Ochratoxin A Inhibits Mouse Embryonic Development by Activating a Mitochondrion-Dependent Apoptotic Signaling Pathway

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
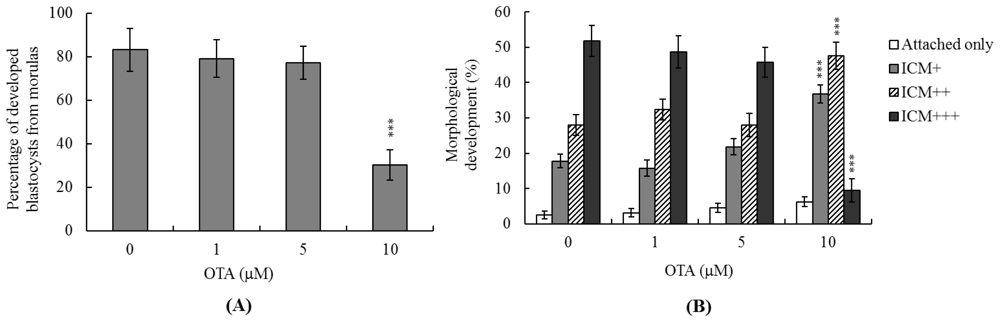
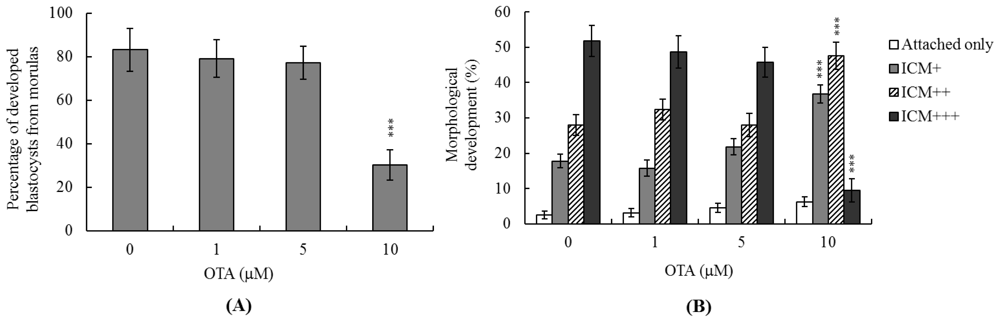
2.1. Effects of OTA on Mouse Blastocysts
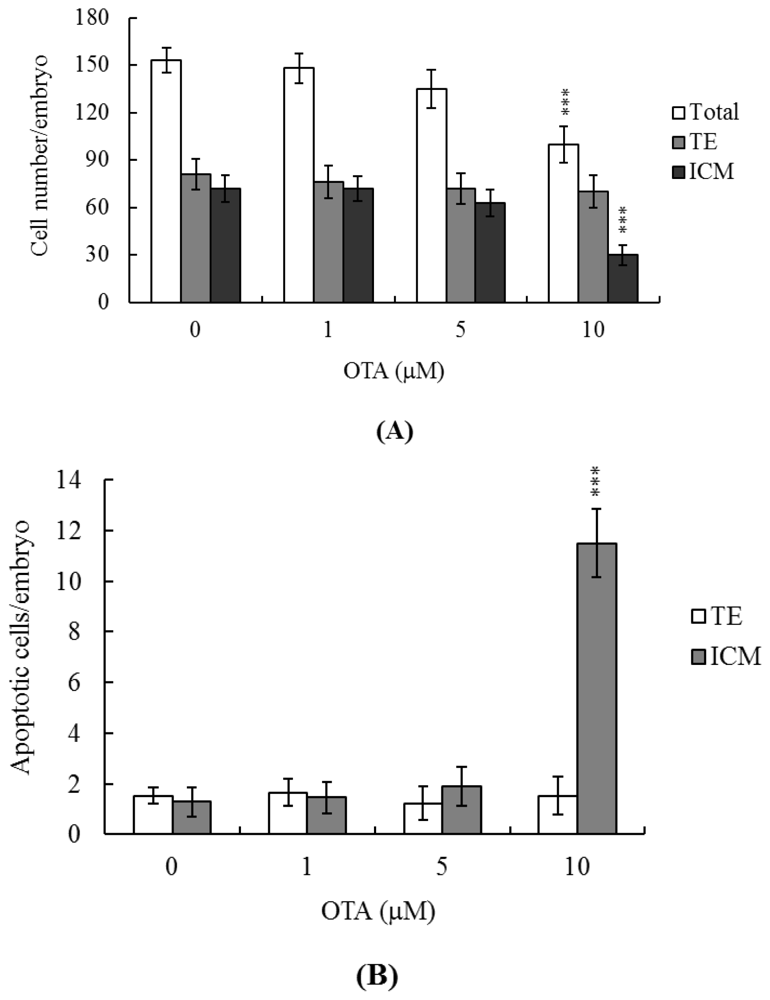
2.2. Effects of OTA on Cell Proliferation
2.3. Effects of OTA on Mouse Embryonic Developmental Potential in Vitro
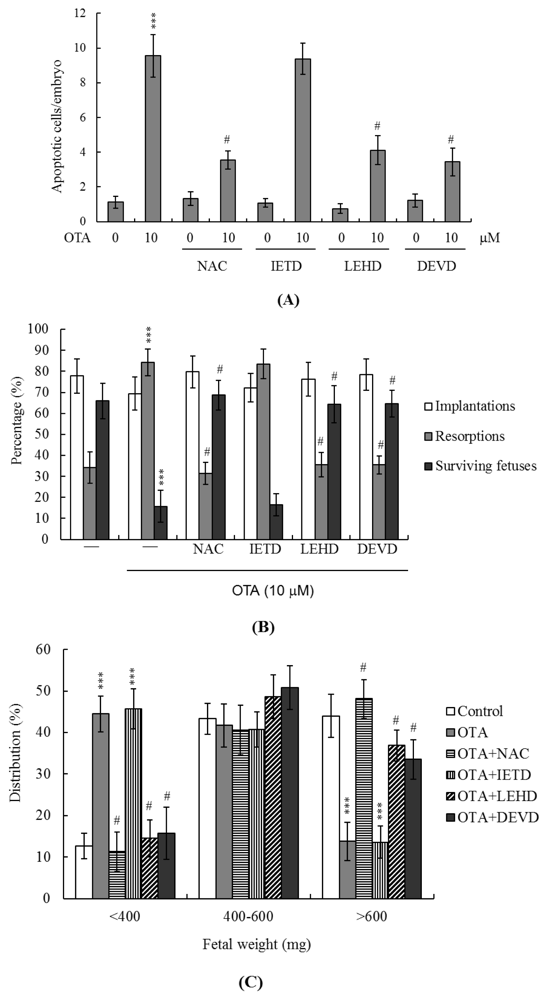
2.4. Effects of OTA on the Developmental Potential of Blastocysts in Vivo
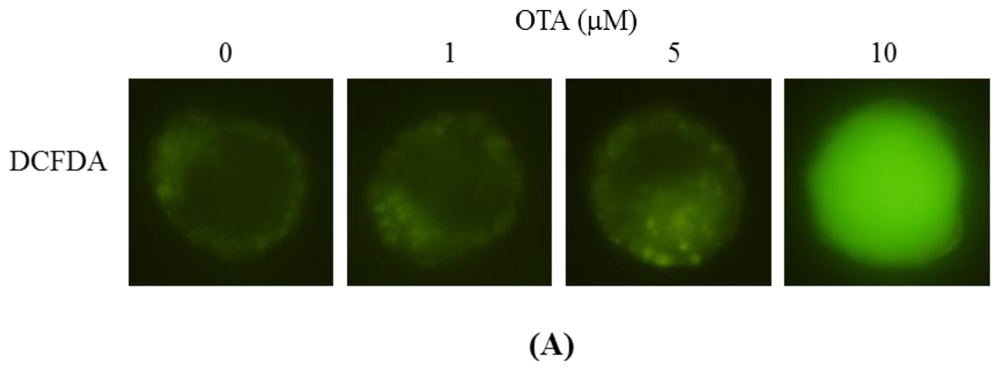
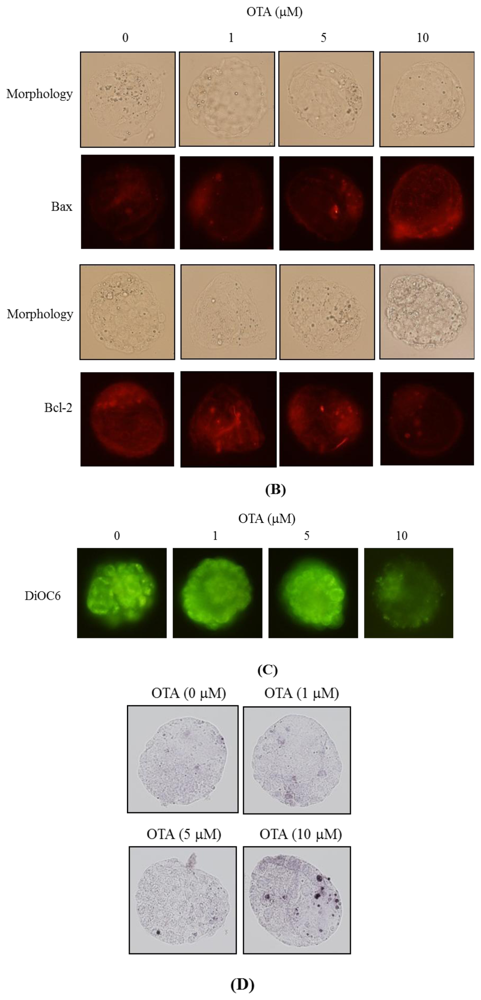
2.5. ROS Generation and Mitochondrion-Dependent Apoptotic Processes Are Involved in Blastocyst Death Induced by OTA
3. Discussion
4. Experimental Section
4.1. Materials
4.2. Collection of Mouse Morulas and Blastocysts
4.3. OTA Treatment and TUNEL Assay
4.4. OTA Treatment and Cell Proliferation
4.5. Annexin V Staining
4.6. Morphological Analysis of Embryonic Development
4.7. Blastocyst Development following Embryo Transfer
4.8. Immunofluorescent Cell Stain
4.9. Statistics
5. Conclusions
Acknowledgment
- Conflict of InterestThe authors declare no conflicts of interest.
References
- Council of Agricultural Science and Technology (CAST), Mycotoxins: Risks in Plant, Animal, and Human Systems; CAST: Ames, IA, USA, 2003.
- IPCS. Safety evaluation of certain mycotoxins in food. WHO Food Addit. Ser. 2001, 47, 103–415.
- Walker, R. Risk assessment of ochratoxin: Current views of the European Scientific Committee on Food, the JECFA and the Codex Committee on Food Additives and Contaminants. Adv. Exp. Med. Biol 2002, 504, 249–255. [Google Scholar]
- Pfohl-Leszkowicz, A.; Manderville, R.A. Ochratoxin A: An overview on toxicity and carcinogenicity in animals and humans. Mol. Nutr. Food Res 2007, 51, 61–99. [Google Scholar]
- Zhang, X.; Boesch-Saadatmandi, C.; Lou, Y.; Wolffram, S.; Huebbe, P.; Rimbach, G. Ochratoxin A induces apoptosis in neuronal cells. Genes Nutr 2009, 4, 41–48. [Google Scholar]
- Sava, V.; Reunova, O.; Velasquez, A.; Harbison, R.; Sanchez-Ramos, J. Acute neurotoxic effects of the fungal metabolite ochratoxin-A. Neurotoxicology 2006, 27, 82–92. [Google Scholar]
- Kanisawa, M.; Suzuki, S. Induction of renal and hepatic tumors in mice by ochratoxin A, a mycotoxin. Gann = Gan 1978, 69, 599–600. [Google Scholar]
- Kanisawa, M. Pathogenesis of human cancer development due to environmental factors. Gan No Rinsho 1984, 30, 1445–1456. [Google Scholar]
- Boorman, G. NTP Technical Report on the Toxicology and Carcinogenesis Studies of Ochratoxin A (CAS No. 303-47-9) in F344/N Rats (Gavage Studies), NTP TR 358, NIH Publication No. 89-2813; National Institute of Health: North Carolina, NC, USA, 1989. [Google Scholar]
- Bouaziz, C.; Sharaf el dein, O.; Martel, C.; Golli, E.E.; Abid-Essefi, S.; Brenner, C.; Lemaire, C.; Bacha, H. Molecular events involved in ochratoxin A induced mitochondrial pathway of apoptosis, modulation by Bcl-2 family members. Environ. Toxicol 2011, 26, 579–590. [Google Scholar]
- Pfohl-Leszkowicz, A.; Manderville, R.A. An update on direct genotoxicity as a molecular mechanism of ochratoxin a carcinogenicity. Chem. Res. Toxicol 2012, 25, 252–262. [Google Scholar]
- Hardy, K. Cell death in the mammalian blastocyst. Mol. Hum. Reprod 1997, 3, 919–925. [Google Scholar]
- Hardy, K.; Stark, J.; Winston, R.M. Maintenance of the inner cell mass in human blastocysts from fragmented embryos. Biol. Reprod 2003, 68, 1165–1169. [Google Scholar]
- Byrne, A.T.; Southgate, J.; Brison, D.R.; Leese, H.J. Analysis of apoptosis in the preimplantation bovine embryo using TUNEL. J. Reprod. Fertil 1999, 117, 97–105. [Google Scholar]
- Hsuuw, Y.D.; Chang, C.K.; Chan, W.H.; Yu, J.S. Curcumin prevents methylglyoxal-induced oxidative stress and apoptosis in mouse embryonic stem cells and blastocysts. J. Cell. Physiol 2005, 205, 379–386. [Google Scholar]
- Chan, W.H. Ginkgolide B induces apoptosis and developmental injury in mouse embryonic stem cells and blastocysts. Hum. Reprod 2006, 21, 2985–2995. [Google Scholar]
- Chan, W.H. Impact of genistein on maturation of mouse oocytes, fertilization, and fetal development. Reprod. Toxicol 2009, 28, 52–58. [Google Scholar]
- Chan, W.H. Effects of citrinin on maturation of mouse oocytes, fertilization, and fetal development in vitro and in vivo. Toxicol. Lett 2008, 180, 28–32. [Google Scholar]
- Chan, W.H. Ginkgolides induce apoptosis and decrease cell numbers in mouse blastocysts. Biochem. Biophys. Res. Commun 2005, 338, 1263–1267. [Google Scholar]
- Jacobson, M.D. Reactive oxygen species and programmed cell death. Trends Biochem. Sci 1996, 21, 83–86. [Google Scholar]
- Thompson, C.B. Apoptosis in the pathogenesis and treatment of disease. Science 1995, 267, 1456–1462. [Google Scholar]
- Brill, A.; Torchinsky, A.; Carp, H.; Toder, V. The role of apoptosis in normal and abnormal embryonic development. J. Assist. Reprod. Genet 1999, 16, 512–519. [Google Scholar]
- Lotz, K.; Proff, P.; Bienengraeber, V.; Fanghaenel, J.; Gedrange, T.; Weingaertner, J. Apoptosis as a creative agent of embryonic development of bucca, mentum and nasolacrimal duct. An in vivo study in rats. J. Craniomaxillofac. Surg 2006, 34, S8–S13. [Google Scholar]
- Weingaertner, J.; Proff, P.; Bienengraeber, V.; Gedrange, T.; Fanghaenel, J.; Lotz, K. In vivo study of apoptosis as a creative agent of embryonic development of the primary nasal duct in rats. J. Craniomaxillofac. Surg 2006, 34, S3–S7. [Google Scholar]
- Huang, F.J.; Shen, C.C.; Chang, S.Y.; Wu, T.C.; Hsuuw, Y.D. Retinoic acid decreases the viability of mouse blastocysts in vitro. Hum. Reprod 2003, 18, 130–136. [Google Scholar]
- Shang, E.H.; Wu, R.S. Aquatic hypoxia is a teratogen and affects fish embryonic development. Environ. Sci. Technol 2004, 38, 4763–4767. [Google Scholar]
- Detmar, J.; Rabaglino, T.; Taniuchi, Y.; Oh, J.; Acton, B.M.; Benito, A.; Nunez, G.; Jurisicova, A. Embryonic loss due to exposure to polycyclic aromatic hydrocarbons is mediated by Bax. Apoptosis 2006, 11, 1413–1425. [Google Scholar]
- Li, J.; Yin, S.; Dong, Y.; Fan, L.; Hu, H. p53 activation inhibits ochratoxin A-induced apoptosis in monkey and human kidney epithelial cells via suppression of JNK activation. Biochem. Biophys. Res. Commun 2011, 411, 458–463. [Google Scholar]
- Golli-Bennour, E.E.; Kouidhi, B.; Bouslimi, A.; Abid-Essefi, S.; Hassen, W.; Bacha, H. Cytotoxicity and genotoxicity induced by aflatoxin B1, ochratoxin A, and their combination in cultured Vero cells. J. Biochem. Mol. Toxicol 2010, 24, 42–50. [Google Scholar]
- Klaric, M.S.; Zeljezic, D.; Rumora, L.; Peraica, M.; Pepeljnjak, S.; Domijan, A.M. A potential role of calcium in apoptosis and aberrant chromatin forms in porcine kidney PK15 cells induced by individual and combined ochratoxin A and citrinin. Arch. Toxicol 2012, 86, 97–107. [Google Scholar]
- Sauvant, C.; Holzinger, H.; Gekle, M. Proximal tubular toxicity of ochratoxin A is amplified by simultaneous inhibition of the extracellular signal-regulated kinases 1/2. J. Pharmacol. Exp. Ther 2005, 313, 234–241. [Google Scholar]
- Hood, R.D.; Naughton, M.J.; Hayes, A.W. Prenatal effects of Ochratoxin A in hamsters. Teratology 1976, 13, 11–14. [Google Scholar]
- Mayura, K.; Reddy, R.V.; Hayes, A.W.; Berndt, W.O. Embryocidal, fetotoxic and teratogenic effects of ochratoxin A in rats. Toxicology 1982, 25, 175–185. [Google Scholar]
- Mayura, K.; Edwards, J.F.; Maull, E.A.; Phillips, T.D. The effects of ochratoxin A on postimplantation rat embryos in culture. Arch. Environ. Contam. Toxicol 1989, 18, 411–415. [Google Scholar]
- Chan, W.H.; Shiao, N.H. Effect of citrinin on mouse embryonic development in vitro and in vivo. Reprod. Toxicol 2007, 24, 120–125. [Google Scholar]
- Chan, W.H. Citrinin induces apoptosis via a mitochondria-dependent pathway and inhibition of survival signals in embryonic stem cells, and causes developmental injury in blastocysts. Biochem. J 2007, 404, 317–326. [Google Scholar]
- Chan, W.H. Citrinin induces apoptosis in mouse embryonic stem cells. IUBMB Life 2008, 60, 171–179. [Google Scholar]
- Yu, F.; Watts, R.N.; Zhang, X.D.; Borrow, J.M.; Hersey, P. Involvement of BH3-only proapoptotic proteins in mitochondrial-dependent Phenoxodiol-induced apoptosis of human melanoma cells. Anticancer Drugs 2006, 17, 1151–1161. [Google Scholar]
- Criollo, A.; Galluzzi, L.; Chiara Maiuri, M.; Tasdemir, E.; Lavandero, S.; Kroemer, G. Mitochondrial control of cell death induced by hyperosmotic stress. Apoptosis 2007, 12, 3–18. [Google Scholar]
- Yoon, S.; Cong, W.T.; Bang, Y.; Lee, S.N.; Yoon, C.S.; Kwack, S.J.; Kang, T.S.; Lee, K.Y.; Choi, J.K.; Choi, H.J. Proteome response to ochratoxin A-induced apoptotic cell death in mouse hippocampal HT22 cells. Neurotoxicology 2009, 30, 666–676. [Google Scholar]
- Sava, V.; Velasquez, A.; Song, S.; Sanchez-Ramos, J. Adult hippocampal neural stem/progenitor cells in vitro are vulnerable to the mycotoxin ochratoxin-A. Toxicol. Sci 2007, 98, 187–197. [Google Scholar]
- El Golli Bennour, E.; Rodriguez-Enfedaque, A.; Bouaziz, C.; Ladjimi, M.; Renaud, F.; Bacha, H. Toxicities induced in cultured human hepatocarcinoma cells exposed to ochratoxin A: Oxidative stress and apoptosis status. J. Biochem. Mol. Toxicol 2009, 23, 87–96. [Google Scholar]
- Wangikar, P.B.; Dwivedi, P.; Sinha, N. Effect in rats of simultaneous prenatal exposure to ochratoxin A and aflatoxin B1. I. Maternal toxicity and fetal malformations. Birth Defects Res. B 2004, 71, 343–351. [Google Scholar]
- Biro, K.; Barna-Vetro, I.; Pecsi, T.; Szabo, E.; Winkler, G.; Fink-Gremmels, J.; Solti, L. Evaluation of spermatological parameters in ochratoxin A—Challenged boars. Theriogenology 2003, 60, 199–207. [Google Scholar]
- Patil, R.D.; Dwivedi, P.; Sharma, A.K. Critical period and minimum single oral dose of ochratoxin A for inducing developmental toxicity in pregnant Wistar rats. Reprod. Toxicol 2006, 22, 679–687. [Google Scholar]
- Jennings-Gee, J.E.; Tozlovanu, M.; Manderville, R.; Miller, M.S.; Pfohl-Leszkowicz, A.; Schwartz, G.G. Ochratoxin A: In utero exposure in mice induces adducts in testicular DNA. Toxins 2010, 2, 1428–1444. [Google Scholar]
- Cross, J.C.; Werb, Z.; Fisher, S.J. Implantation and the placenta: key pieces of the development puzzle. Science 1994, 266, 1508–1518. [Google Scholar]
- Pampfer, S.; de Hertogh, R.; Vanderheyden, I.; Michiels, B.; Vercheval, M. Decreased inner cell mass proportion in blastocysts from diabetic rats. Diabetes 1990, 39, 471–476. [Google Scholar]
- Kelly, S.M.; Robaire, B.; Hales, B.F. Paternal cyclophosphamide treatment causes postimplantation loss via inner cell mass-specific cell death. Teratology 1992, 45, 313–318. [Google Scholar]
- Tam, P.P. Postimplantation development of mitomycin C-treated mouse blastocysts. Teratology 1988, 37, 205–212. [Google Scholar]
- Chen, C.C.; Chan, W.H. Impact effects of puerarin on mouse embryonic development. Reprod. Toxicol 2009, 28, 530–535. [Google Scholar]
- Long, L.H.; Clement, M.V.; Halliwell, B. Artifacts in cell culture: Rapid generation of hydrogen peroxide on addition of (−)-epigallocatechin, (−)-epigallocatechin gallate, (+)-catechin, and quercetin to commonly used cell culture media. Biochem. Biophys. Res. Commun 2000, 273, 50–53. [Google Scholar]
- Halliwell, B. Oxidative stress in cell culture: An under-appreciated problem? FEBS Lett 2003, 540, 3–6. [Google Scholar]
- Liu, J.; Wang, Y.; Cui, J.; Xing, L.; Shen, H.; Wu, S.; Lian, H.; Wang, J.; Yan, X.; Zhang, X. Ochratoxin A induces oxidative DNA damage and G1 phase arrest in human peripheral blood mononuclear cells in vitro. Toxicol. Lett 2012, 211, 164–171. [Google Scholar]
- Kumar, R.; Ansari, K.M.; Chaudhari, B.P.; Dhawan, A.; Dwivedi, P.D.; Jain, S.K.; Das, M. Topical application of ochratoxin a causes DNA damage and tumor initiation in mouse skin. PLoS One 2012, 7, e47280. [Google Scholar]
- Hardy, K.; Handyside, A.H.; Winston, R.M. The human blastocyst: Cell number, death and allocation during late preimplantation development in vitro. Development 1989, 107, 597–604. [Google Scholar]
- Gardner, R.L.; Davies, T.J. Lack of coupling between onset of giant transformation and genome endoreduplication in the mural trophectoderm of the mouse blastocyst. J. Exp. Zool 1993, 265, 54–60. [Google Scholar]
- Huang, F.J.; Wu, T.C.; Tsai, M.Y. Effect of retinoic acid on implantation and post-implantation development of mouse embryos in vitro. Hum. Reprod 2001, 16, 2171–2176. [Google Scholar]
- Witschi, E. Characterization of Developmental Stages. Part II. Rat. In Biology Data Book, 2nd ed; Federation of American Societies of Experimental Biologies: Washington DC, USA, 1972; pp. 178–180. [Google Scholar]
- Armant, D.R.; Kaplan, H.A.; Lennarz, W.J. Fibronectin and laminin promote in vitro attachment and outgrowth of mouse blastocysts. Dev. Biol 1986, 116, 519–523. [Google Scholar]
- Pampfer, S.; Wuu, Y.D.; Vanderheyden, I.; de Hertogh, R. In vitro study of the carry-over effect associated with early diabetic embryopathy in the rat. Diabetologia 1994, 37, 855–862. [Google Scholar]
- Chan, W.H. Embryonic toxicity of sanguinarine through apoptotic processes in mouse blastocysts. Toxicol. Lett 2011, 205, 285–292. [Google Scholar]



© 2013 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Hsuuw, Y.-D.; Chan, W.-H.; Yu, J.-S. Ochratoxin A Inhibits Mouse Embryonic Development by Activating a Mitochondrion-Dependent Apoptotic Signaling Pathway. Int. J. Mol. Sci. 2013, 14, 935-953. https://doi.org/10.3390/ijms14010935
Hsuuw Y-D, Chan W-H, Yu J-S. Ochratoxin A Inhibits Mouse Embryonic Development by Activating a Mitochondrion-Dependent Apoptotic Signaling Pathway. International Journal of Molecular Sciences. 2013; 14(1):935-953. https://doi.org/10.3390/ijms14010935
Chicago/Turabian StyleHsuuw, Yan-Der, Wen-Hsiung Chan, and Jau-Song Yu. 2013. "Ochratoxin A Inhibits Mouse Embryonic Development by Activating a Mitochondrion-Dependent Apoptotic Signaling Pathway" International Journal of Molecular Sciences 14, no. 1: 935-953. https://doi.org/10.3390/ijms14010935