Catalysis of Transesterification Reactions by a Self-Assembled Nanosystem


Abstract
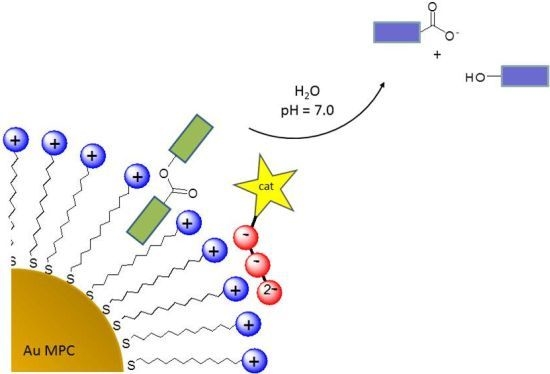
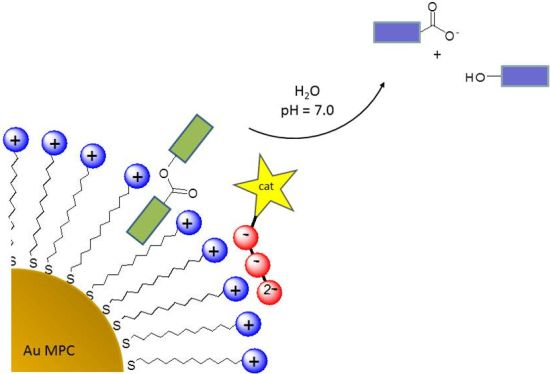
:
1. Introduction
2. Results and Discussion
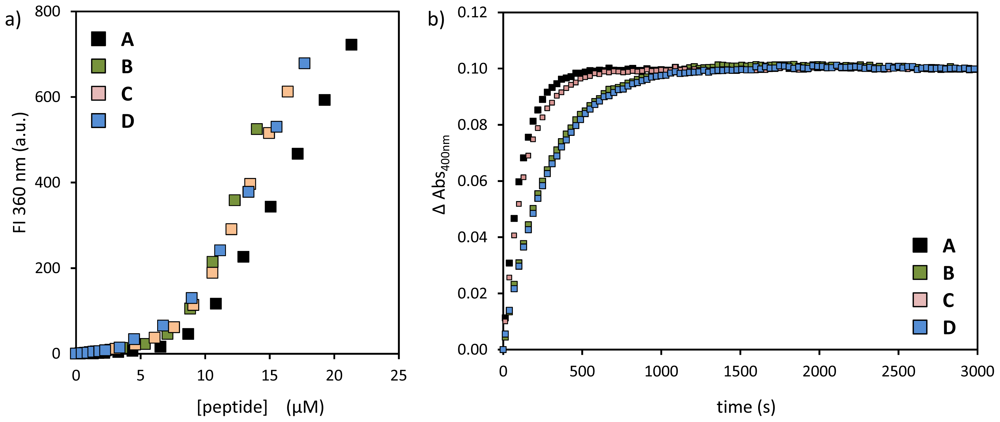
2.1. Structural Modifications in the Catalytic Peptide
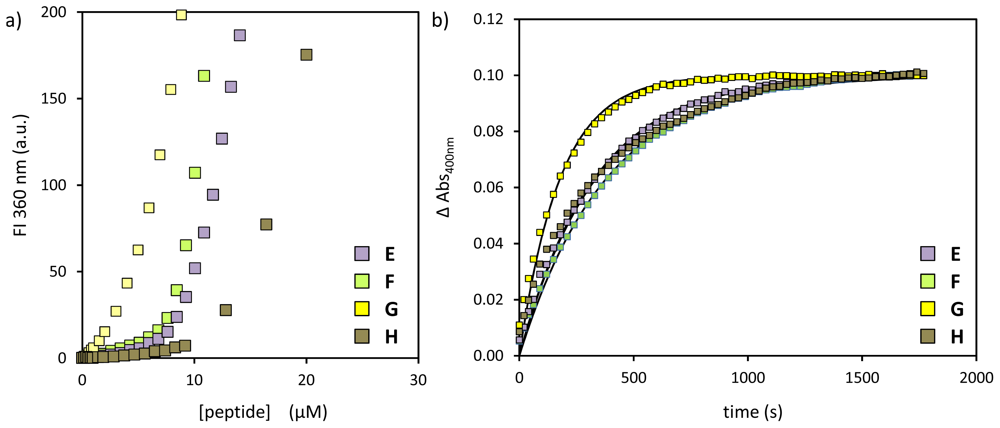
2.2. Substrate Variation
3. Conclusions
Supporting Information
ijms-14-02011-s001.pdfAcknowledgments
- Supporting InformationSupporting information regarding instrumentation, experimental protocols and characterization data for peptides B–H is available.
References and Notes
- Van Leeuwen, P.W.N.M. Supramolecular Catalysis; Wiley-VCH: Weinheim, Germany, 2008. [Google Scholar]
- Gasparini, G.; Dal Molin, M.; Prins, L.J. Dynamic approaches towards catalyst discovery. Eur. J. Org. Chem 2010, 2010, 2429–2440. [Google Scholar]
- Meeuwissen, J.; Reek, J.N.H. Supramolecular catalysis beyond enzyme mimics. Nature Chem 2010, 2, 615–621. [Google Scholar]
- Wiester, M.J.; Ulmann, P.A.; Mirkin, C.A. Enzyme mimics based upon supramolecular coordination chemistry. Angew. Chem. Int. Ed 2011, 50, 114–137. [Google Scholar]
- Breit, B. Supramolecular approaches to generate libraries of chelating bidentate ligands for homogeneous catalysis. Angew. Chem. Int. Ed 2005, 44, 6816–6825. [Google Scholar]
- Sandee, A.J.; Reek, J.N.H. Bidentate ligands by supramolecular chemistry—The future for catalysis? Dalton Trans. 2006, 3385–3391. [Google Scholar]
- Wilkinson, M.J.; van Leeuwen, P.; Reek, J.N.H. New directions in supramolecular transition metal catalysis. Org. Biomol. Chem 2005, 3, 2371–2383. [Google Scholar]
- Koblenz, T.S.; Wassenaar, J.; Reek, J.N.H. Reactivity within a confined self-assembled nanospace. Chem. Soc. Rev 2008, 37, 247–262. [Google Scholar]
- Sanders, J.K.M. Supramolecular catalysis in transition. Chem. Eur. J 1998, 4, 1378–1383. [Google Scholar]
- Prins, L.J.; Mancin, F.; Scrimin, P. Multivalent cooperative catalysts. Curr. Org. Chem 2009, 13, 1050–1064. [Google Scholar]
- Mancin, F.; Scrimin, P.; Tecilla, P.; Tonellato, U. Amphiphilic metalloaggregates: Catalysis, transport and sensing. Coord. Chem. Rev 2009, 253, 2150–2165. [Google Scholar]
- Schatz, A.; Reiser, O.; Stark, W.J. Nanoparticles as semi-heterogeneous catalyst supports. Chem. Eur. J 2010, 16, 8950–8967. [Google Scholar]
- Pieters, G.; Prins, L.J. Catalytic self-assembled monolayers on gold nanoparticles. New. J. Chem 2012, 36, 1931–1939. [Google Scholar]
- Bonomi, R.; Cazzolaro, A.; Prins, L.J. Assessment of the morphology of mixed SAMs on Au nanoparticles using a fluorescent probe. Chem. Commun. 2011, 47, 445–447. [Google Scholar]
- You, C.C.; Miranda, O.R.; Gider, B.; Ghosh, P.S.; Kim, I.B.; Erdogan, B.; Krovi, S.A.; Bunz, U.H.F.; Rotello, V.M. Detection and identification of proteins using nanoparticle-fluorescent polymer ‘chemical nose’ sensors. Nature Nanotechnol 2007, 2, 318–323. [Google Scholar]
- Ghosh, P.S.; Verma, A.; Rotello, V.M. Binding and templation of nanoparticle receptors to peptide alpha-helices through surface recognition. Chem. Commun 2007, 47, 2796–2798. [Google Scholar]
- De, M.; Ghosh, P.S.; Rotello, V.M. Applications of nanoparticles in biology. Adv. Mater 2008, 20, 4225–4241. [Google Scholar]
- De, M.; Rana, S.; Akpinar, H.; Miranda, O.R.; Arvizo, R.R.; Bunz, U.H.F.; Rotello, V.M. Sensing of proteins in human serum using conjugates of nanoparticles and green fluorescent protein. Nature Chem 2009, 1, 461–465. [Google Scholar]
- Bonomi, R.; Cazzolaro, A.; Sansone, A.; Scrimin, P.; Prins, L.J. Detection of enzyme activity through catalytic signal amplification with functionalized gold nanoparticles. Angew. Chem. Int. Ed 2011, 50, 2307–2312. [Google Scholar]
- Pieters, G.; Cazzolaro, A.; Bonomi, R.; Prins, L.J. Self-assembly and selective exchange of oligoanions on the surface of monolayer protected au nanoparticles in water. Chem. Commun 2012, 48, 1916–1918. [Google Scholar]
- Zaramella, D.; Scrimin, P.; Prins, L.J. Self-assembly of a catalytic multivalent peptide-nanoparticle complex. J. Am. Chem. Soc 2012, 134, 8396–8399. [Google Scholar]
- Guarise, C.; Manea, F.; Zaupa, G.; Pasquato, L.; Prins, L.J.; Scrimin, P. Cooperative nanosystems. J. Pep. Sci 2008, 14, 174–183. [Google Scholar]
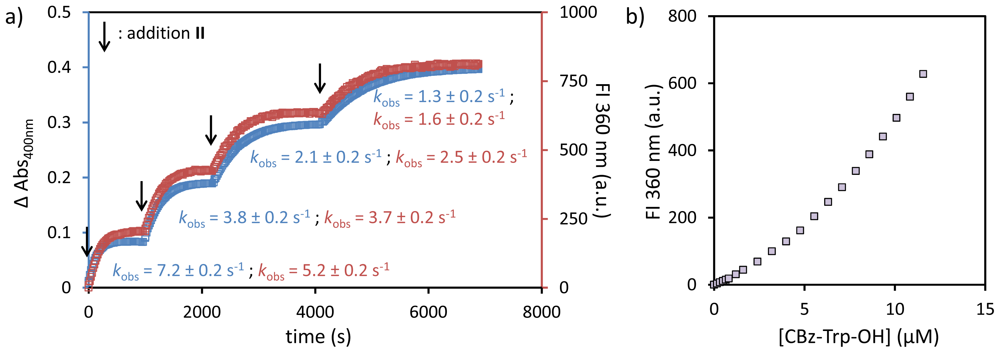
- The SSC and kobs values for peptide A are slightly different then reported before (9.5 against 11 μM for SSC; 8.9 × 10−3 against 6.0 × 10−3·s−1 for kobs), which is ascribed to the use of a different batch of Au MPC 1 and slight variations in the head group concentration).
- Pezzato, C.; Lee, B.; Severin, K.; Prins, L.J. Pattern-based sensing of nucleotides using functionalized gold nanoparticles. Chem. Commun 2013, 49, 469–471. [Google Scholar]
- Pieters, G.; Pezzato, C.; Prins, L.J. Controlling supramolecular complex formation on the surface of a monolayer protected gold nanoparticle in water. Langmuir 2013. [Google Scholar] [CrossRef]
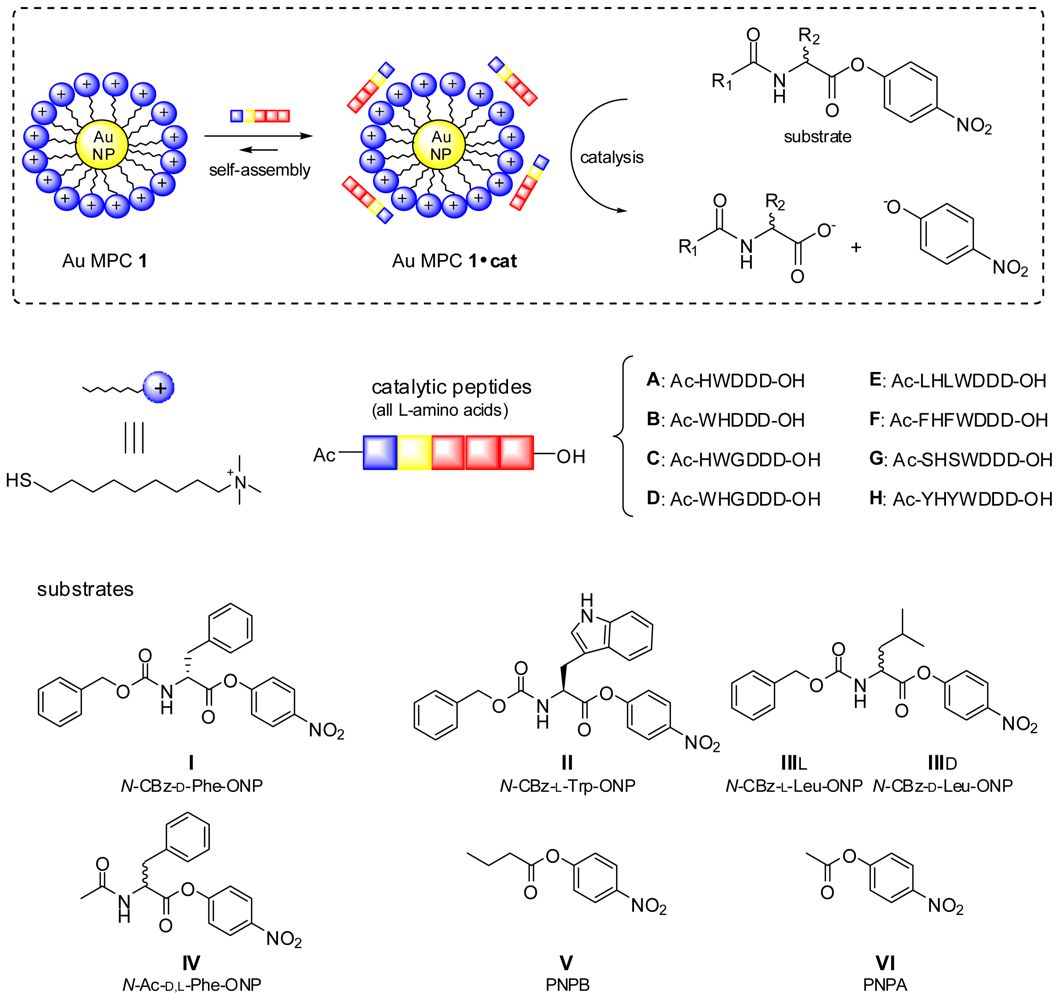





{kind=link}
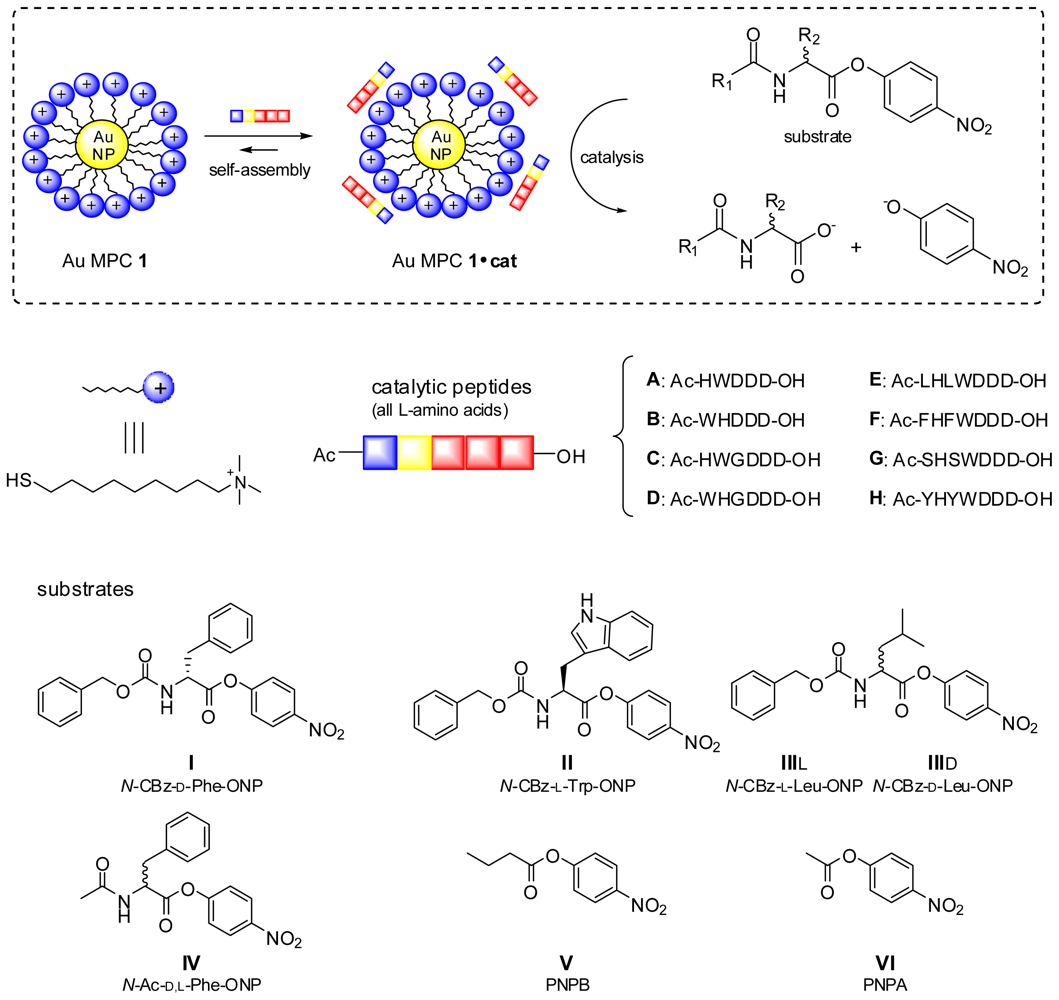{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| peptide | SSC (μM) | kobs (×10−3·s−1) | k2 (×103 L·mol−1·s−1) |
|---|---|---|---|
| A | 9.5 ± 0.3 | 8.9 | 2.1 |
| B | 8.6 ± 0.2 | 3.6 | 0.8 |
| C | 8.5 ± 0.2 | 7.1 | 1.7 |
| D | 7.8 ± 0.2 | 3.5 | 0.9 |
| peptide | SSC (μM) | kobs (×10−3·s−1) | k2 (×103 L·mol−1·s−1) |
|---|---|---|---|
| E | 8.6 ± 0.3 | 3.0 | 620 |
| F | 9.1 ± 0.2 | 2.6 | 490 |
| G | 4.4 ± 0.2 | 5.6 | 1310 |
| H | 15.9 ± 0.2 | 3.0 | 380 |
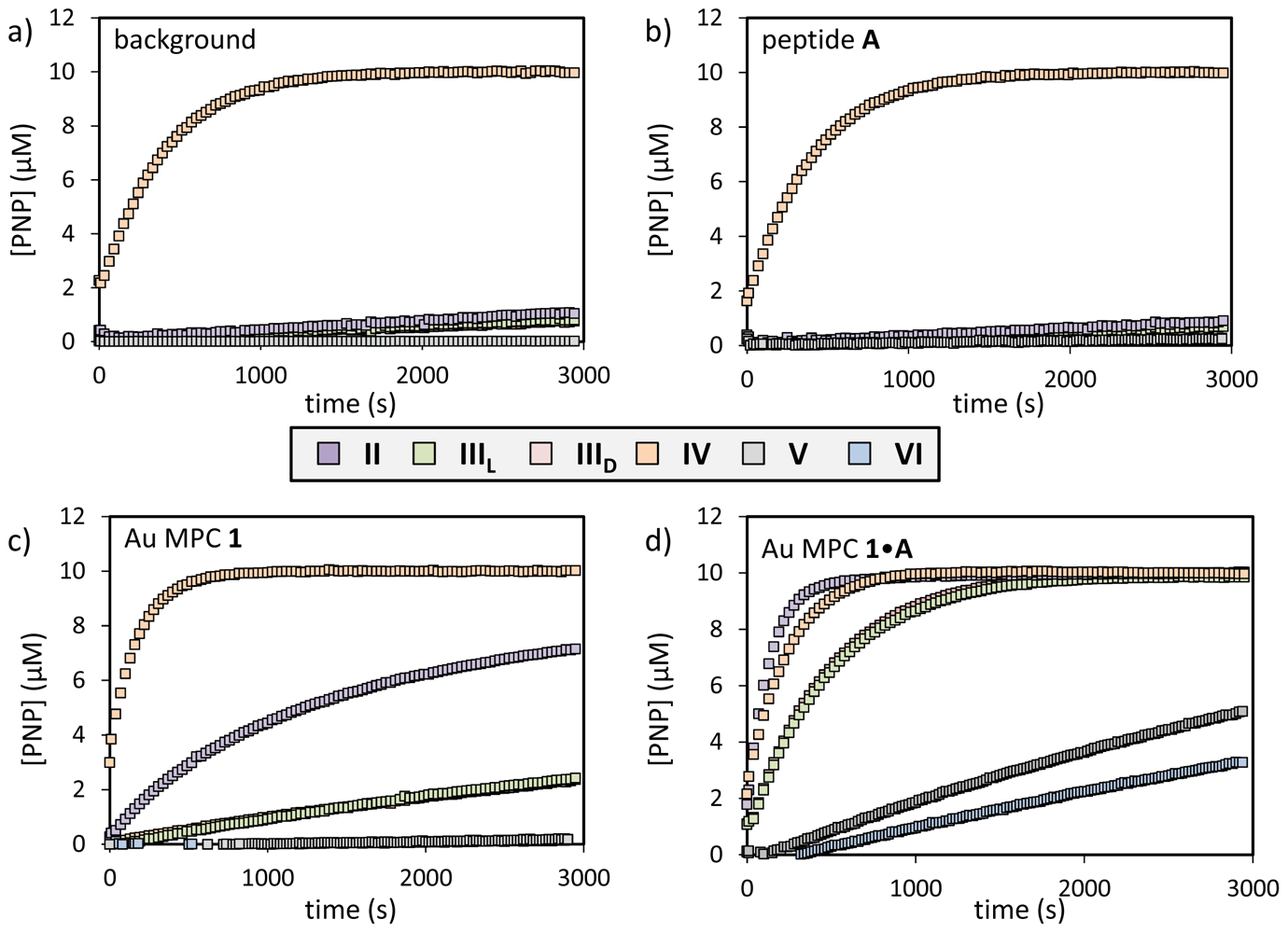
| substrate | background (×10−3·s−1) | catalyst A (×10−3·s−1) | Au MPC 1 (×10−3·s−1) | Au MPC 1•A (×10−3·s−1) | Au MPC 1•WDDD (×10−3·s−1) | Ratio a |
|---|---|---|---|---|---|---|
| I | 0.05 | 0.05 | 1.0 | 8.9 | 0.3 | 30 |
| II | 0.01 | 0.03 | 0.9 | 7.2 | 0.4 | 18 |
| IIIL | 0.05 | 0.04 | 0.2 | 2.0 | 0.1 | 20 |
| IIID | 0.05 | 0.04 | 0.2 | 2.0 | 0.1 | 20 |
| IV | 2.5 | 2.5 | 5.5 | 4.2 | 2.5 | 1.7 |
| V | 0.01 | - | 0.01 | 0.1 | 0.01 | 10b |
| VI | 0.01 | 0.01 | 0.01 | 0.2 | 0.01 | 20b |
© 2013 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Zaramella, D.; Scrimin, P.; Prins, L.J. Catalysis of Transesterification Reactions by a Self-Assembled Nanosystem. Int. J. Mol. Sci. 2013, 14, 2011-2021. https://doi.org/10.3390/ijms14012011
Zaramella D, Scrimin P, Prins LJ. Catalysis of Transesterification Reactions by a Self-Assembled Nanosystem. International Journal of Molecular Sciences. 2013; 14(1):2011-2021. https://doi.org/10.3390/ijms14012011
Chicago/Turabian StyleZaramella, Davide, Paolo Scrimin, and Leonard J. Prins. 2013. "Catalysis of Transesterification Reactions by a Self-Assembled Nanosystem" International Journal of Molecular Sciences 14, no. 1: 2011-2021. https://doi.org/10.3390/ijms14012011