EGFR-Ras-Raf Signaling in Epidermal Stem Cells: Roles in Hair Follicle Development, Regeneration, Tissue Remodeling and Epidermal Cancers

Abstract
:1. Introduction
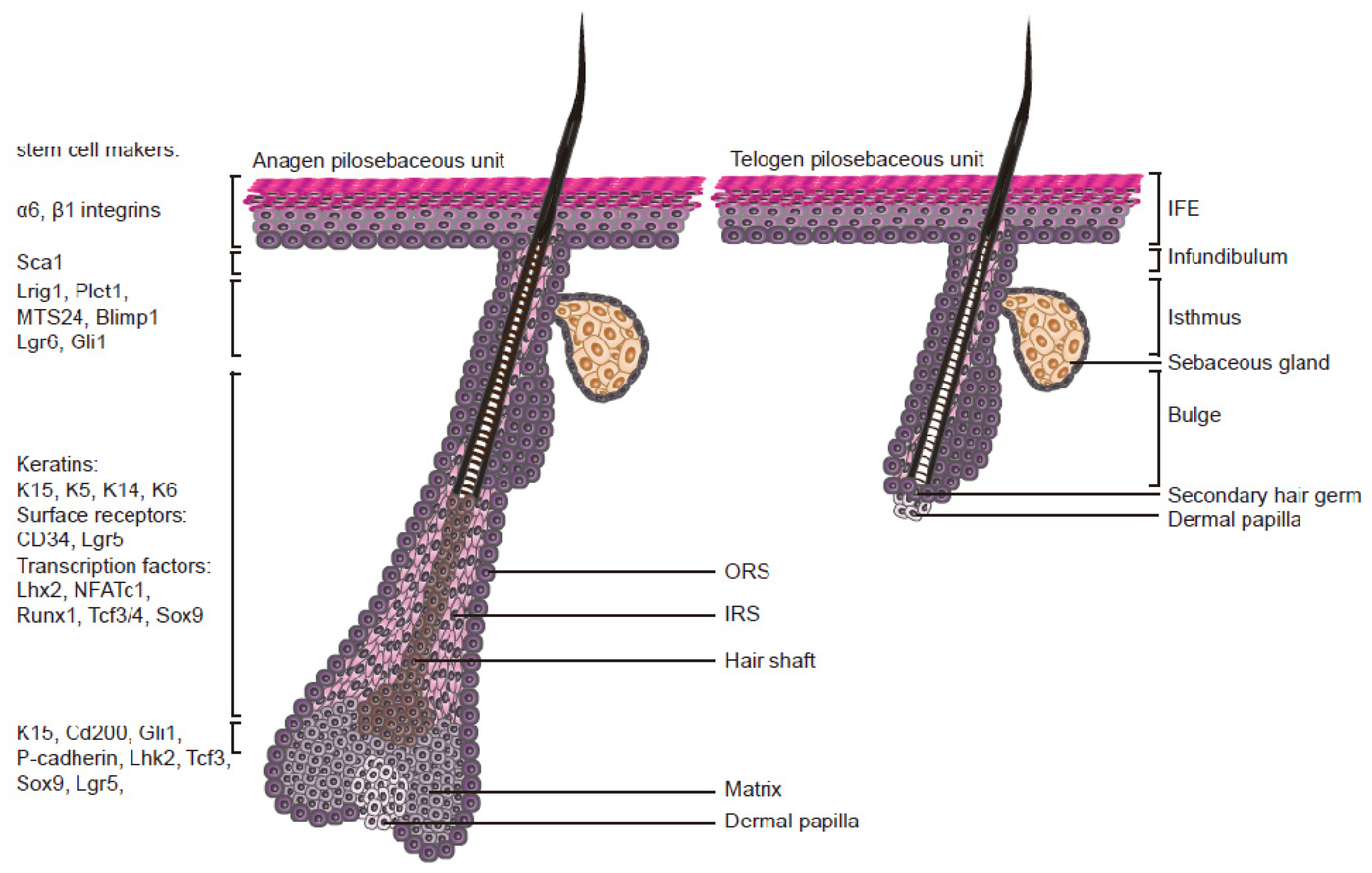
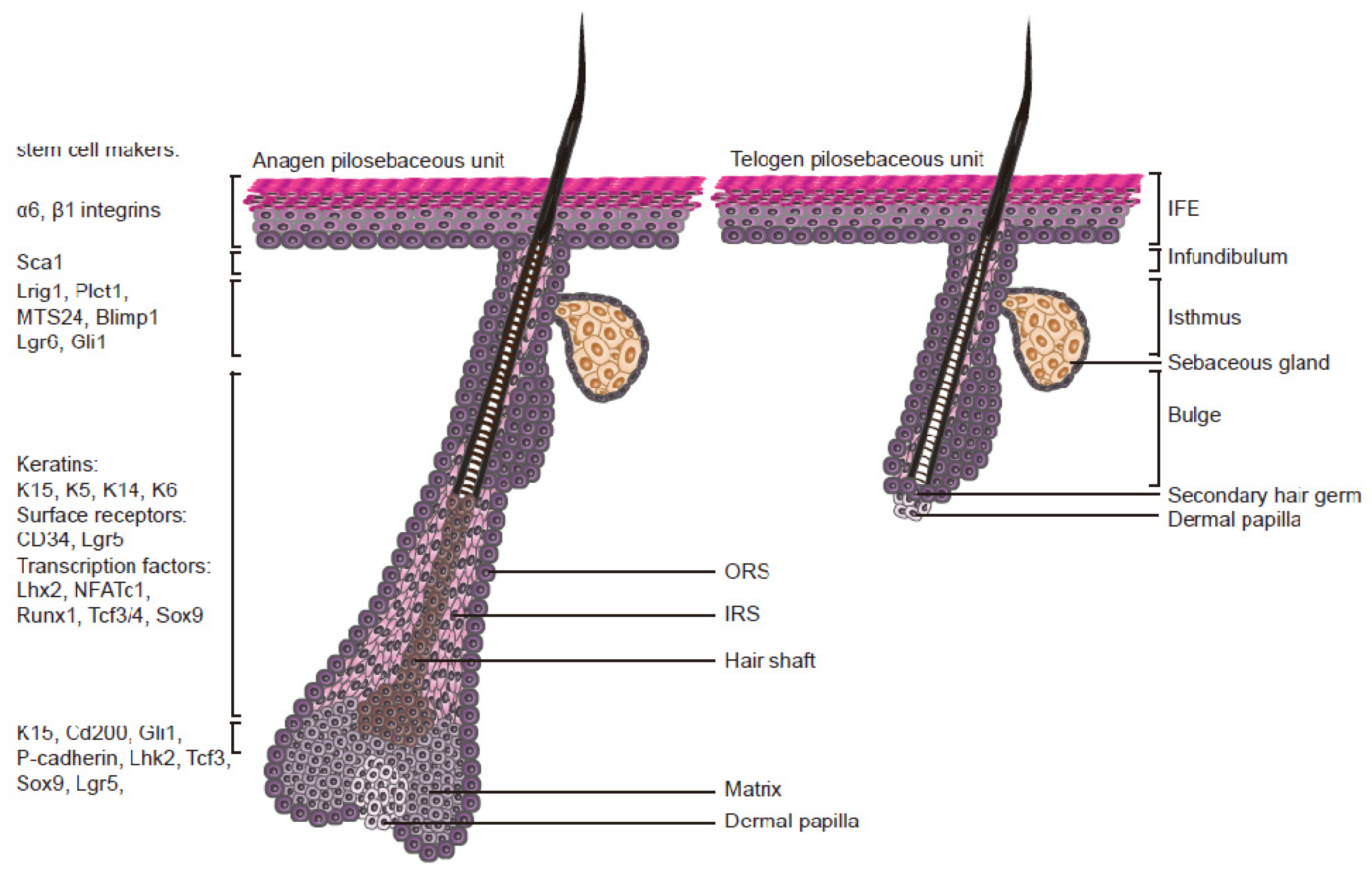
2. Epidermal Stem Cells
2.1. Characteristics
2.2. The Stem Cell Niche
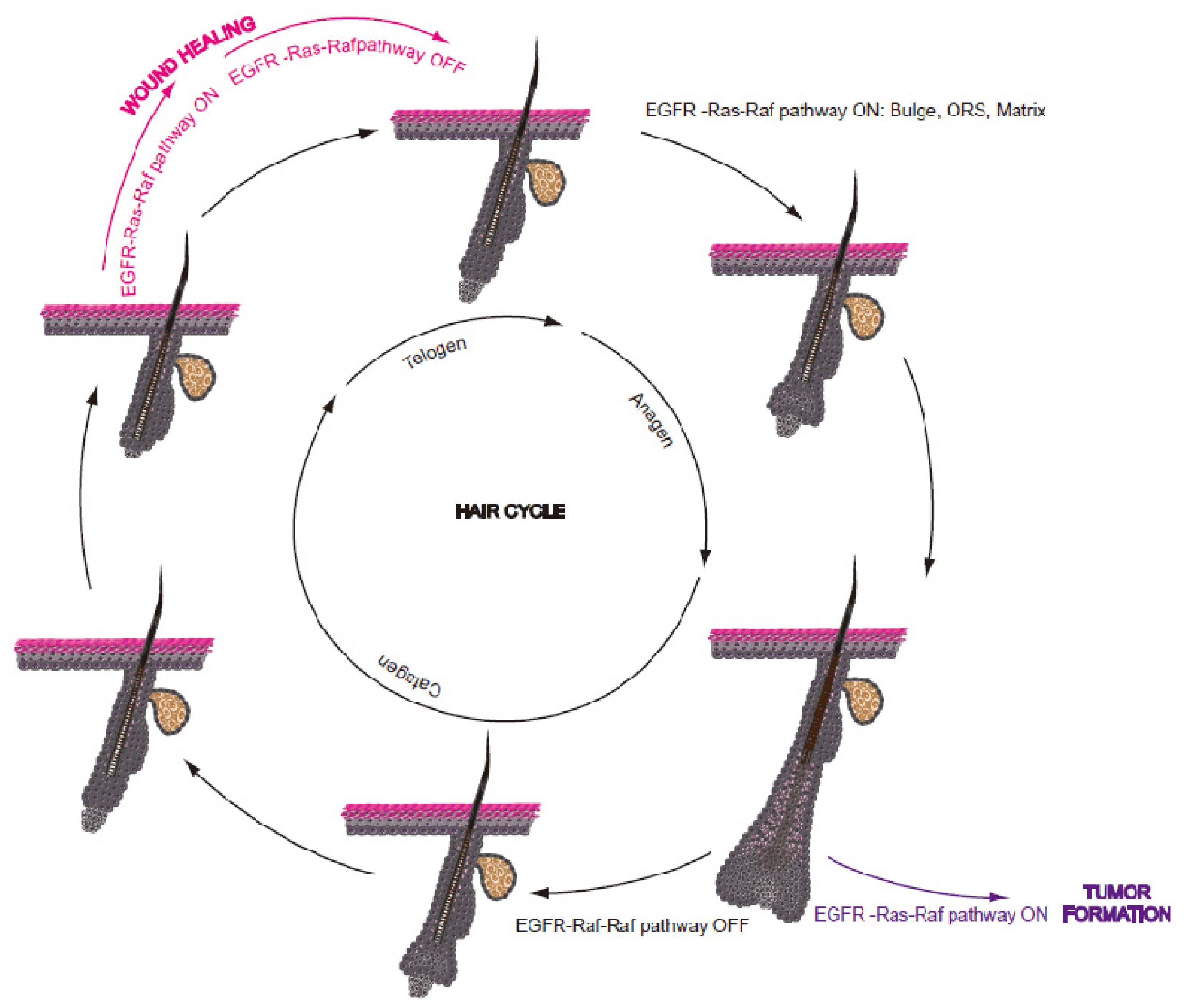
2.3. The Hair Cycle
3. The Role of EGFR-Ras-Raf Pathway in the Pilosebaceous Unit
3.1. The EGFR-Ras-Raf Pathway
3.2. The Effect of EGFR-Ras-Raf Pathway Deregulation on HF Integrity
4. The EGFR-Ras-Raf Pathway in Wound Healing
5. The EGFR-Ras-Raf Pathway during Non-Melanoma Skin Carcinogenesis
5.1. Squamous Cell Carcinomas (SCC)
5.2. Basal Cell Carcinoma (BCC)
6. Conclusions


{kind=link}
{kind=link}
| Model | Phenotypes | References |
|---|---|---|
| Hypomorphic phenotypes | ||
| Epidermal growth factor receptor (EGFR)−/− | Open eyes, rudimentary whiskers, defects in epidermis, delay of hair follicle development, disoriented hair follicles | [79–81] |
| Epidermis restricted dominant negative mutant of EGFR | Short and waved pelage hair, curly whiskers, hairs fail to enter catagen, thinning or loss of the ORS and IRS | [82] |
| Epidermis specific deletion of EGFR exon 3 (K14-Cre, EGFRtmDwt), (abrogated ligand binding) | Wavy coat hair, curly whiskers | [83] |
| Humanized conditional EGFR knock-in (hEGFRKI/KI) (the new allele is not efficiently expressed in the skin) | Homozygotes exhibit skin and hair defects similar to Egfr−/− mutants leading to progressive hair loss | [89] |
| Point mutation (Val 743 Gly) in the EGFR kinase domain (waved-2 allele) | Phenotype of the homozygotes are similar to TGF alpha−/− | [63,90] |
| EGFR waved-2 (see above) Ptpn11+/− (Protein tyrosine phosphatase, non-receptor type11) | Few poorly developed and disordered hair follicles | [91] |
| Dominant negative (Asp 833 Gly) mutation in the EGFR DFG motif in the kinase domain (waved-5/velvet allele) | Heterozygous mice have open eyes and wavy coat and curly whiskers. Homozygous mice die at midgestation owing to placental defects | [84,85] |
| Transforming growth factor-α (TGFα)−/− (or spontaneous TGFα waved 1 mutation) | Wavy coat hair, curly whiskers | [62,92] |
| AR−/−; EGF−/−; TGFα−/− | Wavy coat hair, curly whiskers | [93] |
| A Disintegrin and Metalloproteinase 17 (ADAM17 ) deletion in keratinocytes (A17(ΔKC)(responsible for the TGFα shedding) | Defects in epidermal barrier integrity, chronic dermatitis | [94] |
| Ksr1−/− (Kinase suppressor of Ras1) (positive scaffolding modulator of Ras/MAPK signaling) | Short wavy hair, progressive hair loss, disorganized hair follicles, asynchronous hair growth, IRS separated from the hair shaft | [95] |
| Mek1 (Mitogen activated protein kinase kinase 1)−/− | Reduced hair follicle proliferation | [96] |
| Hypermorphic phenotypes | ||
| Missense mutation ( Leu863Gln ) in the EGFR kinase domain (Dsk5 allele) | Wavy coat, curly vibrissae that becomes less apparent with age and thickened, hyperpigmented epidermis | [97] |
| Continuous expression of EGF in hair follicles | Retarded hair follicle development, reduced hair diameter and increased proliferation in the basal layer | [75,98,99] |
| Skin-specific overexpression of TGFα | Diffuse alopecia, hyperkeratosis, spontaneous SCC development, wrinkled skin | [100] |
| K14 driven TGFα expression | Low hair follicle density, distorted hair follicles, reduced hair growth and thick epidermis | [101] |
| Skin-specific overexpression of human amphiregulin (AR) | Psoriasis-like skin phenotype, alopecia | [102] |
| Ubiquitous overexpression of betacellulin (BTC) | Waved coat and delayed hair follicle morphogenesis and hair cycle induction | [103] |
| Ubiquitous overexpression of human epigen (EPGN) | Enlargement and hyperactivity of the sebaceous glands | [104] |
| Activated Kirsten rat sarcoma viral oncogene homolog (KRasG12D) expression in the midline epidermis | Defective anagen entry, progressive hair loss, overgrown ORS, sHG and matrix cells failing to undergo apoptosis | [105,106] |
Acknowledgments
Conflicts of Interest
References
- Mackenzie, I.C. Relationship between mitosis and the ordered structure of the stratum corneum in mouse epidermis. Nature 1970, 226, 653–655. [Google Scholar]
- Muller-Rover, S.; Handjiski, B.; Van der Veen, C.; Eichmuller, S.; Foitzik, K.; McKay, I.A.; Stenn, K.S.; Paus, R. A comprehensive guide for the accurate classification of murine hair follicles in distinct hair cycle stages. J. Investig. Dermatol 2001, 117, 3–15. [Google Scholar]
- Cotsarelis, G.; Sun, T.T.; Lavker, R.M. Label-retaining cells reside in the bulge area of pilosebaceous unit: Implications for follicular stem cells, hair cycle, and skin carcinogenesis. Cell 1990, 61, 1329–1337. [Google Scholar]
- He, S.; Nakada, D.; Morrison, S.J. Mechanisms of stem cell self-renewal. Annu. Rev. Cell Dev. Biol 2009, 25, 377–406. [Google Scholar]
- Fuchs, E.; Chen, T. A matter of life and death: Self-renewal in stem cells. EMBO Rep 2013, 14, 39–48. [Google Scholar]
- Clayton, E.; Doupe, D.P.; Klein, A.M.; Winton, D.J.; Simons, B.D.; Jones, P.H. A single type of progenitor cell maintains normal epidermis. Nature 2007, 446, 185–189. [Google Scholar]
- Doupe, D.P.; Klein, A.M.; Simons, B.D.; Jones, P.H. The ordered architecture of murine ear epidermis is maintained by progenitor cells with random fate. Dev. Cell 2010, 18, 317–323. [Google Scholar]
- Doupe, D.P.; Alcolea, M.P.; Roshan, A.; Zhang, G.; Klein, A.M.; Simons, B.D.; Jones, P.H. A single progenitor population switches behavior to maintain and repair esophageal epithelium. Science 2012, 337, 1091–1093. [Google Scholar]
- Doupe, D.P.; Jones, P.H. Interfollicular epidermal homeostasis: Dicing with differentiation. Exp. Dermatol 2012, 21, 249–253. [Google Scholar]
- Kaur, P.; Potten, C.S. The interfollicular epidermal stem cell saga: Sensationalism versus reality check. Exp. Dermatol 2011, 20, 697–702. [Google Scholar]
- Morris, R.J.; Fischer, S.M.; Slaga, T.J. Evidence that the centrally and peripherally located cells in the murine epidermal proliferative unit are two distinct cell populations. J. Investig. Dermatol 1985, 84, 277–281. [Google Scholar]
- Potten, C.S. The epidermal proliferative unit: The possible role of the central basal cell. Cell Tissue Kinet 1974, 7, 77–88. [Google Scholar]
- Blanpain, C.; Fuchs, E. Epidermal homeostasis: A balancing act of stem cells in the skin. Nat. Rev. Mol. Cell. Biol 2009, 10, 207–217. [Google Scholar]
- Kobielak, K.; Stokes, N.; De la Cruz, J.; Polak, L.; Fuchs, E. Loss of a quiescent niche but not follicle stem cells in the absence of bone morphogenetic protein signaling. Proc. Natl. Acad. Sci. USA 2007, 104, 10063–10068. [Google Scholar]
- Snippert, H.J.; Haegebarth, A.; Kasper, M.; Jaks, V.; Van Es, J.H.; Barker, N.; Van de Wetering, M.; Van den Born, M.; Begthel, H.; Vries, R.G.; et al. Lgr6 marks stem cells in the hair follicle that generate all cell lineages of the skin. Science 2010, 327, 1385–1389. [Google Scholar]
- Brownell, I.; Guevara, E.; Bai, C.B.; Loomis, C.A.; Joyner, A.L. Nerve-derived sonic hedgehog defines a niche for hair follicle stem cells capable of becoming epidermal stem cells. Cell Stem Cell 2011, 8, 552–565. [Google Scholar]
- Jensen, K.B.; Collins, C.A.; Nascimento, E.; Tan, D.W.; Frye, M.; Itami, S.; Watt, F.M. Lrig1 expression defines a distinct multipotent stem cell population in mammalian epidermis. Cell Stem Cell 2009, 4, 427–439. [Google Scholar]
- Nijhof, J.G.; Braun, K.M.; Giangreco, A.; Van Pelt, C.; Kawamoto, H.; Boyd, R.L.; Willemze, R.; Mullenders, L.H.; Watt, F.M.; De Gruijl, F.R.; et al. The cell-surface marker MTS24 identifies a novel population of follicular keratinocytes with characteristics of progenitor cells. Development 2006, 133, 3027–3037. [Google Scholar]
- Horsley, V.; O’Carroll, D.; Tooze, R.; Ohinata, Y.; Saitou, M.; Obukhanych, T.; Nussenzweig, M.; Tarakhovsky, A.; Fuchs, E. Blimp1 defines a progenitor population that governs cellular input to the sebaceous gland. Cell 2006, 126, 597–609. [Google Scholar]
- Tumbar, T.; Guasch, G.; Greco, V.; Blanpain, C.; Lowry, W.E.; Rendl, M.; Fuchs, E. Defining the epithelial stem cell niche in skin. Science 2004, 303, 359–363. [Google Scholar]
- Morris, R.J.; Liu, Y.; Marles, L.; Yang, Z.; Trempus, C.; Li, S.; Lin, J.S.; Sawicki, J.A.; Cotsarelis, G. Capturing and profiling adult hair follicle stem cells. Nat. Biotechnol 2004, 22, 411–417. [Google Scholar]
- Blanpain, C.; Lowry, W.E.; Geoghegan, A.; Polak, L.; Fuchs, E. Self-renewal, multipotency, and the existence of two cell populations within an epithelial stem cell niche. Cell 2004, 118, 635–648. [Google Scholar]
- Vidal, V.P.; Chaboissier, M.C.; Lutzkendorf, S.; Cotsarelis, G.; Mill, P.; Hui, C.C.; Ortonne, N.; Ortonne, J.P.; Schedl, A. Sox9 is essential for outer root sheath differentiation and the formation of the hair stem cell compartment. Curr. Biol 2005, 15, 1340–1351. [Google Scholar]
- Nowak, J.A.; Polak, L.; Pasolli, H.A.; Fuchs, E. Hair follicle stem cells are specified and function in early skin morphogenesis. Cell Stem Cell 2008, 3, 33–43. [Google Scholar]
- Rhee, H.; Polak, L.; Fuchs, E. Lhx2 maintains stem cell character in hair follicles. Science 2006, 312, 1946–1949. [Google Scholar]
- Nguyen, H.; Merrill, B.J.; Polak, L.; Nikolova, M.; Rendl, M.; Shaver, T.M.; Pasolli, H.A.; Fuchs, E. Tcf3 and Tcf4 are essential for long-term homeostasis of skin epithelia. Nat. Genet 2009, 41, 1068–1075. [Google Scholar]
- Horsley, V.; Aliprantis, A.O.; Polak, L.; Glimcher, L.H.; Fuchs, E. NFATc1 balances quiescence and proliferation of skin stem cells. Cell 2008, 132, 299–310. [Google Scholar]
- Hsu, Y.C.; Pasolli, H.A.; Fuchs, E. Dynamics between stem cells, niche, and progeny in the hair follicle. Cell 2011, 144, 92–105. [Google Scholar]
- Jaks, V.; Barker, N.; Kasper, M.; Van Es, J.H.; Snippert, H.J.; Clevers, H.; Toftgard, R. Lgr5 marks cycling, yet long-lived, hair follicle stem cells. Nat. Genet 2008, 40, 1291–1299. [Google Scholar]
- Greco, V.; Chen, T.; Rendl, M.; Schober, M.; Pasolli, H.A.; Stokes, N.; Dela Cruz-Racelis, J.; Fuchs, E. A two-step mechanism for stem cell activation during hair regeneration. Cell Stem Cell 2009, 4, 155–169. [Google Scholar]
- Morris, R.J.; Potten, C.S. Highly persistent label-retaining cells in the hair follicles of mice and their fate following induction of anagen. J. Investig. Dermatol 1999, 112, 470–475. [Google Scholar]
- Hsu, Y.C.; Fuchs, E. A family business: Stem cell progeny join the niche to regulate homeostasis. Nat. Rev. Mol. Cell. Biol 2012, 13, 103–114. [Google Scholar]
- Rendl, M.; Polak, L.; Fuchs, E. BMP signaling in dermal papilla cells is required for their hair follicle-inductive properties. Genes Dev 2008, 22, 543–557. [Google Scholar]
- Enshell-Seijffers, D.; Lindon, C.; Kashiwagi, M.; Morgan, B.A. beta-catenin activity in the dermal papilla regulates morphogenesis and regeneration of hair. Dev. Cell 2010, 18, 633–642. [Google Scholar]
- Botchkarev, V.A.; Botchkareva, N.V.; Roth, W.; Nakamura, M.; Chen, L.H.; Herzog, W.; Lindner, G.; McMahon, J.A.; Peters, C.; Lauster, R.; et al. Noggin is a mesenchymally derived stimulator of hair-follicle induction. Nat. Cell Biol 1999, 1, 158–164. [Google Scholar]
- Jahoda, C.A.; Horne, K.A.; Oliver, R.F. Induction of hair growth by implantation of cultured dermal papilla cells. Nature 1984, 311, 560–562. [Google Scholar]
- Panteleyev, A.A.; Jahoda, C.A.; Christiano, A.M. Hair follicle predetermination. J. Cell Sci 2001, 114, 3419–3431. [Google Scholar]
- Ito, M.; Kizawa, K.; Hamada, K.; Cotsarelis, G. Hair follicle stem cells in the lower bulge form the secondary germ, a biochemically distinct but functionally equivalent progenitor cell population, at the termination of catagen. Differentiation 2004, 72, 548–557. [Google Scholar]
- Muller-Rover, S.; Tokura, Y.; Welker, P.; Furukawa, F.; Wakita, H.; Takigawa, M.; Paus, R. E- and P-cadherin expression during murine hair follicle morphogenesis and cycling. Exp. Dermatol 1999, 8, 237–246. [Google Scholar]
- Gat, U.; DasGupta, R.; Degenstein, L.; Fuchs, E. De Novo hair follicle morphogenesis and hair tumors in mice expressing a truncated beta-catenin in skin. Cell 1998, 95, 605–614. [Google Scholar]
- Van Mater, D.; Kolligs, F.T.; Dlugosz, A.A.; Fearon, E.R. Transient activation of beta-catenin signaling in cutaneous keratinocytes is sufficient to trigger the active growth phase of the hair cycle in mice. Genes Dev 2003, 17, 1219–1224. [Google Scholar]
- Zhang, J.; He, X.C.; Tong, W.G.; Johnson, T.; Wiedemann, L.M.; Mishina, Y.; Feng, J.Q.; Li, L. Bone morphogenetic protein signaling inhibits hair follicle anagen induction by restricting epithelial stem/progenitor cell activation and expansion. Stem Cells 2006, 24, 2826–2839. [Google Scholar]
- Taylor, G.; Lehrer, M.S.; Jensen, P.J.; Sun, T.T.; Lavker, R.M. Involvement of follicular stem cells in forming not only the follicle but also the epidermis. Cell 2000, 102, 451–461. [Google Scholar]
- Oshima, H.; Rochat, A.; Kedzia, C.; Kobayashi, K.; Barrandon, Y. Morphogenesis and renewal of hair follicles from adult multipotent stem cells. Cell 2001, 104, 233–245. [Google Scholar]
- Plikus, M.V.; Mayer, J.A.; De la Cruz, D.; Baker, R.E.; Maini, P.K.; Maxson, R.; Chuong, C.M. Cyclic dermal BMP signalling regulates stem cell activation during hair regeneration. Nature 2008, 451, 340–344. [Google Scholar] [Green Version]
- Klapper, L.N.; Glathe, S.; Vaisman, N.; Hynes, N.E.; Andrews, G.C.; Sela, M.; Yarden, Y. The ErbB-2/HER2 oncoprotein of human carcinomas may function solely as a shared coreceptor for multiple stroma-derived growth factors. Proc. Natl. Acad. Sci. USA 1999, 96, 4995–5000. [Google Scholar]
- Schneider, M.R.; Werner, S.; Paus, R.; Wolf, E. Beyond wavy hairs: The epidermal growth factor receptor and its ligands in skin biology and pathology. Am. J. Pathol 2008, 173, 14–24. [Google Scholar]
- Shi, F.; Telesco, S.E.; Liu, Y.; Radhakrishnan, R.; Lemmon, M.A. ErbB3/HER3 intracellular domain is competent to bind ATP and catalyze autophosphorylation. Proc. Natl. Acad. Sci. USA 2010, 107, 7692–7697. [Google Scholar]
- Singh, A.B.; Harris, R.C. Autocrine, paracrine and juxtacrine signaling by EGFR ligands. Cell Signal 2005, 17, 1183–1193. [Google Scholar]
- Niault, T.S.; Baccarini, M. Targets of Raf in tumorigenesis. Carcinogenesis 2010, 31, 1165–1174. [Google Scholar]
- Kern, F.; Niault, T.; Baccarini, M. Ras and Raf pathways in epidermis development and carcinogenesis. Br. J. Cancer 2011, 104, 229–234. [Google Scholar]
- Matallanas, D.; Birtwistle, M.; Romano, D.; Zebisch, A.; Rauch, J.; Von Kriegsheim, A.; Kolch, W. Raf family kinases: Old dogs have learned new tricks. Genes Cancer 2011, 2, 232–260. [Google Scholar]
- Wimmer, R.; Baccarini, M. Partner exchange: Protein-protein interactions in the Raf pathway. Trends Biochem. Sci 2010, 35, 660–668. [Google Scholar]
- Castellano, E.; Santos, E. Functional specificity of ras isoforms: So similar but so different. Genes Cancer 2011, 2, 216–231. [Google Scholar]
- Yan, J.; Roy, S.; Apolloni, A.; Lane, A.; Hancock, J.F. Ras isoforms vary in their ability to activate Raf-1 and phosphoinositide 3-kinase. J. Biol. Chem 1998, 273, 24052–24056. [Google Scholar]
- Yoon, S.; Seger, R. The extracellular signal-regulated kinase: Multiple substrates regulate diverse cellular functions. Growth Factors 2006, 24, 21–44. [Google Scholar]
- Ehrenreiter, K.; Piazzolla, D.; Velamoor, V.; Sobczak, I.; Small, J.V.; Takeda, J.; Leung, T.; Baccarini, M. Raf-1 regulates Rho signaling and cell migration. J. Cell Biol 2005, 168, 955–964. [Google Scholar]
- Ehrenreiter, K.; Kern, F.; Velamoor, V.; Meissl, K.; Galabova-Kovacs, G.; Sibilia, M.; Baccarini, M. Raf-1 addiction in Ras-induced skin carcinogenesis. Cancer Cell 2009, 16, 149–160. [Google Scholar]
- Niault, T.; Sobczak, I.; Meissl, K.; Weitsman, G.; Piazzolla, D.; Maurer, G.; Kern, F.; Ehrenreiter, K.; Hamerl, M.; Moarefi, I.; et al. From autoinhibition to inhibition in trans: The Raf-1 regulatory domain inhibits Rok-alpha kinase activity. J. Cell Biol 2009, 187, 335–342. [Google Scholar]
- Honma, M.; Benitah, S.A.; Watt, F.M. Role of LIM kinases in normal and psoriatic human epidermis. Mol. Biol. Cell 2006, 17, 1888–1896. [Google Scholar]
- Miyoshi, K.; Takaishi, M.; Nakajima, K.; Ikeda, M.; Kanda, T.; Tarutani, M.; Iiyama, T.; Asao, N.; DiGiovanni, J.; Sano, S. Stat3 as a therapeutic target for the treatment of psoriasis: A clinical feasibility study with STA-21, a Stat3 inhibitor. J. Investig. Dermatol 2011, 131, 108–117. [Google Scholar]
- Mann, G.B.; Fowler, K.J.; Gabriel, A.; Nice, E.C.; Williams, R.L.; Dunn, A.R. Mice with a null mutation of the TGF alpha gene have abnormal skin architecture, wavy hair, and curly whiskers and often develop corneal inflammation. Cell 1993, 73, 249–261. [Google Scholar]
- Luetteke, N.C.; Phillips, H.K.; Qiu, T.H.; Copeland, N.G.; Earp, H.S.; Jenkins, N.A.; Lee, D.C. The mouse waved-2 phenotype results from a point mutation in the EGF receptor tyrosine kinase. Genes Dev 1994, 8, 399–413. [Google Scholar]
- Stoll, S.W.; Kansra, S.; Peshick, S.; Fry, D.W.; Leopold, W.R.; Wiesen, J.F.; Sibilia, M.; Zhang, T.; Werb, Z.; Derynck, R.; et al. Differential utilization and localization of ErbB receptor tyrosine kinases in skin compared to normal and malignant keratinocytes. Neoplasia 2001, 3, 339–350. [Google Scholar]
- Wang, X.; Bolotin, D.; Chu, D.H.; Polak, L.; Williams, T.; Fuchs, E. AP-2alpha: A regulator of EGF receptor signaling and proliferation in skin epidermis. J. Cell Biol 2006, 172, 409–421. [Google Scholar]
- Jost, M.; Huggett, T.M.; Kari, C.; Rodeck, U. Matrix-independent survival of human keratinocytes through an EGF receptor/MAPK-kinase-dependent pathway. Mol. Biol. Cell 2001, 12, 1519–1527. [Google Scholar]
- Peus, D.; Hamacher, L.; Pittelkow, M.R. EGF-receptor tyrosine kinase inhibition induces keratinocyte growth arrest and terminal differentiation. J. Investig. Dermatol 1997, 109, 751–756. [Google Scholar]
- Getsios, S.; Simpson, C.L.; Kojima, S.; Harmon, R.; Sheu, L.J.; Dusek, R.L.; Cornwell, M.; Green, K.J. Desmoglein 1-dependent suppression of EGFR signaling promotes epidermal differentiation and morphogenesis. J. Cell Biol 2009, 185, 1243–1258. [Google Scholar]
- Robertson, E.D.; Weir, L.; Romanowska, M.; Leigh, I.M.; Panteleyev, A.A. ARNT controls the expression of epidermal differentiation genes through HDAC- and EGFR-dependent pathways. J. Cell Sci 2012, 125, 3320–3332. [Google Scholar]
- Poumay, Y.; Pittelkow, M.R. Cell density and culture factors regulate keratinocyte commitment to differentiation and expression of suprabasal K1/K10 keratins. J. Investig. Dermatol 1995, 104, 271–276. [Google Scholar]
- Dajee, M.; Tarutani, M.; Deng, H.; Cai, T.; Khavari, P.A. Epidermal Ras blockade demonstrates spatially localized Ras promotion of proliferation and inhibition of differentiation. Oncogene 2002, 21, 1527–1538. [Google Scholar]
- Haase, I.; Hobbs, R.M.; Romero, M.R.; Broad, S.; Watt, F.M. A role for mitogen-activated protein kinase activation by integrins in the pathogenesis of psoriasis. J. Clin. Invest 2001, 108, 527–536. [Google Scholar]
- Scholl, F.A.; Dumesic, P.A.; Khavari, P.A. Mek1 alters epidermal growth and differentiation. Cancer Res 2004, 64, 6035–6040. [Google Scholar]
- Tarutani, M.; Cai, T.; Dajee, M.; Khavari, P.A. Inducible activation of Ras and Raf in adult epidermis. Cancer Res 2003, 63, 319–323. [Google Scholar]
- Mak, K.K.; Chan, S.Y. Epidermal growth factor as a biologic switch in hair growth cycle. J. Biol. Chem 2003, 278, 26120–26126. [Google Scholar]
- Sakai, Y.; Nelson, K.G.; Snedeker, S.; Bossert, N.L.; Walker, M.P.; McLachlan, J.; DiAugustine, R.P. Expression of epidermal growth factor in suprabasal cells of stratified squamous epithelia: Implications for a role in differentiation. Cell Growth Differ 1994, 5, 527–535. [Google Scholar]
- Du Cros, D.L.; Isaacs, K.; Moore, G.P. Localization of epidermal growth factor immunoreactivity in sheep skin during wool follicle development. J. Investig. Dermatol 1992, 98, 109–115. [Google Scholar]
- Finzi, E.; Harkins, R.; Horn, T. TGF-alpha is widely expressed in differentiated as well as hyperproliferative skin epithelium. J. Investig. Dermatol 1991, 96, 328–332. [Google Scholar]
- Miettinen, P.J.; Berger, J.E.; Meneses, J.; Phung, Y.; Pedersen, R.A.; Werb, Z.; Derynck, R. Epithelial immaturity and multiorgan failure in mice lacking epidermal growth factor receptor. Nature 1995, 376, 337–341. [Google Scholar]
- Threadgill, D.W.; Dlugosz, A.A.; Hansen, L.A.; Tennenbaum, T.; Lichti, U.; Yee, D.; LaMantia, C.; Mourton, T.; Herrup, K.; Harris, R.C.; et al. Targeted disruption of mouse EGF receptor: Effect of genetic background on mutant phenotype. Science 1995, 269, 230–234. [Google Scholar]
- Sibilia, M.; Wagner, E.F. Strain-dependent epithelial defects in mice lacking the EGF receptor. Science 1995, 269, 234–238. [Google Scholar]
- Murillas, R.; Larcher, F.; Conti, C.J.; Santos, M.; Ullrich, A.; Jorcano, J.L. Expression of a dominant negative mutant of epidermal growth factor receptor in the epidermis of transgenic mice elicits striking alterations in hair follicle development and skin structure. EMBO J 1995, 14, 5216–5223. [Google Scholar]
- Lee, T.C.; Threadgill, D.W. Generation and validation of mice carrying a conditional allele of the epidermal growth factor receptor. Genesis 2009, 47, 85–92. [Google Scholar]
- Lee, D.; Cross, S.H.; Strunk, K.E.; Morgan, J.E.; Bailey, C.L.; Jackson, I.J.; Threadgill, D.W. Wa5 is a novel ENU-induced antimorphic allele of the epidermal growth factor receptor. Mamm. Genome 2004, 15, 525–536. [Google Scholar]
- Du, X.; Tabeta, K.; Hoebe, K.; Liu, H.; Mann, N.; Mudd, S.; Crozat, K.; Sovath, S.; Gong, X.; Beutler, B. Velvet, a dominant Egfr mutation that causes wavy hair and defective eyelid development in mice. Genetics 2004, 166, 331–340. [Google Scholar]
- Hansen, L.A.; Alexander, N.; Hogan, M.E.; Sundberg, J.P.; Dlugosz, A.; Threadgill, D.W.; Magnuson, T.; Yuspa, S.H. Genetically null mice reveal a central role for epidermal growth factor receptor in the differentiation of the hair follicle and normal hair development. Am. J. Pathol 1997, 150, 1959–1975. [Google Scholar]
- Ciardiello, F.; Tortora, G. EGFR antagonists in cancer treatment. N. Engl. J. Med 2008, 358, 1160–1174. [Google Scholar]
- Okamoto, I.; Takahashi, T.; Okamoto, H.; Nakagawa, K.; Watanabe, K.; Nakamatsu, K.; Nishimura, Y.; Fukuoka, M.; Yamamoto, N. Single-agent gefitinib with concurrent radiotherapy for locally advanced non-small cell lung cancer harboring mutations of the epidermal growth factor receptor. Lung Cancer 2011, 72, 199–204. [Google Scholar]
- Sibilia, M.; Wagner, B.; Hoebertz, A.; Elliott, C.; Marino, S.; Jochum, W.; Wagner, E.F. Mice humanised for the EGF receptor display hypomorphic phenotypes in skin, bone and heart. Development 2003, 130, 4515–4525. [Google Scholar]
- Fowler, K.J.; Walker, F.; Alexander, W.; Hibbs, M.L.; Nice, E.C.; Bohmer, R.M.; Mann, G.B.; Thumwood, C.; Maglitto, R.; Danks, J.A.; et al. A mutation in the epidermal growth factor receptor in waved-2 mice has a profound effect on receptor biochemistry that results in impaired lactation. Proc. Natl. Acad. Sci. USA 1995, 92, 1465–1469. [Google Scholar]
- Qu, C.K.; Yu, W.M.; Azzarelli, B.; Feng, G.S. Genetic evidence that Shp-2 tyrosine phosphatase is a signal enhancer of the epidermal growth factor receptor in mammals. Proc. Natl. Acad. Sci. USA 1999, 96, 8528–8533. [Google Scholar]
- Luetteke, N.C.; Qiu, T.H.; Peiffer, R.L.; Oliver, P.; Smithies, O.; Lee, D.C. TGF alpha deficiency results in hair follicle and eye abnormalities in targeted and waved-1 mice. Cell 1993, 73, 263–278. [Google Scholar]
- Luetteke, N.C.; Qiu, T.H.; Fenton, S.E.; Troyer, K.L.; Riedel, R.F.; Chang, A.; Lee, D.C. Targeted inactivation of the EGF and amphiregulin genes reveals distinct roles for EGF receptor ligands in mouse mammary gland development. Development 1999, 126, 2739–2750. [Google Scholar]
- Franzke, C.W.; Cobzaru, C.; Triantafyllopoulou, A.; Loffek, S.; Horiuchi, K.; Threadgill, D.W.; Kurz, T.; Van Rooijen, N.; Bruckner-Tuderman, L.; Blobel, C.P. Epidermal ADAM17 maintains the skin barrier by regulating EGFR ligand-dependent terminal keratinocyte differentiation. J. Exp. Med 2012, 209, 1105–1119. [Google Scholar]
- Lozano, J.; Xing, R.; Cai, Z.; Jensen, H.L.; Trempus, C.; Mark, W.; Cannon, R.; Kolesnick, R. Deficiency of kinase suppressor of Ras1 prevents oncogenic ras signaling in mice. Cancer Res 2003, 63, 4232–4238. [Google Scholar]
- Scholl, F.A.; Dumesic, P.A.; Barragan, D.I.; Harada, K.; Bissonauth, V.; Charron, J.; Khavari, P.A. Mek1/2 MAPK kinases are essential for Mammalian development, homeostasis, and Raf-induced hyperplasia. Dev. Cell 2007, 12, 615–629. [Google Scholar]
- Fitch, K.R.; McGowan, K.A.; Van Raamsdonk, C.D.; Fuchs, H.; Lee, D.; Puech, A.; Herault, Y.; Threadgill, D.W.; Hrabe de Angelis, M.; Barsh, G.S. Genetics of dark skin in mice. Genes Dev 2003, 17, 214–228. [Google Scholar]
- Moore, G.P.; Panaretto, B.A.; Robertson, D. Epidermal growth factor delays the development of the epidermis and hair follicles of mice during growth of the first coat. Anat Rec 1983, 205, 47–55. [Google Scholar]
- Richardson, G.D.; Bazzi, H.; Fantauzzo, K.A.; Waters, J.M.; Crawford, H.; Hynd, P.; Christiano, A.M.; Jahoda, C.A. KGF and EGF signalling block hair follicle induction and promote interfollicular epidermal fate in developing mouse skin. Development 2009, 136, 2153–2164. [Google Scholar]
- Dominey, A.M.; Wang, X.J.; King, L.E., Jr.; Nanney, L.B.; Gagne, T.A.; Sellheyer, K.; Bundman, D.S.; Longley, M.A.; Rothnagel, J.A.; Greenhalgh, D.A.; et al. Targeted overexpression of transforming growth factor alpha in the epidermis of transgenic mice elicits hyperplasia, hyperkeratosis, and spontaneous, squamous papillomas. Cell Growth Differ 1993, 4, 1071–1082. [Google Scholar]
- Vassar, R.; Fuchs, E. Transgenic mice provide new insights into the role of TGF-alpha during epidermal development and differentiation. Genes Dev 1991, 5, 714–727. [Google Scholar]
- Cook, P.W.; Brown, J.R.; Cornell, K.A.; Pittelkow, M.R. Suprabasal expression of human amphiregulin in the epidermis of transgenic mice induces a severe, early-onset, psoriasis-like skin pathology: Expression of amphiregulin in the basal epidermis is also associated with synovitis. Exp. Dermatol 2004, 13, 347–356. [Google Scholar]
- Schneider, M.R.; Antsiferova, M.; Feldmeyer, L.; Dahlhoff, M.; Bugnon, P.; Hasse, S.; Paus, R.; Wolf, E.; Werner, S. Betacellulin regulates hair follicle development and hair cycle induction and enhances angiogenesis in wounded skin. J. Investig. Dermatol 2008, 128, 1256–1265. [Google Scholar]
- Dahlhoff, M.; Muller, A.K.; Wolf, E.; Werner, S.; Schneider, M.R. Epigen transgenic mice develop enlarged sebaceous glands. J. Investig. Dermatol 2010, 130, 623–626. [Google Scholar]
- Mukhopadhyay, A.; Krishnaswami, S.R.; Yu, B.D. Activated Kras alters epidermal homeostasis of mouse skin, resulting in redundant skin and defective hair cycling. J. Investig. Dermatol 2011, 131, 311–319. [Google Scholar]
- Mukhopadhyay, A.; Krishnaswami, S.R.; Cowing-Zitron, C.; Hung, N.J.; Reilly-Rhoten, H.; Burns, J.; Yu, B.D. Negative regulation of Shh levels by Kras and Fgfr2 during hair follicle development. Dev. Biol 2013, 373, 373–382. [Google Scholar]
- Philpott, M.P.; Kealey, T. Effects of EGF on the morphology and patterns of DNA synthesis in isolated human hair follicles. J. Investig. Dermatol 1994, 102, 186–191. [Google Scholar]
- Tidyman, W.E.; Rauen, K.A. The RASopathies: Developmental syndromes of Ras/MAPK pathway dysregulation. Curr. Opin. Genet. Dev 2009, 19, 230–236. [Google Scholar]
- Siegel, D.H.; Mann, J.A.; Krol, A.L.; Rauen, K.A. Dermatological phenotype in Costello syndrome: Consequences of Ras dysregulation in development. Br. J. Dermatol 2012, 166, 601–607. [Google Scholar]
- Kerr, B.; Allanson, J.; Delrue, M.A.; Gripp, K.W.; Lacombe, D.; Lin, A.E.; Rauen, K.A. The diagnosis of Costello syndrome: Nomenclature in Ras/MAPK pathway disorders. Am. J. Med. Genet. Part A 2008, 146A, 1218–1220. [Google Scholar]
- Roberts, A.; Allanson, J.; Jadico, S.K.; Kavamura, M.I.; Noonan, J.; Opitz, J.M.; Young, T.; Neri, G. The cardiofaciocutaneous syndrome. J. Med. Genet 2006, 43, 833–842. [Google Scholar]
- St-Jacques, B.; Dassule, H.R.; Karavanova, I.; Botchkarev, V.A.; Li, J.; Danielian, P.S.; McMahon, J.A.; Lewis, P.M.; Paus, R.; McMahon, A.P. Sonic hedgehog signaling is essential for hair development. Curr. Biol 1998, 8, 1058–1068. [Google Scholar]
- Chiang, C.; Swan, R.Z.; Grachtchouk, M.; Bolinger, M.; Litingtung, Y.; Robertson, E.K.; Cooper, M.K.; Gaffield, W.; Westphal, H.; Beachy, P.A.; et al. Essential role for Sonic hedgehog during hair follicle morphogenesis. Dev. Biol 1999, 205, 1–9. [Google Scholar]
- Sato, N.; Leopold, P.L.; Crystal, R.G. Induction of the hair growth phase in postnatal mice by localized transient expression of Sonic hedgehog. J. Clin. Invest 1999, 104, 855–864. [Google Scholar]
- Wang, L.C.; Liu, Z.Y.; Gambardella, L.; Delacour, A.; Shapiro, R.; Yang, J.; Sizing, I.; Rayhorn, P.; Garber, E.A.; Benjamin, C.D.; et al. Regular articles: Conditional disruption of hedgehog signaling pathway defines its critical role in hair development and regeneration. J. Investig. Dermatol 2000, 114, 901–908. [Google Scholar]
- Raguz, J.; Baccarini, M. University of Vienna: Vienna Austria, Unpublished work; 2013.
- McMullan, R.; Lax, S.; Robertson, V.H.; Radford, D.J.; Broad, S.; Watt, F.M.; Rowles, A.; Croft, D.R.; Olson, M.F.; Hotchin, N.A. Keratinocyte differentiation is regulated by the Rho and ROCK signaling pathway. Curr. Biol 2003, 13, 2185–2189. [Google Scholar]
- Watanabe, K.; Ueno, M.; Kamiya, D.; Nishiyama, A.; Matsumura, M.; Wataya, T.; Takahashi, J.B.; Nishikawa, S.; Muguruma, K.; Sasai, Y. A ROCK inhibitor permits survival of dissociated human embryonic stem cells. Nat. Biotechnol 2007, 25, 681–686. [Google Scholar]
- Chapman, S.; Liu, X.; Meyers, C.; Schlegel, R.; McBride, A.A. Human keratinocytes are efficiently immortalized by a Rho kinase inhibitor. J. Clin. Invest 2010, 120, 2619–2626. [Google Scholar]
- Singer, A.J.; Clark, R.A. Cutaneous wound healing. N. Engl. J. Med 1999, 341, 738–746. [Google Scholar]
- Levy, V.; Lindon, C.; Harfe, B.D.; Morgan, B.A. Distinct stem cell populations regenerate the follicle and interfollicular epidermis. Dev. Cell 2005, 9, 855–861. [Google Scholar]
- Ito, M.; Liu, Y.; Yang, Z.; Nguyen, J.; Liang, F.; Morris, R.J.; Cotsarelis, G. Stem cells in the hair follicle bulge contribute to wound repair but not to homeostasis of the epidermis. Nat. Med 2005, 11, 1351–1354. [Google Scholar]
- Langton, A.K.; Herrick, S.E.; Headon, D.J. An extended epidermal response heals cutaneous wounds in the absence of a hair follicle stem cell contribution. J. Investig. Dermatol 2008, 128, 1311–1318. [Google Scholar]
- Levy, V.; Lindon, C.; Zheng, Y.; Harfe, B.D.; Morgan, B.A. Epidermal stem cells arise from the hair follicle after wounding. FASEB J 2007, 21, 1358–1366. [Google Scholar]
- Watt, F.M.; Driskell, R.R. The therapeutic potential of stem cells. Philos. Trans. R. Soc. Lond. B Biol. Sci 2010, 365, 155–163. [Google Scholar]
- Marikovsky, M.; Breuing, K.; Liu, P.Y.; Eriksson, E.; Higashiyama, S.; Farber, P.; Abraham, J.; Klagsbrun, M. Appearance of heparin-binding EGF-like growth factor in wound fluid as a response to injury. Proc. Natl. Acad. Sci. USA 1993, 90, 3889–3893. [Google Scholar]
- Shirakata, Y.; Kimura, R.; Nanba, D.; Iwamoto, R.; Tokumaru, S.; Morimoto, C.; Yokota, K.; Nakamura, M.; Sayama, K.; Mekada, E.; et al. Heparin-binding EGF-like growth factor accelerates keratinocyte migration and skin wound healing. J. Cell Sci 2005, 118, 2363–2370. [Google Scholar]
- Yoshioka, R.; Shiraishi, A.; Kobayashi, T.; Morita, S.; Hayashi, Y.; Higashiyama, S.; Ohashi, Y. Corneal epithelial wound healing impaired in keratinocyte-specific HB-EGF-deficient mice in vivo and in vitro. Invest. Ophthalmol. Vis. Sci. 2010, 51, 5630–5639. [Google Scholar]
- Repertinger, S.K.; Campagnaro, E.; Fuhrman, J.; El-Abaseri, T.; Yuspa, S.H.; Hansen, L.A. EGFR enhances early healing after cutaneous incisional wounding. J. Investig. Dermatol 2004, 123, 982–989. [Google Scholar]
- Gonda, T.A.; Tu, S.; Wang, T.C. Chronic inflammation, the tumor microenvironment and carcinogenesis. Cell Cycle 2009, 8, 2005–2013. [Google Scholar]
- Pedersen, T.X.; Leethanakul, C.; Patel, V.; Mitola, D.; Lund, L.R.; Dano, K.; Johnsen, M.; Gutkind, J.S.; Bugge, T.H. Laser capture microdissection-based in vivo genomic profiling of wound keratinocytes identifies similarities and differences to squamous cell carcinoma. Oncogene 2003, 22, 3964–3976. [Google Scholar]
- Arwert, E.N.; Hoste, E.; Watt, F.M. Epithelial stem cells, wound healing and cancer. Nat. Rev. Cancer 2012, 12, 170–180. [Google Scholar]
- Schafer, M.; Werner, S. Cancer as an overhealing wound: An old hypothesis revisited. Nat. Rev. Mol. Cell. Biol 2008, 9, 628–638. [Google Scholar]
- Alam, M.; Ratner, D. Cutaneous squamous-cell carcinoma. N. Engl. J. Med 2001, 344, 975–983. [Google Scholar]
- Thompson, S.C.; Jolley, D.; Marks, R. Reduction of solar keratoses by regular sunscreen use. N. Engl. J. Med 1993, 329, 1147–1151. [Google Scholar]
- Marks, R.; Rennie, G.; Selwood, T.S. Malignant transformation of solar keratoses to squamous cell carcinoma. Lancet 1988, 1, 795–797. [Google Scholar]
- Bamford, S.; Dawson, E.; Forbes, S.; Clements, J.; Pettett, R.; Dogan, A.; Flanagan, A.; Teague, J.; Futreal, P.A.; Stratton, M.R.; et al. The COSMIC (Catalogue of Somatic Mutations in Cancer) database and website. Br. J. Cancer 2004, 91, 355–358. [Google Scholar]
- Lazarov, M.; Kubo, Y.; Cai, T.; Dajee, M.; Tarutani, M.; Lin, Q.; Fang, M.; Tao, S.; Green, C.L.; Khavari, P.A. CDK4 coexpression with Ras generates malignant human epidermal tumorigenesis. Nat. Med 2002, 8, 1105–1114. [Google Scholar]
- Dajee, M.; Lazarov, M.; Zhang, J.Y.; Cai, T.; Green, C.L.; Russell, A.J.; Marinkovich, M.P.; Tao, S.; Lin, Q.; Kubo, Y.; et al. NF-kappaB blockade and oncogenic Ras trigger invasive human epidermal neoplasia. Nature 2003, 421, 639–643. [Google Scholar]
- Ratushny, V.; Gober, M.D.; Hick, R.; Ridky, T.W.; Seykora, J.T. From keratinocyte to cancer: The pathogenesis and modeling of cutaneous squamous cell carcinoma. J. Clin. Invest 2012, 122, 464–472. [Google Scholar]
- Uribe, P.; Gonzalez, S. Epidermal growth factor receptor (EGFR) and squamous cell carcinoma of the skin: Molecular bases for EGFR-targeted therapy. Pathol. Res. Pract 2011, 207, 337–342. [Google Scholar]
- Bollag, G.; Hirth, P.; Tsai, J.; Zhang, J.; Ibrahim, P.N.; Cho, H.; Spevak, W.; Zhang, C.; Zhang, Y.; Habets, G.; et al. Clinical efficacy of a RAF inhibitor needs broad target blockade in BRAF-mutant melanoma. Nature 2010, 467, 596–599. [Google Scholar]
- Oberholzer, P.A.; Kee, D.; Dziunycz, P.; Sucker, A.; Kamsukom, N.; Jones, R.; Roden, C.; Chalk, C.J.; Ardlie, K.; Palescandolo, E.; et al. RAS mutations are associated with the development of cutaneous squamous cell tumors in patients treated with RAF inhibitors. J. Clin. Oncol 2011, 30, 316–321. [Google Scholar]
- Su, F.; Viros, A.; Milagre, C.; Trunzer, K.; Bollag, G.; Spleiss, O.; Reis-Filho, J.S.; Kong, X.; Koya, R.C.; Flaherty, K.T.; et al. RAS mutations in cutaneous squamous-cell carcinomas in patients treated with BRAF inhibitors. N. Engl. J. Med 2012, 366, 207–215. [Google Scholar]
- Anforth, R.M.; Blumetti, T.C.; Kefford, R.F.; Sharma, R.; Scolyer, R.A.; Kossard, S.; Long, G.V.; Fernandez-Penas, P. Cutaneous manifestations of dabrafenib (GSK2118436): A selective inhibitor of mutant BRAF in patients with metastatic melanoma. Br. J. Dermatol 2012, 167, 1153–1160. [Google Scholar]
- Poulikakos, P.I.; Zhang, C.; Bollag, G.; Shokat, K.M.; Rosen, N. RAF inhibitors transactivate RAF dimers and ERK signalling in cells with wild-type BRAF. Nature 2010, 464, 427–430. [Google Scholar]
- Hatzivassiliou, G.; Song, K.; Yen, I.; Brandhuber, B.J.; Anderson, D.J.; Alvarado, R.; Ludlam, M.J.; Stokoe, D.; Gloor, S.L.; Vigers, G.; et al. RAF inhibitors prime wild-type RAF to activate the MAPK pathway and enhance growth. Nature 2010, 464, 431–435. [Google Scholar]
- Molina-Arcas, M.; Downward, J. How to fool a wonder drug: Truncate and dimerize. Cancer Cell 2012, 21, 7–9. [Google Scholar]
- Lacouture, M.E.; O’Reilly, K.; Rosen, N.; Solit, D.B. Induction of cutaneous squamous cell carcinomas by RAF inhibitors: Cause for concern? J. Clin. Oncol 2012, 30, 329–330. [Google Scholar]
- Quintanilla, M.; Brown, K.; Ramsden, M.; Balmain, A. Carcinogen-specific mutation and amplification of Ha-ras during mouse skin carcinogenesis. Nature 1986, 322, 78–80. [Google Scholar]
- Abel, E.L.; Angel, J.M.; Kiguchi, K.; DiGiovanni, J. Multi-stage chemical carcinogenesis in mouse skin: Fundamentals and applications. Nat. Protoc 2009, 4, 1350–1362. [Google Scholar]
- Morris, R.J. Keratinocyte stem cells: Targets for cutaneous carcinogens. J. Clin. Invest 2000, 106, 3–8. [Google Scholar]
- Lapouge, G.; Youssef, K.K.; Vokaer, B.; Achouri, Y.; Michaux, C.; Sotiropoulou, P.A.; Blanpain, C. Identifying the cellular origin of squamous skin tumors. Proc. Natl. Acad. Sci. USA 2011, 108, 7431–7436. [Google Scholar]
- White, A.C.; Tran, K.; Khuu, J.; Dang, C.; Cui, Y.; Binder, S.W.; Lowry, W.E. Defining the origins of Ras/p53-mediated squamous cell carcinoma. Proc. Natl. Acad. Sci. USA 2011, 108, 7425–7430. [Google Scholar]
- Kemp, C.J.; Donehower, L.A.; Bradley, A.; Balmain, A. Reduction of p53 gene dosage does not increase initiation or promotion but enhances malignant progression of chemically induced skin tumors. Cell 1993, 74, 813–822. [Google Scholar]
- Argyris, T.S.; Slaga, T.J. Promotion of carcinomas by repeated abrasion in initiated skin of mice. Cancer Res 1981, 41, 5193–5195. [Google Scholar]
- Brown, K.; Strathdee, D.; Bryson, S.; Lambie, W.; Balmain, A. The malignant capacity of skin tumours induced by expression of a mutant H-ras transgene depends on the cell type targeted. Curr. Biol 1998, 8, 516–524. [Google Scholar]
- Bailleul, B.; Surani, M.A.; White, S.; Barton, S.C.; Brown, K.; Blessing, M.; Jorcano, J.; Balmain, A. Skin hyperkeratosis and papilloma formation in transgenic mice expressing a ras oncogene from a suprabasal keratin promoter. Cell 1990, 62, 697–708. [Google Scholar]
- Malanchi, I.; Peinado, H.; Kassen, D.; Hussenet, T.; Metzger, D.; Chambon, P.; Huber, M.; Hohl, D.; Cano, A.; Birchmeier, W.; et al. Cutaneous cancer stem cell maintenance is dependent on beta-catenin signalling. Nature 2008, 452, 650–653. [Google Scholar]
- Lapouge, G.; Beck, B.; Nassar, D.; Dubois, C.; Dekoninck, S.; Blanpain, C. Skin squamous cell carcinoma propagating cells increase with tumour progression and invasiveness. EMBO J 2012, 31, 4563–4575. [Google Scholar]
- Driessens, G.; Beck, B.; Caauwe, A.; Simons, B.D.; Blanpain, C. Defining the mode of tumour growth by clonal analysis. Nature 2012, 488, 527–530. [Google Scholar]
- Lomas, A.; Leonardi-Bee, J.; Bath-Hextall, F. A systematic review of worldwide incidence of nonmelanoma skin cancer. Br. J. Dermatol 2012, 166, 1069–1080. [Google Scholar]
- Kasper, M.; Jaks, V.; Hohl, D.; Toftgard, R. Basal cell carcinoma-molecular biology and potential new therapies. J. Clin. Invest 2012, 122, 455–463. [Google Scholar]
- Epstein, E.H. Basal cell carcinomas: Attack of the hedgehog. Nat. Rev. Cancer 2008, 8, 743–754. [Google Scholar]
- Cirrone, F.; Harris, C.S. Vismodegib and the hedgehog pathway: A new treatment for basal cell carcinoma. Clin. Ther 2012, 34, 2039–2050. [Google Scholar]
- Blanpain, C. Tracing the cellular origin of cancer. Nat. Cell Biol 2013, 15, 126–134. [Google Scholar]
- Youssef, K.K.; Van Keymeulen, A.; Lapouge, G.; Beck, B.; Michaux, C.; Achouri, Y.; Sotiropoulou, P.A.; Blanpain, C. Identification of the cell lineage at the origin of basal cell carcinoma. Nat. Cell Biol 2010, 12, 299–305. [Google Scholar]
- Wong, S.Y.; Reiter, J.F. Wounding mobilizes hair follicle stem cells to form tumors. Proc. Natl. Acad. Sci. USA 2011, 108, 4093–4098. [Google Scholar]
- Kasper, M.; Jaks, V.; Are, A.; Bergström, Å.; Schwäger, A.; Svärd, J.; Teglund, S.; Barker, N.; Toftgård, R. Wounding enhances epidermal tumorigenesis by recruiting hair follicle keratinocytes. Proc. Natl. Acad. Sci. USA 2011, 108, 4099–4104. [Google Scholar]
- Wang, G.Y.; Wang, J.; Mancianti, M.-L.; Epstein, E.H., Jr. Basal cell carcinomas arise from hair follicle stem cells in Ptch1+/− mice. Cancer Cell 2011, 19, 114–124. [Google Scholar]
- Youssef, K.K.; Lapouge, G.; Bouvree, K.; Rorive, S.; Brohee, S.; Appelstein, O.; Larsimont, J.C.; Sukumaran, V.; Van de Sande, B.; Pucci, D.; et al. Adult interfollicular tumour-initiating cells are reprogrammed into an embryonic hair follicle progenitor-like fate during basal cell carcinoma initiation. Nat. Cell Biol 2012, 14, 1282–1294. [Google Scholar]
- Hoseong Yang, S.; Andl, T.; Grachtchouk, V.; Wang, A.; Liu, J.; Syu, L.-J.; Ferris, J.; Wang, T.S.; Glick, A.B.; Millar, S.E.; et al. Pathological responses to oncogenic Hedgehog signaling in skin are dependent on canonical Wnt/[beta]-catenin signaling. Nat. Genet 2008, 40, 1130–1135. [Google Scholar]
- Eberl, M.; Klingler, S.; Mangelberger, D.; Loipetzberger, A.; Damhofer, H.; Zoidl, K.; Schnidar, H.; Hache, H.; Bauer, H.C.; Solca, F.; et al. Hedgehog-EGFR cooperation response genes determine the oncogenic phenotype of basal cell carcinoma and tumour-initiating pancreatic cancer cells. EMBO Mol. Med 2012, 4, 218–233. [Google Scholar] [Green Version]
- Schnidar, H.; Eberl, M.; Klingler, S.; Mangelberger, D.; Kasper, M.; Hauser-Kronberger, C.; Regl, G.; Kroismayr, R.; Moriggl, R.; Sibilia, M.; et al. Epidermal growth factor receptor signaling synergizes with Hedgehog/GLI in oncogenic transformation via activation of the MEK/ERK/JUN pathway. Cancer Res 2009, 69, 1284–1292. [Google Scholar]
- Sugawara, K.; Schneider, M.R.; Dahlhoff, M.; Kloepper, J.E.; Paus, R. Cutaneous consequences of inhibiting EGF receptor signaling in vivo: Normal hair follicle development, but retarded hair cycle induction and inhibition of adipocyte growth in Egfr(Wa5) mice. J. Dermatol. Sci 2010, 57, 155–161. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Doma, E.; Rupp, C.; Baccarini, M. EGFR-Ras-Raf Signaling in Epidermal Stem Cells: Roles in Hair Follicle Development, Regeneration, Tissue Remodeling and Epidermal Cancers. Int. J. Mol. Sci. 2013, 14, 19361-19384. https://doi.org/10.3390/ijms141019361
Doma E, Rupp C, Baccarini M. EGFR-Ras-Raf Signaling in Epidermal Stem Cells: Roles in Hair Follicle Development, Regeneration, Tissue Remodeling and Epidermal Cancers. International Journal of Molecular Sciences. 2013; 14(10):19361-19384. https://doi.org/10.3390/ijms141019361
Chicago/Turabian StyleDoma, Eszter, Christian Rupp, and Manuela Baccarini. 2013. "EGFR-Ras-Raf Signaling in Epidermal Stem Cells: Roles in Hair Follicle Development, Regeneration, Tissue Remodeling and Epidermal Cancers" International Journal of Molecular Sciences 14, no. 10: 19361-19384. https://doi.org/10.3390/ijms141019361