MicroRNA Expression Profiling of the Porcine Developing Hypothalamus and Pituitary Tissue

Abstract
:1. Introduction
2. Results
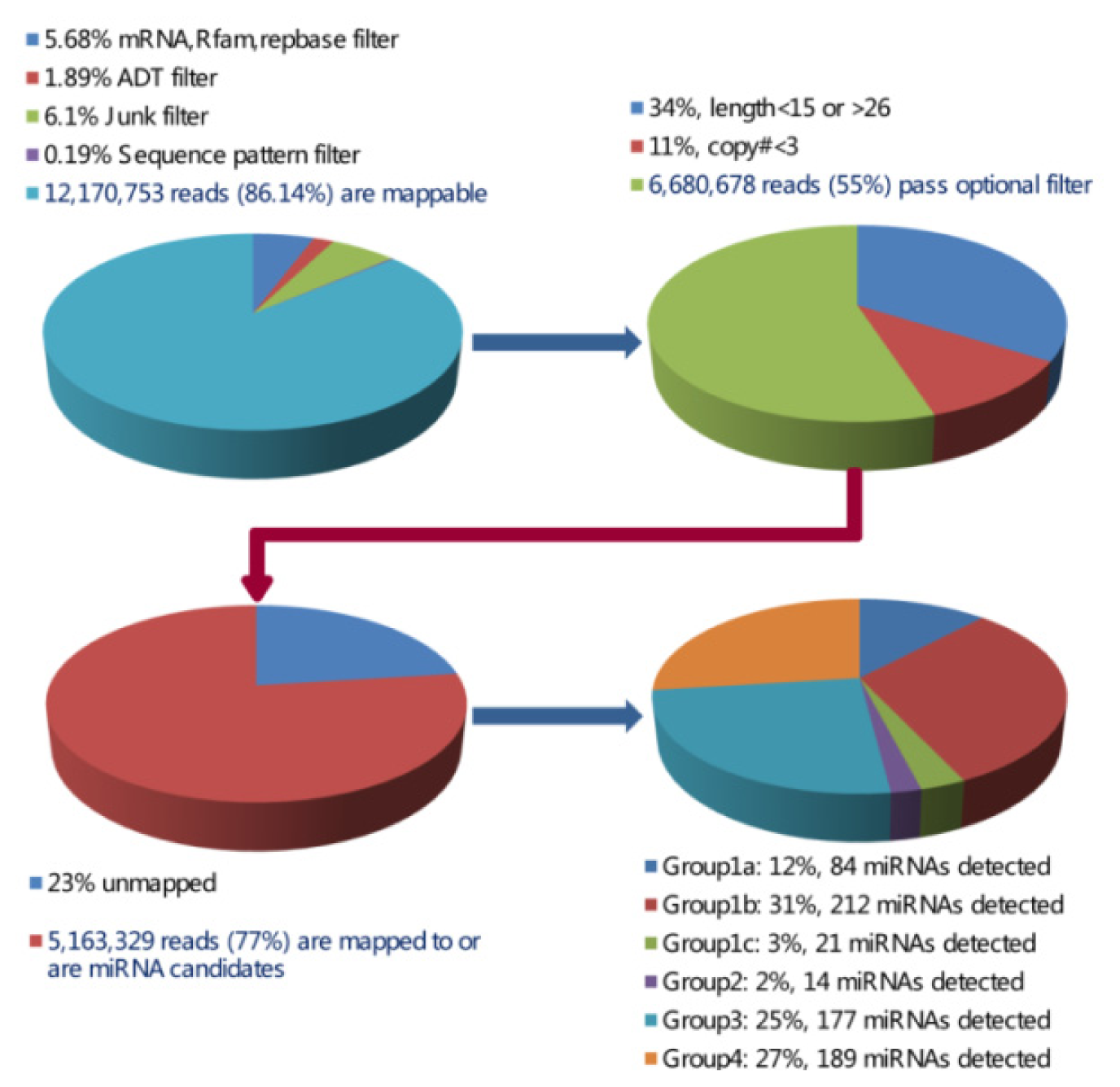
2.1. Solexa Sequencing Analysis of Small RNAs
2.2. Identification of Porcine miRNA Candidates by Microarray Assay
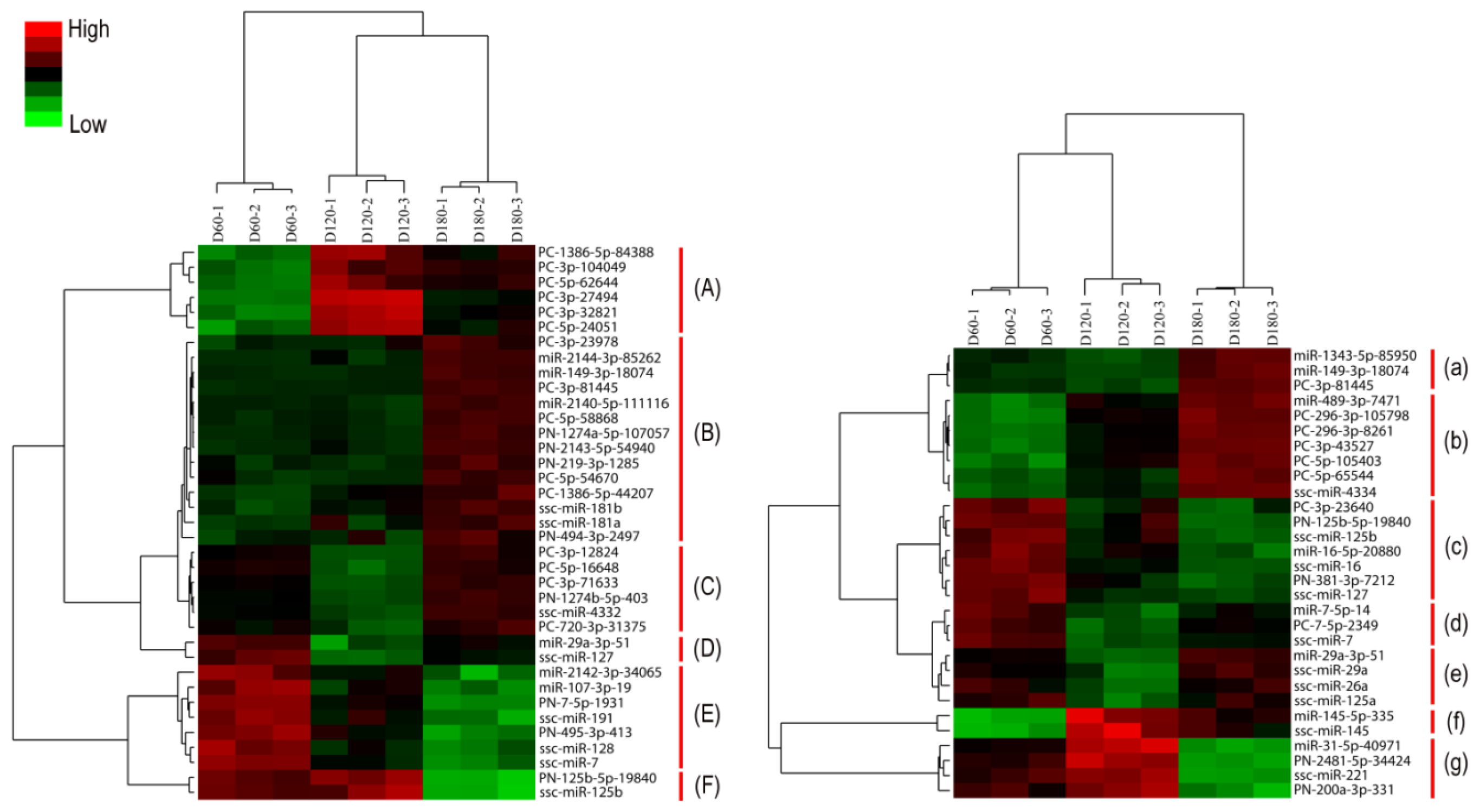
2.3. Discovery of Differentially Expressed miRNAs during Hypothalamus and Pituitary Development
2.4. Expression Models of Differentially Expressed miRNAs
2.5. Prediction and Analysis of miRNA Target Genes
2.6. Assessment of miRNA Expression by Real-Time RT-PCR
3. Discussion
4. Experimental Section
4.1. Ethics Statement and Sample Collection
4.2. RNA Isolation and Solexa Sequencing
4.3. Data Processing
4.4. μ-Paraflo MicroRNA Microarray Assay
4.5. Stem-Loop Real-Time RT-PCR
4.6. Target Prediction and Gene Ontology Analysis
5. Conclusions


{kind=link}
{kind=link}
{kind=link}
| miRNA name | Hypothalamus D120/D60 | D180/D120 | D180/D60 | miRNA name | Pituitary D120/D60 | D180/D120 | D180/D60 |
|---|---|---|---|---|---|---|---|
| PC-1386-5p-84388 | 2.46 | - | 3.14 | miR-1343-5p-85950 | 0.26 | 14.10 | 3.73 |
| PC-3p-104049 | 2.23 | - | 3.82 | miR-149-3p-18074 | 0.32 | 13.70 | 4.39 |
| PC-3p-27494 | 5.79 | - | 4.04 | PC-3p-81445 | 0.27 | 16.94 | 4.62 |
| PC-3p-32821 | 3.99 | - | 3.73 | miR-489-3p-7471 | - | 11.36 | 14.48 |
| PC-5p-24051 | 3.11 | - | 3.18 | PC-296-3p-105798 | - | 9.33 | 13.00 |
| PC-5p-62644 | 2.59 | - | 4.05 | PC-296-3p-8261 | - | 11.22 | 12.57 |
| miR-149-3p-18074 | - | 12.20 | 8.18 | PC-5p-105403 | - | 6.38 | 8.16 |
| miR-2140-5p-111116 | - | 6.90 | 4.52 | PC-3p-43527 | - | 13.93 | 16.35 |
| miR-2144-3p-85262 | - | 4.87 | 4.09 | PC-5p-65544 | - | 24.12 | 13.78 |
| PC-1386-5p-44207 | - | 4.08 | 4.41 | ssc-miR-4334 | - | 13.17 | 9.34 |
| PC-3p-23978 | - | 3.00 | 2.75 | PC-3p-23640 | 0.44 | - | 0.48 |
| PC-3p-81445 | - | 21.58 | 13.83 | PN-125b-5p-19840 | 0.48 | - | 0.44 |
| PC-5p-54670 | - | 6.11 | 3.99 | ssc-miR-125b | 0.46 | - | 0.42 |
| PC-5p-58868 | - | 21.86 | 12.39 | miR-16-5p-20880 | 0.35 | - | 0.34 |
| PN-1274a-5p-107057 | - | 15.13 | 9.86 | ssc-miR-16 | 0.21 | - | 0.23 |
| PN-2143-5p-54940 | - | 6.32 | 5.06 | PN-381-3p-7212 | 0.28 | - | 0.29 |
| PN-219-3p-1285 | - | 4.55 | 3.21 | ssc-miR-127 | 0.42 | - | 0.50 |
| PN-494-3p-2497 | - | 2.72 | 2.40 | miR-7-5p-14 | 0.11 | 3.47 | 0.39 |
| ssc-miR-181a | - | 2.75 | 2.85 | PC-7-5p-2349 | 0.04 | 9.54 | 0.36 |
| ssc-miR-181b | - | 3.08 | 3.20 | ssc-miR-7 | 0.03 | 6.98 | 0.21 |
| PC-3p-12824 | 0.34 | 7.86 | 2.65 | miR-29a-3p-51 | 0.40 | 3.20 | - |
| PC-3p-71633 | 0.36 | 8.03 | 2.90 | ssc-miR-29a | 0.39 | 3.13 | - |
| PC-5p-16648 | 0.19 | 14.62 | 2.85 | ssc-miR-26a | 0.42 | 2.29 | - |
| PC-720-3p-31375 | 0.48 | 4.77 | 2.28 | ssc-miR-125a | 0.38 | 2.30 | - |
| PN-1274b-5p-403 | 0.19 | 49.38 | 9.61 | miR-145-5p-335 | 2.35 | - | 2.46 |
| ssc-miR-4332 | 0.21 | 42.20 | 8.81 | ssc-miR-145 | 2.65 | - | 2.43 |
| miR-29a-3p-51 | 0.43 | 2.31 | - | miR-31-5p-40971 | - | 0.15 | 0.20 |
| ssc-miR-127 | 0.33 | 2.55 | - | PN-2481-5p-34424 | - | 0.11 | 0.09 |
| miR-107-3p-19 | 0.48 | - | 0.48 | ssc-miR-221 | - | 0.18 | 0.12 |
| miR-2142-3p-34065 | 0.46 | - | 0.46 | PN-200a-3p-331 | - | 0.38 | 0.30 |
| PN-495-3p-413 | 0.46 | - | 0.41 | ||||
| PN-7-5p-1931 | 0.10 | - | 0.07 | ||||
| ssc-miR-128 | 0.33 | - | 0.37 | ||||
| ssc-miR-191 | 0.48 | - | 0.45 | ||||
| ssc-miR-7 | 0.07 | - | 0.08 | ||||
| PN-125b-5p-19840 | - | 0.37 | 0.29 | ||||
| ssc-miR-125b | - | 0.39 | 0.30 |
Acknowledgments
Conflicts of Interest
References
- Kunej, T.; Godnic, I.; Horvat, S.; Zorc, M.; Calin, G.A. Cross talk between microRNA and coding cancer genes. Cancer J 2012, 18, 223–231. [Google Scholar]
- Lee, R.C.; Feinbaum, R.L.; Ambros, V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell 1993, 75, 843–854. [Google Scholar]
- Friedman, R.C.; Farh, K.K.; Burge, C.B.; Bartel, D.P. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res 2009, 19, 92–105. [Google Scholar]
- Xiao, C.; Rajewsky, K. MicroRNA control in the immune system: Basic principles. Cell 2009, 136, 26–36. [Google Scholar]
- Li, M.; Xia, Y.; Gu, Y.; Zhang, K.; Lang, Q.; Chen, L.; Guan, J.; Luo, Z.; Chen, H.; Li, Y.; et al. MicroRNAome of porcine pre- and postnatal development. PLoS One 2010, 5, e11541. [Google Scholar]
- Li, G.; Li, Y.; Li, X.; Ning, X.; Li, M.; Yang, G. MicroRNA identity and abundance in developing swine adipose tissue as determined by Solexa sequencing. J. Cell. Biochem 2011, 112, 1318–1328. [Google Scholar]
- Li, M.; Liu, Y.; Wang, T.; Guan, J.; Luo, Z.; Chen, H.; Wang, X.; Chen, L.; Ma, J.; Mu, Z.; et al. Repertoire of porcine microRNAs in adult ovary and testis by deep sequencing. Int. J. Biol. Sci 2011, 7, 1045–1055. [Google Scholar]
- Podolska, A.; Kaczkowski, B.; Kamp Busk, P.; Søkilde, R.; Litman, T.; Fredholm, M.; Cirera, S. MicroRNA expression profiling of the porcine developing brain. PLoS One 2011, 6, e14494. [Google Scholar]
- Xie, S.S.; Li, X.Y.; Liu, T.; Cao, J.H.; Zhong, Q.; Zhao, S.H. Discovery of porcine microRNAs in multiple tissues by a Solexa deep sequencing approach. PLoS One 2011, 6, e16235. [Google Scholar]
- Spenrath, M.A.; Clarke, M.E.; Kutcher, S. The science of brain and biological development: Implications for mental health research, practice and policy. J. Can. Acad. Child Adolesc. Psychiatry 2011, 20, 298–304. [Google Scholar]
- Sahu, A. Minireview: A hypothalamic role in energy balance with special emphasis on leptin. Endocrinology 2004, 145, 2613–2620. [Google Scholar]
- Minokoshi, Y.; Shiuchi, T.; Lee, S.; Suzuki, A.; Okamoto, S. Role of hypothalamic AMP-kinase in food intake regulation. Nutrition 2008, 24, 786–790. [Google Scholar]
- Simpson, K.A.; Martin, N.M.; Bloom, S.R. Hypothalamic regulation of food intake and clinical therapeutic applications. Arq. Bras. Endocrinol. Metabol 2009, 53, 120–128. [Google Scholar]
- Bilezikjian, L.M.; Vale, W.W. The local control of the pituitary by activin signaling and modulation. Open Neuroendocrinol. J 2011, 4, 90–101. [Google Scholar]
- Li, H.; Xi, Q.; Xiong, Y.; Cheng, X.; Qi, Q.; Yang, L.; Shu, G.; Wang, S.; Wang, L.; Gao, P.; et al. A comprehensive expression profile of microRNAs in porcine pituitary. PLoS One 2011, 6, e24883. [Google Scholar]
- Wei, Z.; Liu, X.; Feng, T.; Chang, Y. Novel and conserved micrornas in Dalian purple urchin (Strongylocentrotus nudus) identified by next generation sequencing. Int. J. Biol. Sci 2011, 7, 180–192. [Google Scholar]
- Li, A.; Song, T.; Wang, F.; Liu, D.; Fan, Z.; Zhang, C.; He, J.; Wang, S. MicroRNAome and expression profile of developing tooth germ in miniature pigs. PLoS One 2012, 7, e52256. [Google Scholar]
- Sharbati, S.; Friedländer, M.R.; Sharbati, J.; Hoeke, L.; Chen, W.; Keller, A.; Stähler, P.F.; Rajewsky, N.; Einspanier, R. Deciphering the porcine intestinal microRNA transcriptome. BMC Genomics 2010, 11, 275. [Google Scholar]
- Chen, C.; Ai, H.; Ren, J.; Li, W.; Li, P.; Qiao, R.; Ouyang, J.; Yang, M.; Ma, J.; Huang, L. A global view of porcine transcriptome in three tissues from a full-sib pair with extreme phenotypes in growth and fat deposition by paired-end RNA sequencing. BMC Genomics 2011, 12, 448. [Google Scholar]
- Chen, C.; Deng, B.; Qiao, M.; Zheng, R.; Chai, J.; Ding, Y.; Peng, J.; Jiang, S. Solexa sequencing identification of conserved and novel microRNAs in backfat of Large White and Chinese Meishan pigs. PLoS One 2012, 7, e31426. [Google Scholar]
- Hou, X.; Tang, Z.; Liu, H.; Wang, N.; Ju, H.; Li, K. Discovery of MicroRNAs associated with myogenesis by deep sequencing of serial developmental skeletal muscles in pigs. PLoS One 2012, 7, e52123. [Google Scholar]
- Lian, C.; Sun, B.; Niu, S.; Yang, R.; Liu, B.; Lu, C.; Meng, J.; Qiu, Z.; Zhang, L.; Zhao, Z. A comparative profile of the microRNA transcriptome in immature and mature porcine testes using Solexa deep sequencing. FEBS J 2012, 279, 964–975. [Google Scholar]
- Li, H.Y.; Xi, Q.Y.; Xiong, Y.Y.; Liu, X.L.; Cheng, X.; Shu, G.; Wang, S.B.; Wang, L.N.; Gao, P.; Zhu, X.T.; et al. Identification and comparison of microRNAs from skeletal muscle and adipose tissues from two porcine breeds. Anim. Genet 2012, 43, 704–713. [Google Scholar]
- Liu, Y.; Li, M.; Ma, J.; Zhang, J.; Zhou, C.; Wang, T.; Gao, X.; Li, X. Identification of differences in microRNA transcriptomes between porcine oxidative and glycolytic skeletal muscles. BMC Mol. Biol 2013, 14, 7. [Google Scholar]
- Guo, Y.; Mo, D.; Zhang, Y.; Zhang, Y.; Cong, P.; Xiao, S.; He, Z.; Liu, X.; Chen, Y. MicroRNAome comparison between intramuscular and subcutaneous vascular stem cell adipogenesis. PLoS One 2012, 7, e45410. [Google Scholar]
- Zhou, Y.; Tang, X.; Song, Q.; Ji, Y.; Wang, H.; Wang, H.; Jiao, H.; Ouyang, H.; Pang, D. Identification and characterization of pig embryo microRNAs by Solexa sequencing. Reprod. Domest. Anim 2013, 48, 112–120. [Google Scholar]
- Miao, Z.G.; Wang, L.J.; Xu, Z.R.; Huang, J.F.; Wang, Y.R. Developmental changes of carcass composition, meat quality and organs in the Jinhua pig and Landrace. Animal 2009, 3, 468–473. [Google Scholar]
- Zhong, T.M.; Zhao, Q. Study on analysis and fitting of growth curves in Jinhua pig line I. Chin. J. Anim. Sci 2008, 44, 9–11. [Google Scholar]
- Zhao, X.F.; Xu, N.Y.; Hu, X.X.; Li, N. Effects of microsatellite in the regulatory region of IGF1 on growth traits in Jinhua swine. Yi Chuan 2007, 29, 206–210. [Google Scholar]
- Shan, T.; Ren, Y.; Liu, Y.; Zhu, L.; Wang, Y. Breed difference and regulation of the porcine Sirtuin 1 by insulin. J. Anim. Sci 2010, 88, 3909–3917. [Google Scholar]
- Lau, N.C.; Lim, L.P.; Weinstein, E.G.; Bartel, D.P. An abundant class of tiny RNAs with probable regulatory roles in Caenorhabditis elegans. Science 2001, 294, 858–862. [Google Scholar]
- Richards, M.P.; Proszkowiec-Weglarz, M. Mechanisms regulating feed intake, energy expenditure, and body weight in poultry. Poult. Sci 2007, 86, 1478–1490. [Google Scholar]
- Bak, M.; Silahtaroglu, A.; Møller, M.; Christensen, M.; Rath, M.F.; Skryabin, B.; Tommerup, N.; Kauppinen, S. MicroRNA expression in the adult mouse central nervous system. RNA 2008, 14, 432–444. [Google Scholar]
- Sun, G.R.; Li, M.; Li, G.X.; Tian, Y.D.; Han, R.L.; Kang, X.T. Identification and abundance of miRNA in chicken hypothalamus tissue determined by Solexa sequencing. Genet. Mol. Res 2012, 11, 4682–4694. [Google Scholar]
- Bottoni, A.; Piccin, D.; Tagliati, F.; Luchin, A.; Zatelli, M.C.; degli Uberti, E.C. miR-15a and miR-16–1 down-regulation in pituitary adenomas. J. Cell. Physiol 2005, 204, 280–285. [Google Scholar]
- Gentilin, E.; Tagliati, F.; Filieri, C.; Molè, D.; Minoia, M.; Rosaria Ambrosio, M.; Degli Uberti, E.C.; Zatelli, M.C. miR-26a plays an important role in cell cycle regulation in ACTH-secreting pituitary adenomas by modulating protein kinase Cδ. Endocrinology 2013, 154, 1690–1700. [Google Scholar]
- Amaral, F.C.; Torres, N.; Saggioro, F.; Neder, L.; Machado, H.R.; Silva, W.A., Jr.; Moreira, A.C.; Castro, M. MicroRNAs differentially expressed in ACTH-secreting pituitary tumors. J. Clin. Endocrinol. Metab 2009, 94, 320–323. [Google Scholar]
- Marsh, E.E.; Lin, Z.; Yin, P.; Milad, M.; Chakravarti, D.; Bulun, S.E. Differential expression of microRNA species in human uterine leiomyoma versus normal myometrium. Fertil. Steril 2008, 89, 1771–1776. [Google Scholar]
- Li, Y.; Wang, F.; Xu, J.; Ye, F.; Shen, Y.; Zhou, J.; Lu, W.; Wan, X.; Ma, D.; Xie, X. Progressive miRNA expression profiles in cervical carcinogenesis and identification of HPVrelated target genes for miR-29. J. Pathol 2011, 224, 484–495. [Google Scholar]
- Gao, X.; Gulari, E.; Zhou, X. In situ synthesis of oligonucleotide microarrays. Biopolymers 2004, 73, 579–596. [Google Scholar]
- Tusher, V.G.; Tibshirani, R.; Chu, G. Significance analysis of microarrays applied to the ionizing radiation response. Proc. Natl. Acad. Sci. USA 2001, 98, 5116–5121. [Google Scholar]
- Castruita, M.; Casero, D.; Karpowicz, S.J.; Kropat, J.; Vieler, A.; Hsieh, S.I.; Yan, W.; Cokus, S.; Loo, J.A.; Benning, C.; et al. Systems biology approach in Chlamydomonas reveals connections between copper nutrition and multiple metabolic steps. Plant Cell 2011, 23, 1273–1292. [Google Scholar]
- Huang, T.H.; Zhu, M.J.; Li, X.Y.; Zhao, S.H. Discovery of porcine microRNAs and profiling from skeletal muscle tissues during development. PLoS One 2008, 3, e3225. [Google Scholar]
- Lewis, B.P.; Burge, C.B.; Bartel, D.P. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell 2005, 120, 15–20. [Google Scholar]
- Garcia, D.M.; Baek, D.; Shin, C.; Bell, G.W.; Grimson, A.; Bartel, D.P. Weak seed-pairing stability and high target-site abundance decrease the proficiency of lsy-6 and other microRNAs. Nat. Struct. Mol. Biol 2011, 18, 1139–1146. [Google Scholar]
- Yehya, N.; Yerrapureddy, A.; Tobias, J.; Margulies, S.S. MicroRNA modulate alveolar epithelial response to cyclic stretch. BMC Genomics 2012, 13, 154. [Google Scholar]
- Eden, E.; Navon, R.; Steinfeld, I.; Lipson, D.; Yakhini, Z. GOrilla: A tool for discovery and visualization of enriched GO terms in ranked gene lists. BMC Bioinforma 2009, 10, 48. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Zhang, L.; Cai, Z.; Wei, S.; Zhou, H.; Zhou, H.; Jiang, X.; Xu, N. MicroRNA Expression Profiling of the Porcine Developing Hypothalamus and Pituitary Tissue. Int. J. Mol. Sci. 2013, 14, 20326-20339. https://doi.org/10.3390/ijms141020326
Zhang L, Cai Z, Wei S, Zhou H, Zhou H, Jiang X, Xu N. MicroRNA Expression Profiling of the Porcine Developing Hypothalamus and Pituitary Tissue. International Journal of Molecular Sciences. 2013; 14(10):20326-20339. https://doi.org/10.3390/ijms141020326
Chicago/Turabian StyleZhang, Lifan, Zhaowei Cai, Shengjuan Wei, Huiyun Zhou, Hongmei Zhou, Xiaoling Jiang, and Ningying Xu. 2013. "MicroRNA Expression Profiling of the Porcine Developing Hypothalamus and Pituitary Tissue" International Journal of Molecular Sciences 14, no. 10: 20326-20339. https://doi.org/10.3390/ijms141020326