Role of Hepatic Progenitor Cells in Nonalcoholic Fatty Liver Disease Development: Cellular Cross-Talks and Molecular Networks




Abstract
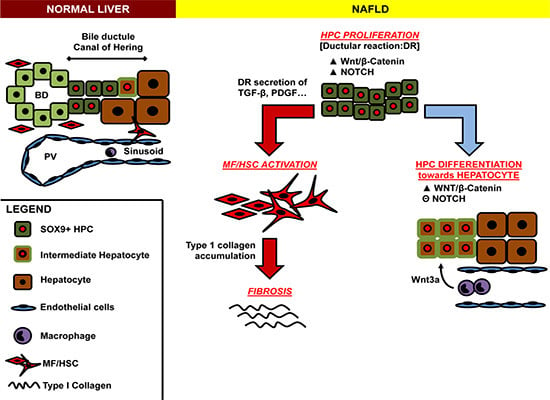
:
{kind=link}
{kind=link}
{kind=link}
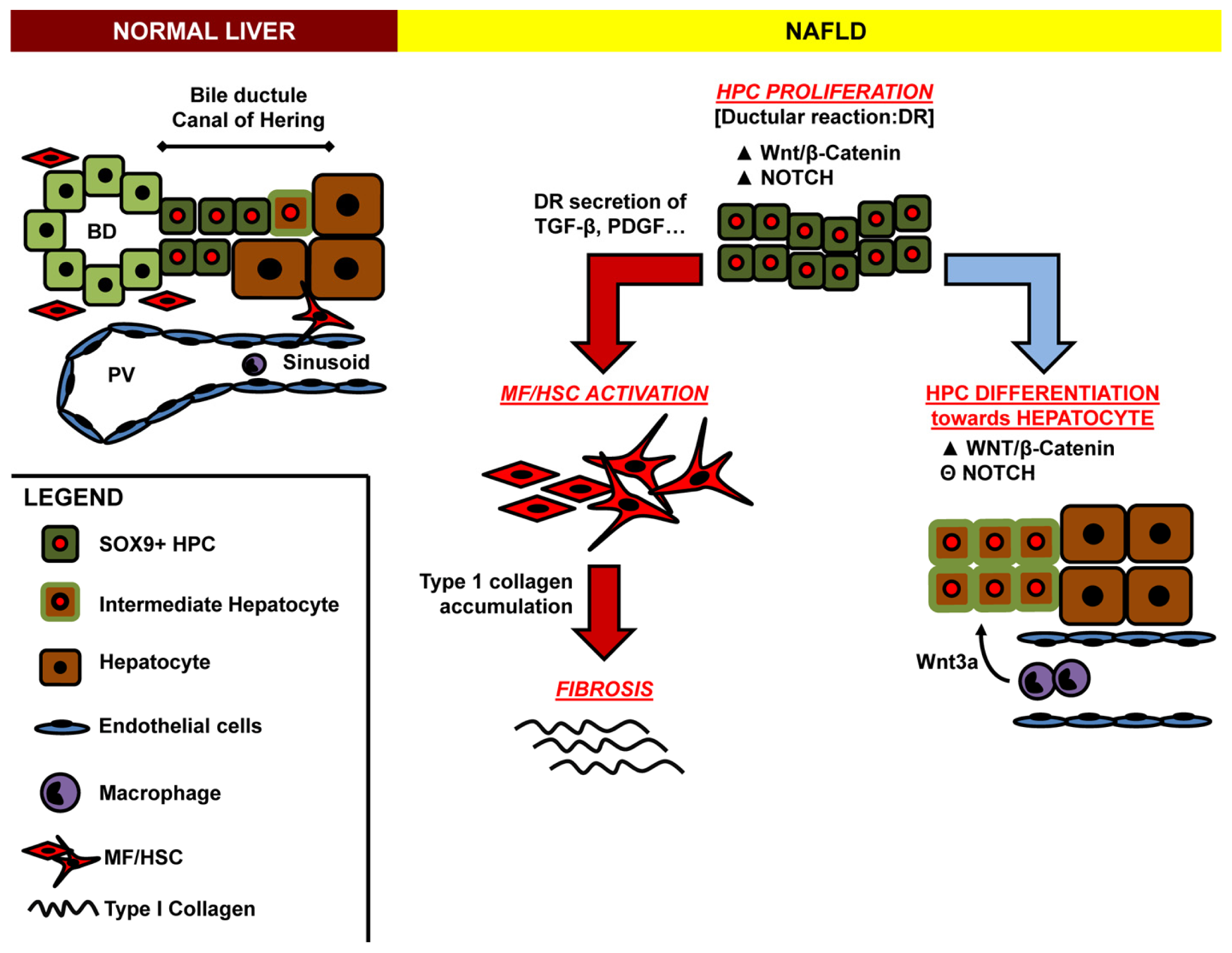{kind=link}
1. Introduction
2. Histo-Pathological Aspects of NAFLD
3. Liver Fibrosis in NAFLD and Hepatic Stellate Cell Activation
4. HPC Niches within Adult Intrahepatic Bile Duct Systems
5. HPC Microenvironment and Niche Modulation
6. Cellular Cross-Talk between HPC and HSC in Fibrogenesis
7. Role of Kupffer Cells and Macrophages and Their Cross-Talk with HPC and HSC in NAFLD
8. Adipokines as a New Tool in HPC and HSC Cross-Talk in NAFLD
9. NAFLD and Atherosclerosis: Possible Molecular Mechanisms
10. Conclusions
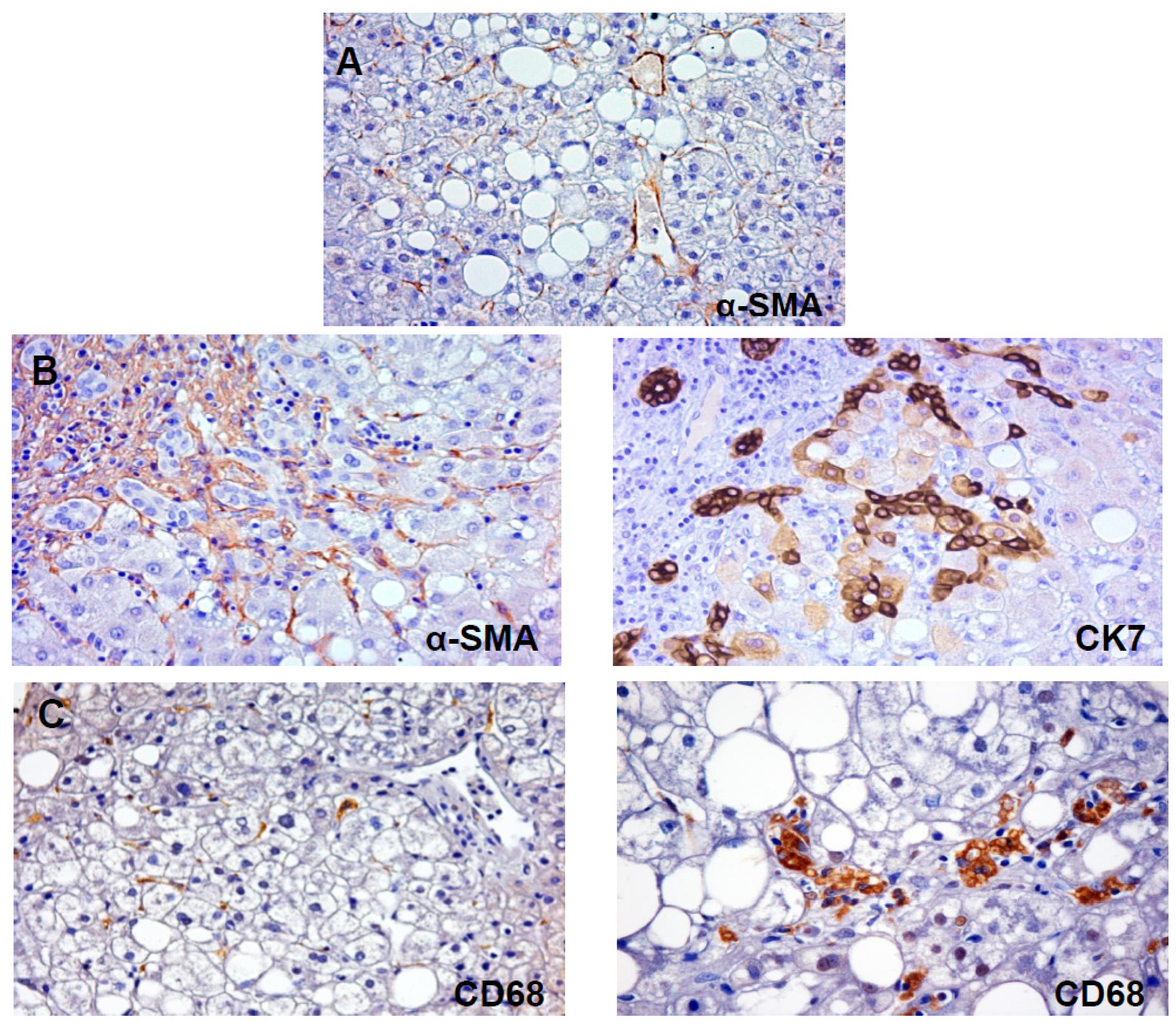
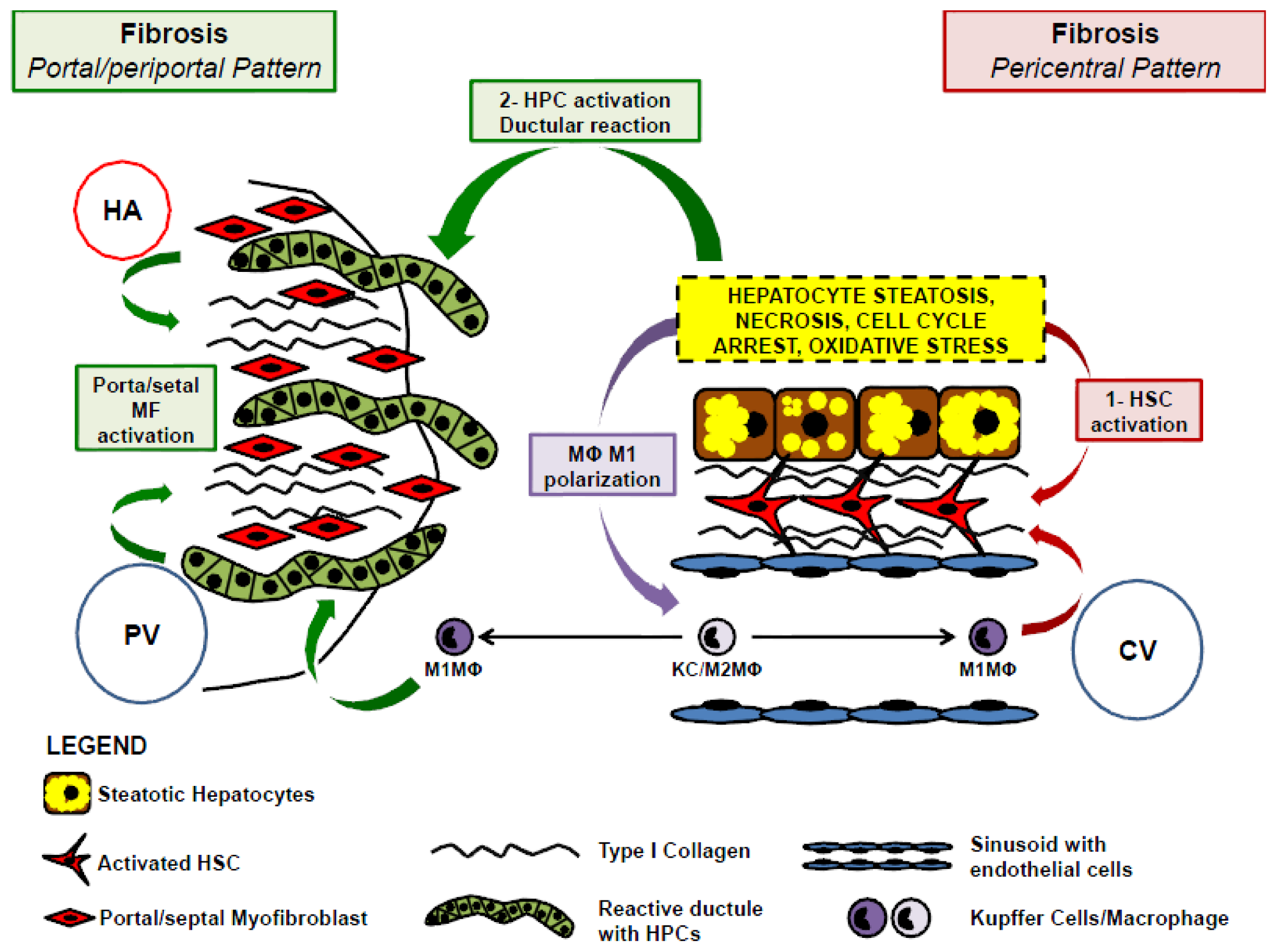
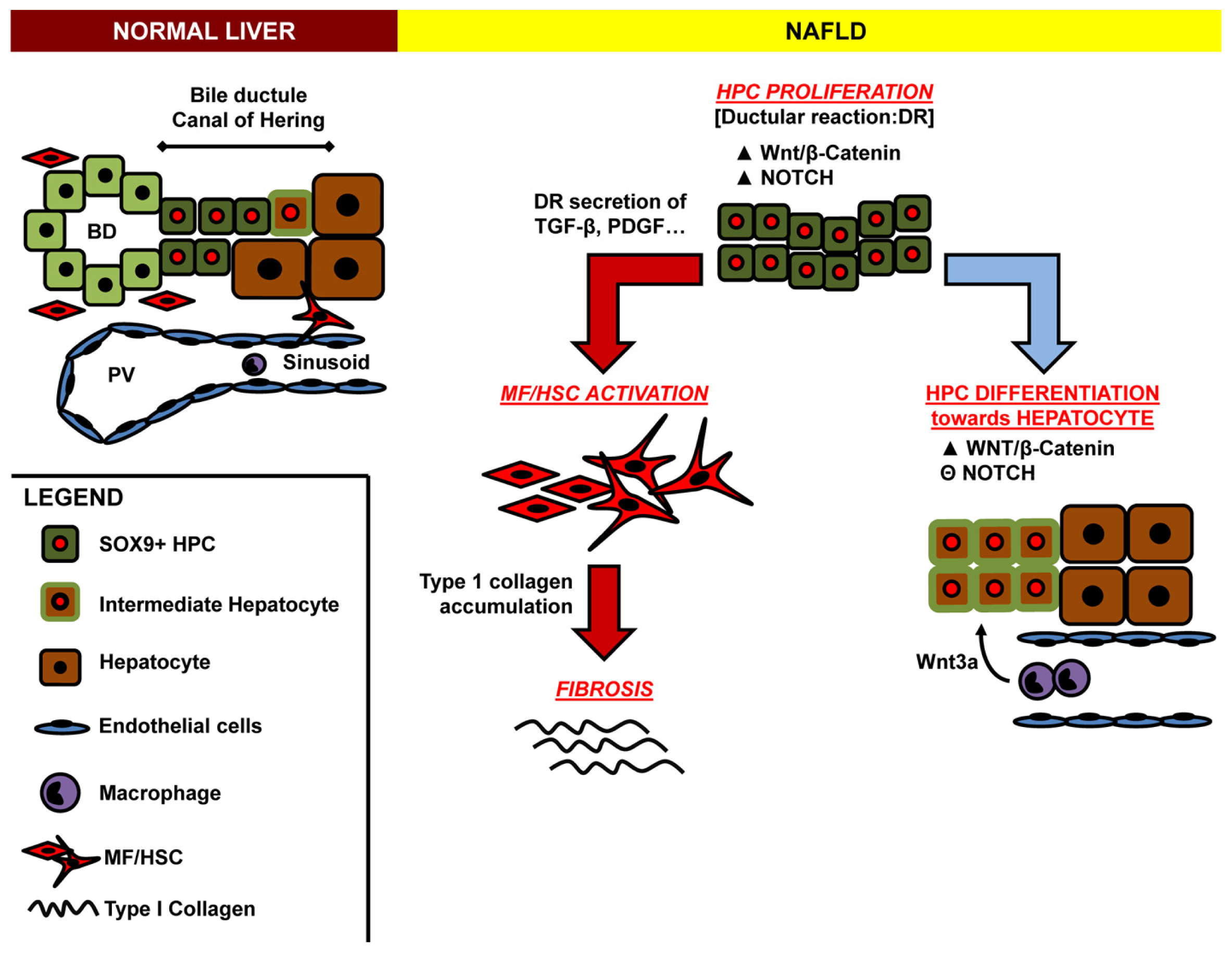
Acknowledgments
Conflicts of Interest
References
- Angulo, P. Nonalcoholic fatty liver disease. N. Engl. J. Med 2002, 346, 1221–1231. [Google Scholar]
- Gaudio, E.; Nobili, V.; Franchitto, A.; Onori, P.; Carpino, G. Nonalcoholic fatty liver disease and atherosclerosis. Intern. Emerg. Med 2012, 7, S297–S305. [Google Scholar]
- Alisi, A.; Locatelli, M.; Nobili, V. Nonalcoholic fatty liver disease in children. Curr. Opin. Clin. Nutr. Metab. Care 2010, 13, 397–402. [Google Scholar]
- Bieghs, V.; Rensen, P.C.; Hofker, M.H.; Shiri-Sverdlov, R. NASH and atherosclerosis are two aspects of a shared disease: Central role for macrophages. Atherosclerosis 2012, 220, 287–293. [Google Scholar]
- Sookoian, S.; Gianotti, T.F.; Rosselli, M.S.; Burgueno, A.L.; Castano, G.O.; Pirola, C.J. Liver transcriptional profile of atherosclerosis-related genes in human nonalcoholic fatty liver disease. Atherosclerosis 2011, 218, 378–385. [Google Scholar]
- Brunt, E.M.; Kleiner, D.E.; Wilson, L.A.; Belt, P.; Neuschwander-Tetri, B.A. Nonalcoholic fatty liver disease (NAFLD) activity score and the histopathologic diagnosis in NAFLD: Distinct clinicopathologic meanings. Hepatology 2011, 53, 810–820. [Google Scholar]
- Kleiner, D.E.; Brunt, E.M.; van Natta, M.; Behling, C.; Contos, M.J.; Cummings, O.W.; Ferrell, L.D.; Liu, Y.C.; Torbenson, M.S.; Unalp-Arida, A.; et al. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology 2005, 41, 1313–1321. [Google Scholar]
- Brunt, E.M. Nonalcoholic steatohepatitis: Definition and pathology. Semin. Liver Dis 2001, 21, 3–16. [Google Scholar]
- Brunt, E.M. Pathology of nonalcoholic steatohepatitis. Hepatol. Res 2005, 33, 68–71. [Google Scholar]
- Falck-Ytter, Y.; Younossi, Z.M.; Marchesini, G.; McCullough, A.J. Clinical features and natural history of nonalcoholic steatosis syndromes. Semin. Liver Dis 2001, 21, 17–26. [Google Scholar]
- Brunt, E.M. Pathology of nonalcoholic fatty liver disease. Nat. Rev. Gastroenterol. Hepatol 2010, 7, 195–203. [Google Scholar]
- Lee, U.E.; Friedman, S.L. Mechanisms of hepatic fibrogenesis. Best Pract. Res. Clin. Gastroenterol 2011, 25, 195–206. [Google Scholar]
- Friedman, S.L. Hepatic stellate cells: Protean, multifunctional, and enigmatic cells of the liver. Physiol. Rev 2008, 88, 125–172. [Google Scholar]
- Blaner, W.S.; O’Byrne, S.M.; Wongsiriroj, N.; Kluwe, J.; D’Ambrosio, D.M.; Jiang, H.; Schwabe, R.F.; Hillman, E.M.; Piantedosi, R.; Libien, J. Hepatic stellate cell lipid droplets: A specialized lipid droplet for retinoid storage. Biochim. Biophys. Acta 2009, 1791, 467–473. [Google Scholar]
- Carotti, S.; Morini, S.; Corradini, S.G.; Burza, M.A.; Molinaro, A.; Carpino, G.; Merli, M.; de Santis, A.; Muda, A.O.; Rossi, M.; et al. Glial fibrillary acidic protein as an early marker of hepatic stellate cell activation in chronic and posttransplant recurrent hepatitis C. Liver Transpl 2008, 14, 806–814. [Google Scholar]
- Carpino, G.; Morini, S.; Ginanni Corradini, S.; Franchitto, A.; Merli, M.; Siciliano, M.; Gentili, F.; Onetti Muda, A.; Berloco, P.; Rossi, M.; et al. Alpha-SMA expression in hepatic stellate cells and quantitative analysis of hepatic fibrosis in cirrhosis and in recurrent chronic hepatitis after liver transplantation. Dig. Liver Dis 2005, 37, 349–356. [Google Scholar]
- Carpino, G.; Franchitto, A.; Morini, S.; Corradini, S.G.; Merli, M.; Gaudio, E. Activated hepatic stellate cells in liver cirrhosis. A morphologic and morphometrical study. Ital. J. Anat. Embryol 2004, 109, 225–238. [Google Scholar]
- Fujii, H.; Kawada, N. Inflammation and fibrogenesis in steatohepatitis. J. Gastroenterol 2012, 47, 215–225. [Google Scholar]
- Tsukamoto, H.; She, H.; Hazra, S.; Cheng, J.; Miyahara, T. Anti-adipogenic regulation underlies hepatic stellate cell transdifferentiation. J. Gastroenterol. Hepatol 2006, 21, S102–S105. [Google Scholar]
- Tsukamoto, H.; Zhu, N.L.; Asahina, K.; Mann, D.A.; Mann, J. Epigenetic cell fate regulation of hepatic stellate cells. Hepatol. Res 2011, 41, 675–682. [Google Scholar]
- Aziz-Seible, R.S.; McVicker, B.L.; Kharbanda, K.K.; Casey, C.A. Cellular fibronectin stimulates hepatocytes to produce factors that promote alcohol-induced liver injury. World J. Hepatol 2011, 3, 45–55. [Google Scholar]
- Pellicoro, A.; Ramachandran, P.; Iredale, J.P. Reversibility of liver fibrosis. Fibrogenesis Tissue Repair 2012, 5, S26. [Google Scholar]
- Guy, C.D.; Suzuki, A.; Zdanowicz, M.; Abdelmalek, M.F.; Burchette, J.; Unalp, A.; Diehl, A.M. Hedgehog pathway activation parallels histologic severity of injury and fibrosis in human nonalcoholic fatty liver disease. Hepatology 2012, 55, 1711–1721. [Google Scholar]
- Xie, G.; Karaca, G.; Swiderska-Syn, M.; Michelotti, G.A.; Kruger, L.; Chen, Y.; Premont, R.T.; Choi, S.S.; Diehl, A.M. Cross-talk between notch and hedgehog regulates hepatic stellate cell fate. Hepatology 2013. [Google Scholar] [CrossRef]
- Marra, F.; Aleffi, S.; Bertolani, C.; Petrai, I.; Vizzutti, F. Adipokines and liver fibrosis. Eur. Rev. Med. Pharmacol. Sci 2005, 9, 279–284. [Google Scholar]
- Seki, E.; Brenner, D.A. Toll-like receptors and adaptor molecules in liver disease: Update. Hepatology 2008, 48, 322–335. [Google Scholar]
- Vespasiani-Gentilucci, U.; Carotti, S.; Onetti-Muda, A.; Perrone, G.; Ginanni-Corradini, S.; Latasa, M.U.; Avila, M.A.; Carpino, G.; Picardi, A.; Morini, S. Toll-like receptor-4 expression by hepatic progenitor cells and biliary epithelial cells in HCV-related chronic liver disease. Mod. Pathol 2012, 25, 576–589. [Google Scholar]
- Kaji, K.; Yoshiji, H.; Kitade, M.; Ikenaka, Y.; Noguchi, R.; Shirai, Y.; Aihara, Y.; Namisaki, T.; Yoshii, J.; Yanase, K.; et al. Combination treatment of angiotensin II type I receptor blocker and new oral iron chelator attenuates progression of nonalcoholic steatohepatitis in rats. Am. J. Physiol. Gastrointest. Liver Physiol 2011, 300, G1094–G1104. [Google Scholar]
- Marra, F.; Aleffi, S.; Bertolani, C.; Petrai, I.; Vizzutti, F. Review article: The pathogenesis of fibrosis in non-alcoholic steatohepatitis. Aliment. Pharmacol. Ther 2005, 22, 44–47. [Google Scholar]
- Schwimmer, J.B.; Behling, C.; Newbury, R.; Deutsch, R.; Nievergelt, C.; Schork, N.J.; Lavine, J.E. Histopathology of pediatric nonalcoholic fatty liver disease. Hepatology 2005, 42, 641–649. [Google Scholar]
- Neuschwander-Tetri, B.A. Hepatic lipotoxicity and the pathogenesis of nonalcoholic steatohepatitis: The central role of nontriglyceride fatty acid metabolites. Hepatology 2010, 52, 774–788. [Google Scholar]
- Kaplowitz, N. Mechanisms of liver cell injury. J. Hepatol 2000, 32, 39–47. [Google Scholar]
- Tilg, H.; Diehl, A.M. Cytokines in alcoholic and nonalcoholic steatohepatitis. N. Engl. J. Med 2000, 343, 1467–1476. [Google Scholar]
- Alkhouri, N.; de Vito, R.; Alisi, A.; Yerian, L.; Lopez, R.; Feldstein, A.E.; Nobili, V. Development and validation of a new histological score for pediatric non-alcoholic fatty liver disease. J. Hepatol 2012, 57, 1312–1318. [Google Scholar]
- Carter-Kent, C.; Yerian, L.M.; Brunt, E.M.; Angulo, P.; Kohli, R.; Ling, S.C.; Xanthakos, S.A.; Whitington, P.F.; Charatcharoenwitthaya, P.; Yap, J.; et al. Nonalcoholic steatohepatitis in children: A multicenter clinicopathological study. Hepatology 2009, 50, 1113–1120. [Google Scholar]
- Skoien, R.; Richardson, M.M.; Jonsson, J.R.; Powell, E.E.; Brunt, E.M.; Neuschwander-Tetri, B.A.; Bhathal, P.S.; Dixon, J.B.; O’Brien, P.E.; Tilg, H.; et al. Heterogeneity of fibrosis patterns in non-alcoholic fatty liver disease supports the presence of multiple fibrogenic pathways. Liver Int 2013, 33, 624–632. [Google Scholar]
- Clouston, A.D.; Powell, E.E.; Walsh, M.J.; Richardson, M.M.; Demetris, A.J.; Jonsson, J.R. Fibrosis correlates with a ductular reaction in hepatitis C: Roles of impaired replication, progenitor cells and steatosis. Hepatology 2005, 41, 809–818. [Google Scholar]
- Nobili, V.; Carpino, G.; Alisi, A.; Franchitto, A.; Alpini, G.; de Vito, R.; Onori, P.; Alvaro, D.; Gaudio, E. Hepatic progenitor cells activation, fibrosis and adipokines production in pediatric nonalcoholic fatty liver disease. Hepatology 2012, 56, 2142–2153. [Google Scholar]
- Richardson, M.M.; Jonsson, J.R.; Powell, E.E.; Brunt, E.M.; Neuschwander-Tetri, B.A.; Bhathal, P.S.; Dixon, J.B.; Weltman, M.D.; Tilg, H.; Moschen, A.R.; et al. Progressive fibrosis in nonalcoholic steatohepatitis: Association with altered regeneration and a ductular reaction. Gastroenterology 2007, 133, 80–90. [Google Scholar]
- Gaudio, E.; Carpino, G.; Cardinale, V.; Franchitto, A.; Onori, P.; Alvaro, D. New insights into liver stem cells. Dig. Liver Dis 2009, 41, 455–462. [Google Scholar]
- Roskams, T.A.; Theise, N.D.; Balabaud, C.; Bhagat, G.; Bhathal, P.S.; Bioulac-Sage, P.; Brunt, E.M.; Crawford, J.M.; Crosby, H.A.; Desmet, V.; et al. Nomenclature of the finer branches of the biliary tree: Canals, ductules, and ductular reactions in human livers. Hepatology 2004, 39, 1739–1745. [Google Scholar]
- Alison, M.R.; Golding, M.H.; Sarraf, C.E. Pluripotential liver stem cells: Facultative stem cells located in the biliary tree. Cell Prolif 1996, 29, 373–402. [Google Scholar]
- Cai, X.; Zhai, J.; Kaplan, D.E.; Zhang, Y.; Zhou, L.; Chen, X.; Qian, G.; Zhao, Q.; Li, Y.; Gao, L.; et al. Background progenitor activation is associated with recurrence after hepatectomy of combined hepatocellular-cholangiocarcinoma. Hepatology 2012, 56, 1804–1816. [Google Scholar]
- Gouw, A.S.; Clouston, A.D.; Theise, N.D. Ductular reactions in human liver: Diversity at the interface. Hepatology 2011, 54, 1853–1863. [Google Scholar]
- Mancino, M.G.; Carpino, G.; Onori, P.; Franchitto, A.; Alvaro, D.; Gaudio, E. Hepatic “stem” cells: State of the art. Ital. J. Anat. Embryol 2007, 112, 93–109. [Google Scholar]
- Huch, M.; Dorrell, C.; Boj, S.F.; van Es, J.H.; Li, V.S.; van de Wetering, M.; Sato, T.; Hamer, K.; Sasaki, N.; Finegold, M.J.; et al. In vitro expansion of single Lgr5+ liver stem cells induced by Wnt-driven regeneration. Nature 2013, 494, 247–250. [Google Scholar]
- Cardinale, V.; Wang, Y.; Carpino, G.; Alvaro, D.; Reid, L.; Gaudio, E. Multipotent stem cells in the biliary tree. Ital. J. Anat. Embryol 2010, 115, 85–90. [Google Scholar]
- Carpino, G.; Cardinale, V.; Onori, P.; Franchitto, A.; Berloco, P.B.; Rossi, M.; Wang, Y.; Semeraro, R.; Anceschi, M.; Brunelli, R.; et al. Biliary tree stem/progenitor cells in glands of extrahepatic and intraheptic bile ducts: An anatomical in situ study yielding evidence of maturational lineages. J. Anat 2012, 220, 186–199. [Google Scholar]
- Boulter, L.; Govaere, O.; Bird, T.G.; Radulescu, S.; Ramachandran, P.; Pellicoro, A.; Ridgway, R.A.; Seo, S.S.; Spee, B.; van Rooijen, N.; et al. Macrophage-derived Wnt opposes Notch signaling to specify hepatic progenitor cell fate in chronic liver disease. Nat. Med 2012, 18, 572–579. [Google Scholar]
- Boulter, L.; Lu, W.Y.; Forbes, S.J. Differentiation of progenitors in the liver: A matter of local choice. J. Clin. Invest 2013, 123, 1867–1873. [Google Scholar]
- Furuyama, K.; Kawaguchi, Y.; Akiyama, H.; Horiguchi, M.; Kodama, S.; Kuhara, T.; Hosokawa, S.; Elbahrawy, A.; Soeda, T.; Koizumi, M.; et al. Continuous cell supply from a Sox9-expressing progenitor zone in adult liver, exocrine pancreas and intestine. Nat. Genet 2011, 43, 34–41. [Google Scholar]
- Fausto, N. Liver regeneration and repair: Hepatocytes, progenitor cells, and stem cells. Hepatology 2004, 39, 1477–1487. [Google Scholar]
- Malato, Y.; Naqvi, S.; Schurmann, N.; Ng, R.; Wang, B.; Zape, J.; Kay, M.A.; Grimm, D.; Willenbring, H. Fate tracing of mature hepatocytes in mouse liver homeostasis and regeneration. J. Clin. Invest 2011, 121, 4850–4860. [Google Scholar]
- Carpentier, R.; Suner, R.E.; van Hul, N.; Kopp, J.L.; Beaudry, J.B.; Cordi, S.; Antoniou, A.; Raynaud, P.; Lepreux, S.; Jacquemin, P.; et al. Embryonic ductal plate cells give rise to cholangiocytes, periportal hepatocytes, and adult liver progenitor cells. Gastroenterology 2011, 141, 1432–1438. [Google Scholar]
- Theise, N.D.; Dolle, L.; Kuwahara, R. Low hepatocyte repopulation from stem cells: A matter of hepatobiliary linkage not massive production. Gastroenterology 2013, 145, 253–254. [Google Scholar]
- Espanol-Suner, R.; Carpentier, R.; van Hul, N.; Legry, V.; Achouri, Y.; Cordi, S.; Jacquemin, P.; Lemaigre, F.; Leclercq, I.A. Liver progenitor cells yield functional hepatocytes in response to chronic liver injury in mice. Gastroenterology 2012, 143, 1564–1575. [Google Scholar]
- Clevers, H. The intestinal crypt, a prototype stem cell compartment. Cell 2013, 154, 274–284. [Google Scholar]
- Spee, B.; Carpino, G.; Schotanus, B.A.; Katoonizadeh, A.; Vander Borght, S.; Gaudio, E.; Roskams, T. Characterisation of the liver progenitor cell niche in liver diseases: Potential involvement of Wnt and Notch signalling. Gut 2010, 59, 247–257. [Google Scholar]
- Kodama, Y.; Hijikata, M.; Kageyama, R.; Shimotohno, K.; Chiba, T. The role of notch signaling in the development of intrahepatic bile ducts. Gastroenterology 2004, 127, 1775–1786. [Google Scholar]
- McCright, B.; Lozier, J.; Gridley, T. A mouse model of Alagille syndrome: Notch2 as a genetic modifier of Jag1 haploinsufficiency. Development 2002, 129, 1075–1082. [Google Scholar]
- Burke, Z.D.; Reed, K.R.; Phesse, T.J.; Sansom, O.J.; Clarke, A.R.; Tosh, D. Liver zonation occurs through a beta-catenin-dependent, c-Myc-independent mechanism. Gastroenterology 2009, 136, 2316–2324. [Google Scholar]
- Roskams, T.; Yang, S.Q.; Koteish, A.; Durnez, A.; DeVos, R.; Huang, X.; Achten, R.; Verslype, C.; Diehl, A.M. Oxidative stress and oval cell accumulation in mice and humans with alcoholic and nonalcoholic fatty liver disease. Am. J. Pathol 2003, 163, 1301–1311. [Google Scholar]
- Tilg, H.; Moschen, A.R. Evolution of inflammation in nonalcoholic fatty liver disease: The multiple parallel hits hypothesis. Hepatology 2010, 52, 1836–1846. [Google Scholar]
- Franchitto, A.; Onori, P.; Renzi, A.; Carpino, G.; Mancinelli, R.; Alvaro, D.; Gaudio, E. Expression of vascular endothelial growth factors and their receptors by hepatic progenitor cells in human liver diseases. Hepatobiliary Surg. Nutr 10, 11. [CrossRef]
- Glaser, S.S.; Gaudio, E.; Miller, T.; Alvaro, D.; Alpini, G. Cholangiocyte proliferation and liver fibrosis. Expert Rev. Mol. Med 2009, 11, e7. [Google Scholar]
- Theise, N.D.; Kuwahara, R. The tissue biology of ductular reactions in human chronic liver disease. Gastroenterology 2007, 133, 350–352. [Google Scholar]
- Omenetti, A.; Choi, S.; Michelotti, G.; Diehl, A.M. Hedgehog signaling in the liver. J. Hepatol 2011, 54, 366–373. [Google Scholar]
- Omenetti, A.; Porrello, A.; Jung, Y.; Yang, L.; Popov, Y.; Choi, S.S.; Witek, R.P.; Alpini, G.; Venter, J.; Vandongen, H.M.; et al. Hedgehog signaling regulates epithelial-mesenchymal transition during biliary fibrosis in rodents and humans. J. Clin. Invest 2008, 118, 3331–3342. [Google Scholar]
- Libbrecht, L.; Roskams, T. Hepatic progenitor cells in human liver diseases. Semin. Cell Dev. Biol 2002, 13, 389–396. [Google Scholar]
- Grappone, C.; Pinzani, M.; Parola, M.; Pellegrini, G.; Caligiuri, A.; DeFranco, R.; Marra, F.; Herbst, H.; Alpini, G.; Milani, S. Expression of platelet-derived growth factor in newly formed cholangiocytes during experimental biliary fibrosis in rats. J. Hepatol 1999, 31, 100–109. [Google Scholar]
- Marra, F.; DeFranco, R.; Grappone, C.; Milani, S.; Pastacaldi, S.; Pinzani, M.; Romanelli, R.G.; Laffi, G.; Gentilini, P. Increased expression of monocyte chemotactic protein-1 during active hepatic fibrogenesis: Correlation with monocyte infiltration. Am. J. Pathol 1998, 152, 423–430. [Google Scholar]
- Marra, F.; Romanelli, R.G.; Giannini, C.; Failli, P.; Pastacaldi, S.; Arrighi, M.C.; Pinzani, M.; Laffi, G.; Montalto, P.; Gentilini, P. Monocyte chemotactic protein-1 as a chemoattractant for human hepatic stellate cells. Hepatology 1999, 29, 140–148. [Google Scholar]
- Malizia, G.; Brunt, E.M.; Peters, M.G.; Rizzo, A.; Broekelmann, T.J.; McDonald, J.A. Growth factor and procollagen type I gene expression in human liver disease. Gastroenterology 1995, 108, 145–156. [Google Scholar]
- Kiss, A.; Schnur, J.; Szabo, Z.; Nagy, P. Immunohistochemical analysis of atypical ductular reaction in the human liver, with special emphasis on the presence of growth factors and their receptors. Liver 2001, 21, 237–246. [Google Scholar]
- Sakaguchi, S.; Takahashi, S.; Sasaki, T.; Kumagai, T.; Nagata, K. Progression of alcoholic and non-alcoholic steatohepatitis: Common metabolic aspects of innate immune system and oxidative stress. Drug Metab. Pharmacokinet 2011, 26, 30–46. [Google Scholar]
- Wan, J.; Benkdane, M.; Teixeira-Clerc, F.; Bonnafous, S.; Louvet, A.; Lafdil, F.; Pecker, F.; Tran, A.; Gual, P.; Mallat, A.; et al. M2 Kupffer cells promote M1 Kupffer cell apoptosis: A protective mechanism against alcoholic and non-alcoholic fatty liver disease. Hepatology 2013. [Google Scholar] [CrossRef]
- Ley, K.; Miller, Y.I.; Hedrick, C.C. Monocyte and macrophage dynamics during atherogenesis. Arterioscler. Thromb. Vasc. Biol 2011, 31, 1506–1516. [Google Scholar]
- Moore, K.J.; Tabas, I. Macrophages in the pathogenesis of atherosclerosis. Cell 2011, 145, 341–355. [Google Scholar]
- Gordon, S.; Taylor, P.R. Monocyte and macrophage heterogeneity. Nat. Rev. Immunol 2005, 5, 953–964. [Google Scholar]
- Gordon, S.; Martinez, F.O. Alternative activation of macrophages: Mechanism and functions. Immunity 2010, 32, 593–604. [Google Scholar]
- Hunter, M.M.; Wang, A.; Parhar, K.S.; Johnston, M.J.; van Rooijen, N.; Beck, P.L.; McKay, D.M. In vitro-derived alternatively activated macrophages reduce colonic inflammation in mice. Gastroenterology 2010, 138, 1395–1405. [Google Scholar]
- Olefsky, J.M.; Glass, C.K. Macrophages, inflammation, and insulin resistance. Annu. Rev. Physiol 2010, 72, 219–246. [Google Scholar]
- Van Hul, N.; Lanthier, N.; Espanol Suner, R.; Abarca Quinones, J.; van Rooijen, N.; Leclercq, I. Kupffer cells influence parenchymal invasion and phenotypic orientation, but not the proliferation, of liver progenitor cells in a murine model of liver injury. Am. J. Pathol 2011, 179, 1839–1850. [Google Scholar]
- Ramachandran, P.; Pellicoro, A.; Vernon, M.A.; Boulter, L.; Aucott, R.L.; Ali, A.; Hartland, S.N.; Snowdon, V.K.; Cappon, A.; Gordon-Walker, T.T.; et al. Differential Ly-6C expression identifies the recruited macrophage phenotype, which orchestrates the regression of murine liver fibrosis. Proc. Natl. Acad. Sci. USA 2012, 109, E3186–E3195. [Google Scholar]
- Marra, F.; Bertolani, C. Adipokines in liver diseases. Hepatology 2009, 50, 957–969. [Google Scholar]
- Bertolani, C.; Sancho-Bru, P.; Failli, P.; Bataller, R.; Aleffi, S.; DeFranco, R.; Mazzinghi, B.; Romagnani, P.; Milani, S.; Gines, P.; et al. Resistin as an intrahepatic cytokine: Overexpression during chronic injury and induction of proinflammatory actions in hepatic stellate cells. Am. J. Pathol 2006, 169, 2042–2053. [Google Scholar]
- Lumeng, C.N.; Saltiel, A.R. Inflammatory links between obesity and metabolic disease. J. Clin. Invest 2011, 121, 2111–2117. [Google Scholar]
- Beier, J.I.; Guo, L.; von Montfort, C.; Kaiser, J.P.; Joshi-Barve, S.; Arteel, G.E. New role of resistin in lipopolysaccharide-induced liver damage in mice. J. Pharmacol. Exp. Ther 2008, 325, 801–808. [Google Scholar]
- Marra, F.; Gastaldelli, A.; Svegliati Baroni, G.; Tell, G.; Tiribelli, C. Molecular basis and mechanisms of progression of non-alcoholic steatohepatitis. Trends Mol. Med 2008, 14, 72–81. [Google Scholar]
- Samuel, V.T.; Shulman, G.I. Mechanisms for insulin resistance: Common threads and missing links. Cell 2012, 148, 852–871. [Google Scholar]
- Peyrou, M.; Bourgoin, L.; Foti, M. PTEN in non-alcoholic fatty liver disease/non-alcoholic steatohepatitis and cancer. Dig. Dis 2010, 28, 236–246. [Google Scholar]
- Targher, G.; Day, C.P.; Bonora, E. Risk of cardiovascular disease in patients with nonalcoholic fatty liver disease. N. Engl. J. Med 2011, 363, 1341–1350. [Google Scholar]
- Targher, G.; Chonchol, M.; Miele, L.; Zoppini, G.; Pichiri, I.; Muggeo, M. Nonalcoholic fatty liver disease as a contributor to hypercoagulation and thrombophilia in the metabolic syndrome. Semin. Thromb. Hemost 2009, 35, 277–287. [Google Scholar]
- Targher, G.; Bertolini, L.; Rodella, S.; Lippi, G.; Franchini, M.; Zoppini, G.; Muggeo, M.; Day, C.P. NASH predicts plasma inflammatory biomarkers independently of visceral fat in men. Obesity (Silver Spring) 2008, 16, 1394–1399. [Google Scholar]
- Day, C.P.; James, O.F. Steatohepatitis: A tale of two “hits”? Gastroenterology 1998, 114, 842–845. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Carpino, G.; Renzi, A.; Onori, P.; Gaudio, E. Role of Hepatic Progenitor Cells in Nonalcoholic Fatty Liver Disease Development: Cellular Cross-Talks and Molecular Networks. Int. J. Mol. Sci. 2013, 14, 20112-20130. https://doi.org/10.3390/ijms141020112
Carpino G, Renzi A, Onori P, Gaudio E. Role of Hepatic Progenitor Cells in Nonalcoholic Fatty Liver Disease Development: Cellular Cross-Talks and Molecular Networks. International Journal of Molecular Sciences. 2013; 14(10):20112-20130. https://doi.org/10.3390/ijms141020112
Chicago/Turabian StyleCarpino, Guido, Anastasia Renzi, Paolo Onori, and Eugenio Gaudio. 2013. "Role of Hepatic Progenitor Cells in Nonalcoholic Fatty Liver Disease Development: Cellular Cross-Talks and Molecular Networks" International Journal of Molecular Sciences 14, no. 10: 20112-20130. https://doi.org/10.3390/ijms141020112
APA StyleCarpino, G., Renzi, A., Onori, P., & Gaudio, E. (2013). Role of Hepatic Progenitor Cells in Nonalcoholic Fatty Liver Disease Development: Cellular Cross-Talks and Molecular Networks. International Journal of Molecular Sciences, 14(10), 20112-20130. https://doi.org/10.3390/ijms141020112