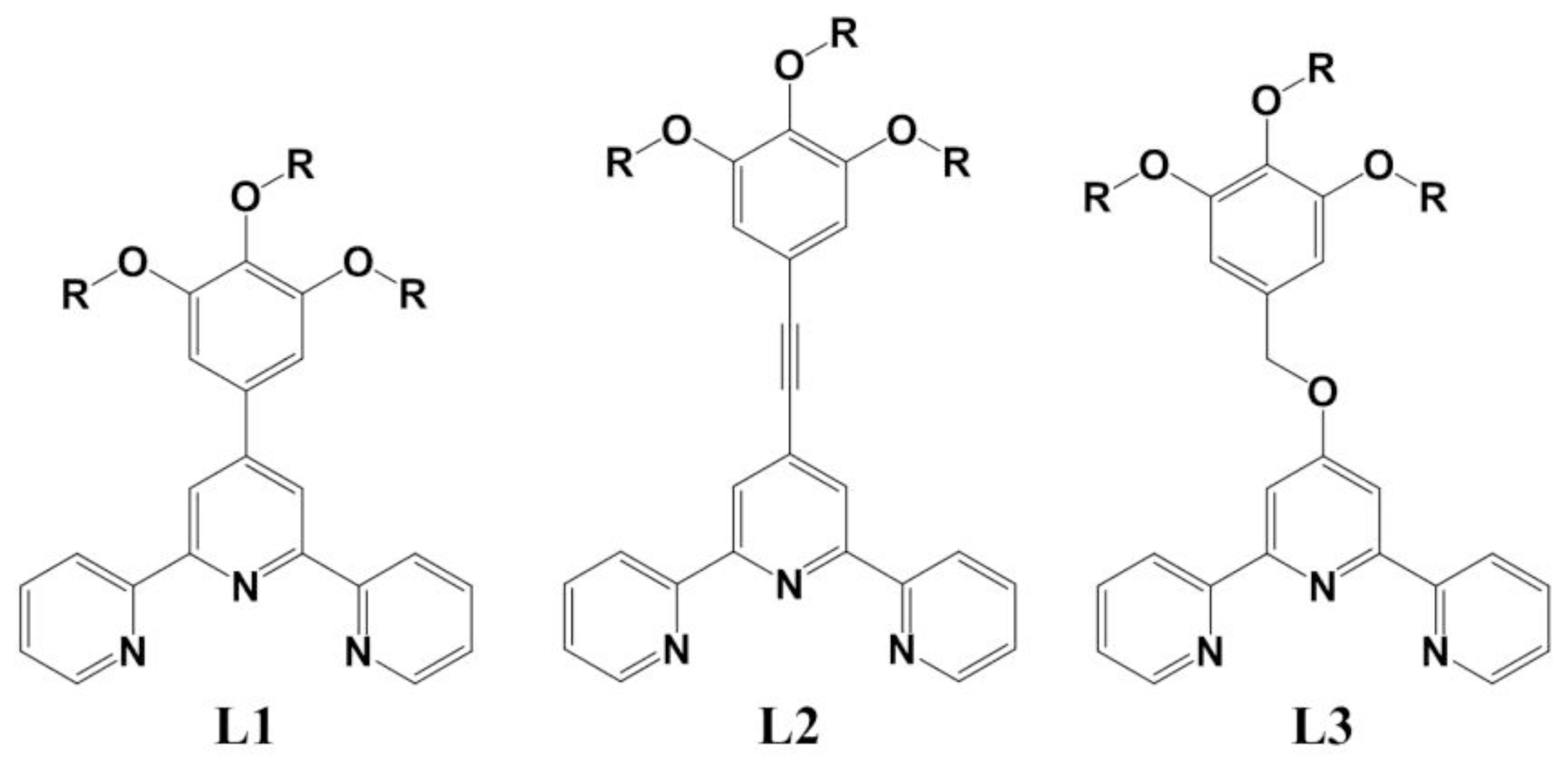





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Bond | 100(2) K (low spin) | 294(2) K (high spin) |
|---|---|---|
| Co–N1 | 1.983(1) | 2.138(3) |
| Co–N2 | 1.871(1) | 1.996(3) |
| Co–N3 | 2.002(1) | 2.119(4) |
| Co–N4 | 2.163(1) | 2.163(4) |
| Co–N5 | 1.935(1) | 1.997(3) |
| Co–N6 | 2.169(1) | 2.146(3) |
| [Fe(L1)2](ClO4)2·CH3CN | [Ni(L1)2](ClO4)2·CH3CN | [Co(L2)2](BF4)2·H2O | [Cd(L2)2](ClO4)2·CH3CN·Et2O | [Cu(L3)2](ClO4)2·CH3CN | [Co(L3)2](BF4)2 | [Co(L3)2](BF4)2 | |
|---|---|---|---|---|---|---|---|
| Empirical formula | C50H45Cl2FeN7O14 | C50H45Cl2N7NiO14 | C52H42B2CoF8N6O7 | C58H55CdCl2N7O15 | C54H52Cl2CuN8O16 | C50H46B2CoF8N6O8 | C50H46B2CoF8N6O8 |
| Formula weight | 1094.68 | 1097.54 | 1095.47 | 1273.39 | 1203.47 | 1091.48 | 1091.48 |
| Crystal system | Triclinic | Triclinic | Triclinic | Triclinic | Triclinic | Triclinic | Triclinic |
| Space group | P-1 | P-1 | P-1 | P-1 | P-1 | P-1 | P-1 |
| Color | Violet | Brown | Brown | Green | Blue | Violet | Violet |
| Crystal size/mm3 | 0.30 × 0.20 × 0.10 | 0.20 × 0.15 × 0.05 | 0.60 × 0.20 × 0.20 | 0.40 × 0.20 ×0.20 | 0.25 × 0.05 × 0.05 | 0.15 × 0.10 × 0.04 | 0.15 × 0.10 × 0.04 |
| a/Å | 9.5687(1) | 9.524(2) | 12.1236(7) | 11.679(2) | 9.468(2) | 12.913(3) | 13.195(3) |
| b/Å | 12.8164(2) | 12.744(3) | 14.1679(7) | 14.919(3) | 11.740(2) | 13.071(3) | 13.245(3) |
| c/Å | 20.6974(3) | 20.896(4) | 18.7880(9) | 18.546(4) | 25.516(5) | 15.749(3) | 16.037(3) |
| α/° | 91.228(1) | 91.40(3) | 69.932(1) | 70.86(3) | 80.38(3) | 102.03(3) | 98.96(3) |
| β/° | 101.151(1) | 100.88(3) | 89.557(2) | 80.39(3) | 89.84(3) | 98.33(3) | 101.43(3) |
| γ/° | 107.052(1) | 106.62(3) | 71.613(2) | 74.71(3) | 76.99(3) | 107.56(3) | 108.45(3) |
| V/Å3 | 2372.58(6) | 2378.3(9) | 2,858.1(3) | 2933.3(10) | 2722.7(10) | 2416.3(8) | 2532.8(9) |
| Z | 2 | 2 | 2 | 2 | 2 | 2 | 2 |
| ρcalc/g cm−3 | 1.532 | 1.533 | 1.273 | 1.442 | 1.468 | 1.500 | 1.431 |
| λ/Å | 0.71073 | 0.62988 | 0.71073 | 0.6300 | 0.8000 | 0.65000 | 0.65000 |
| λ/K | 173(2) | 102(2) | 153(2) | 102(2) | 102(2) | 100(2) | 294(2) |
| μ/mm−1 | 0.509 | 0.432 | 0.378 | 0.389 | 0.796 | 0.448 | 0.428 |
| F(000) | 1132 | 1136 | 1122 | 1308 | 1246 | 1122 | 1122 |
| θmin-max | 1.67–28.35 | 1.765–33.388 | 3.00–27.47 | 1.83–29.49 | 0.46–29.59 | 1.55–33.28 | 1.22–27.09 |
| Reflections collected | 41,675 | 39,046 | 28,444 | 40,426 | 18,452 | 30,929 | 14,656 |
| Independent reflections (Rint) | 11,681 (0.0481) | 19,720 (0.0215) | 13,012 (0.0594) | 20,465 (0.0102) | 9463 (0.0367) | 16.065 (0.0227) | 7942 (0.0336) |
| Reflections with I > 2σ(I) | 8029 | 16,887 | 7570 | 19,871 | 7544 | 13,193 | 5658 |
| Goodness-of-fit on F2 | 1.041 | 1.074 | 1.086 | 1.026 | 1.053 | 1.108 | 1.030 |
| Final R indices [I > 2σ(I)]a | R1 = 0.0475 | 0.0433 | R1 = 0.0744 | 0.0443 | 0.0536 | R1 = 0.0414 | 0.0710 |
| wR2 = 0.1131 | 0.1258 | wR2 = 0.1977 | 0.1257 | 0.1493 | wR2 = 0.1208 | 0.2077 | |
| Final R indices [all data] a | R1 = 0.0802 | 0.0505 | R1 = 0.1227 | 0.0451 | 0.0660 | R1 = 0.0524 | 0.0942 |
| wR2 = 0.1279 | 0.1304 | wR2 = 0.2379 | 0.1581 | wR2 = 0.1259 | 0.2346 |
Acknowledgments
Conflicts of Interest
References and Notes
- Schubert, U.S.; Hofmeier, H.; Newkome, G.R. Modern Terpyridine Chemistry; Wiley-VCH: Weinheim, Baden-Wurtemberg, Germany, 2006. [Google Scholar]
- Wild, A.; Winter, A.; Schlütter, F.; Schubert, U.S. Advances in the field of π-conjugated 2,2′:6′,2″-terpyridines. Chem. Soc. Rev 2011, 40, 1459–1511. [Google Scholar]
- Hayami, S.; Komatsu, Y.; Shimuzu, T.; Kamihata, H.; Lee, Y.H. Spin-crossover in cobalt(II) compounds containing terpyridine and its derivatives. Coord. Chem. Rev 2011, 255, 1981–1990. [Google Scholar]
- Hayami, S. Spin-Crossover Materials: Properties and Applications; Halcrow, M.A., Ed.; Wiley-VCH: Weinheim, Baden-Wurtemberg, Germany, 2013; Volume Ch.12. [Google Scholar]
- Lee, Y.H.; Karim, M.R.; Ikeda, Y.; Shimizu, T.; Kawata, S.; Fuyuhiro, A.; Hayami, S. Tris-Alkoxyphenylterpyridine cobalt(II) complexes: Synthesis, structure, and magnetic and mesomorphic behaviors. J. Inorg. Organomet. Polym 2013, 23, 186–192. [Google Scholar]
- Alexeev, Y.E.; Kharisov, B.I.; Hernandez Garcia, T.C.; Garnovskii, A.D. Coordination motifs in modern supramolecular chemistry. Coord. Chem. Rev 2010, 254, 794–831. [Google Scholar]
- Imrie, C.T.; Henderson, P.A.; Yeap, G.-T. Liquid crystal oligomers: going beyond dimers. Liquid Crystals 2009, 36, 755–777. [Google Scholar]
- Scudder, M.L.; Goodwin, H.A.; Dance, I.G. Crystal supramolecular motifs: two-dimensional grids of terpy embraces in [ML2]z complexes (L = terpy or aromatic N3-tridentate ligand). New J. Chem 1999, 23, 695–705. [Google Scholar]
- McMurtrie, J.; Dance, I. Alternative metal grid structures formed by [M(terpy)2]2+ and [M(terpyOH)2]2+ complexes with small and large tetrahedral dianions, and by [Ru(terpy)2]0. CrystEngComm 2005, 7, 216–229. [Google Scholar]
- For recent reviews of aromatic interactions in general, see Waters, M.L. Aromatic Interactions (Guest Editorial). Acc. Chem. Res 2013, 46, 873, and following articles (e.g. Reference 14 below).. [Google Scholar]
- Lee, Y.H.; Won, M.S.; Harrowfield, J.M.; Kawata, S.; Hayami, S.; Kim, Y. Spin crossover in Co(II) metallorods—Replacing aliphatic tails by aromatic. Dalton Trans 2013, 42, 11507–11521. [Google Scholar]
- Spackman, M.A.; Jayatilaka, D. Hirshfeld surface analysis. CrystEngComm 2009, 11, 19–32. [Google Scholar]
- Ballester, P. Experimental quantification of anion-π interactions in solution using neutral host-guest model systems. Acc. Chem. Res 2013, 46, 874–884. [Google Scholar]
- Chifotides, H.T.; Dunbar, K.R. Anion-π interactions in supramolecular architectures. Acc. Chem. Res 2013, 46, 894–906. [Google Scholar]
- Gale, P.A. Anion receptor chemistry. Chem. Commun 2010, 47, 82–86. [Google Scholar]
- Gale, P.A.; Gunnlaugson, T. Preface: supramolecular chemistry of anionic species themed issue. Chem. Soc. Rev 2010, 39, 3595–3596, and following articles. [Google Scholar]
- Alcock, N.W.; Parker, P.R.; Haider, J.M.; Hannon, M.J.; Painting, C.L.; Pikramenou, Z.; Plummer, E.A.; Rissanen, K.; Saarenketo, P. Red and blue luminescent metallo-supramolecular coordination polymers assembled through π-π interactions. J. Chem. Soc., Dalton Trans 2000, 29, 1447–1461. [Google Scholar]
- CrystalMaker, version 8.7.2; CrystalMaker Software Ltd: Begbroke, Oxfordshire, OX5 1PF, United Kingdom, 2013.
- Constable, E.C.; Ward, M.D. Synthesis and coordination behaviour of 6′,6″-bis(2-pyridyl)-2,2′:4,4″:2″,2‴-quaterpyridine; ‘back-to-back’ 2,2′:6′,2″-terpyridine. J. Chem. Soc., Dalton Trans 1990, 1405–1409. [Google Scholar]
- Potts, K.T.; Konwar, D. Synthesis of 4′-vinyl-2,2′:6′,2″-terpyridine. J. Org. Chem 1991, 56, 4815–4816. [Google Scholar]
- Rasolofonjatovo, E.; Provot, O.; Hamze, A.; Bignon, J.; Thoret, S.; Brion, J.-D.; Alami, M. Regioselective hydrostannation of diarylalkynes directed by a labile ortho bromine atom: An easy acces to stereodefined triarylolefins, hybrids of combretastatin A-4 and isocombretastatin A-4. Eur. J. Med. Chem 2010, 45, 3617–3626. [Google Scholar]
- Li, H.; Yang, L.; Liu, J.; Wang, C.; Gao, F.; Zhang, S. Two-photon optical properties of novel branched conjugated derivatives carrying benzophenone moiety with various electron donor-acceptor substituent groups. J. Fluorescence 2011, 21, 393–407. [Google Scholar]
- ADSC Quantum-210 ADX Program; Arvai, A.J.; Nielsen,, C. Area Detector System Corporation: Poway, CA, USA, 1983. [Google Scholar]
- Otwinowski, Z.; Minor, W. Methods in Enzymology; Carter, C.W., Jr., Sweet, R.M., Eds.; Academic Press: New York, NY, USA, 1997; Volume 276, p. 307. [Google Scholar]
- Bruker-Nonius, APEX v. 2, SAINT v. 7 and XPREP v. 6.14; Bruker AXS, Inc.: Madison, WI, USA, 2003.
- Sheldrick, G.M. Phase annealing in SHELX-90: direct methods for larger structures. Acta Crystallogr. A 1990, 46, 467–473. [Google Scholar]
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. A 2008, 64, 112–122. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Lee, Y.H.; Harrowfield, J.M.; Shin, J.W.; Won, M.S.; Rukmini, E.; Hayami, S.; Min, K.S.; Kim, Y. Supramolecular Interactions of Terpyridine-Derived Cores of Metallomesogen Precursors. Int. J. Mol. Sci. 2013, 14, 20729-20743. https://doi.org/10.3390/ijms141020729
Lee YH, Harrowfield JM, Shin JW, Won MS, Rukmini E, Hayami S, Min KS, Kim Y. Supramolecular Interactions of Terpyridine-Derived Cores of Metallomesogen Precursors. International Journal of Molecular Sciences. 2013; 14(10):20729-20743. https://doi.org/10.3390/ijms141020729
Chicago/Turabian StyleLee, Young Hoon, Jack M. Harrowfield, Jong Won Shin, Mi Seon Won, Elisabeth Rukmini, Shinya Hayami, Kil Sik Min, and Yang Kim. 2013. "Supramolecular Interactions of Terpyridine-Derived Cores of Metallomesogen Precursors" International Journal of Molecular Sciences 14, no. 10: 20729-20743. https://doi.org/10.3390/ijms141020729