Inhibition of GTRAP3-18 May Increase Neuroprotective Glutathione (GSH) Synthesis

{kind=link}
{kind=link}
Abstract
:1. Introduction
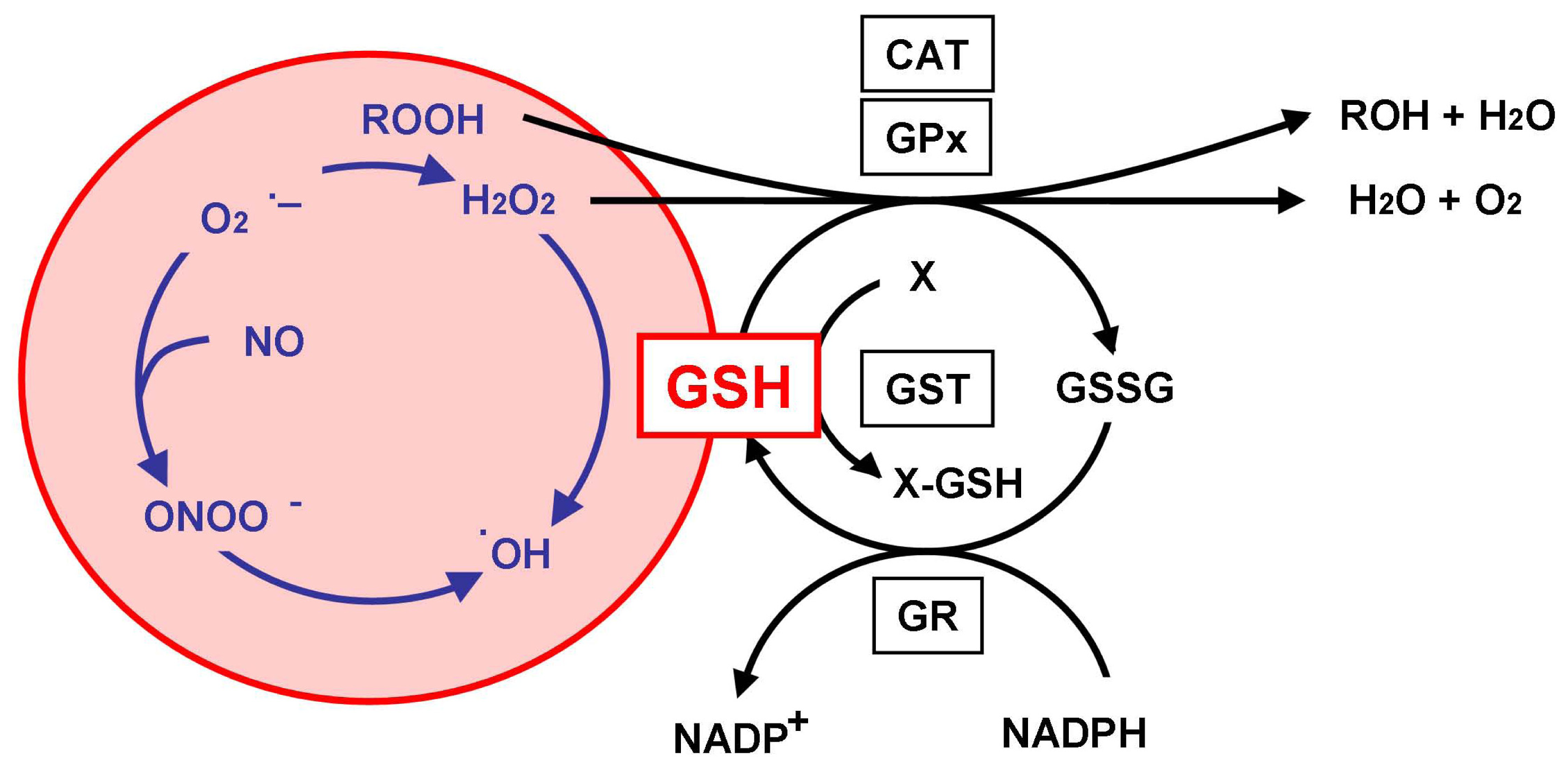
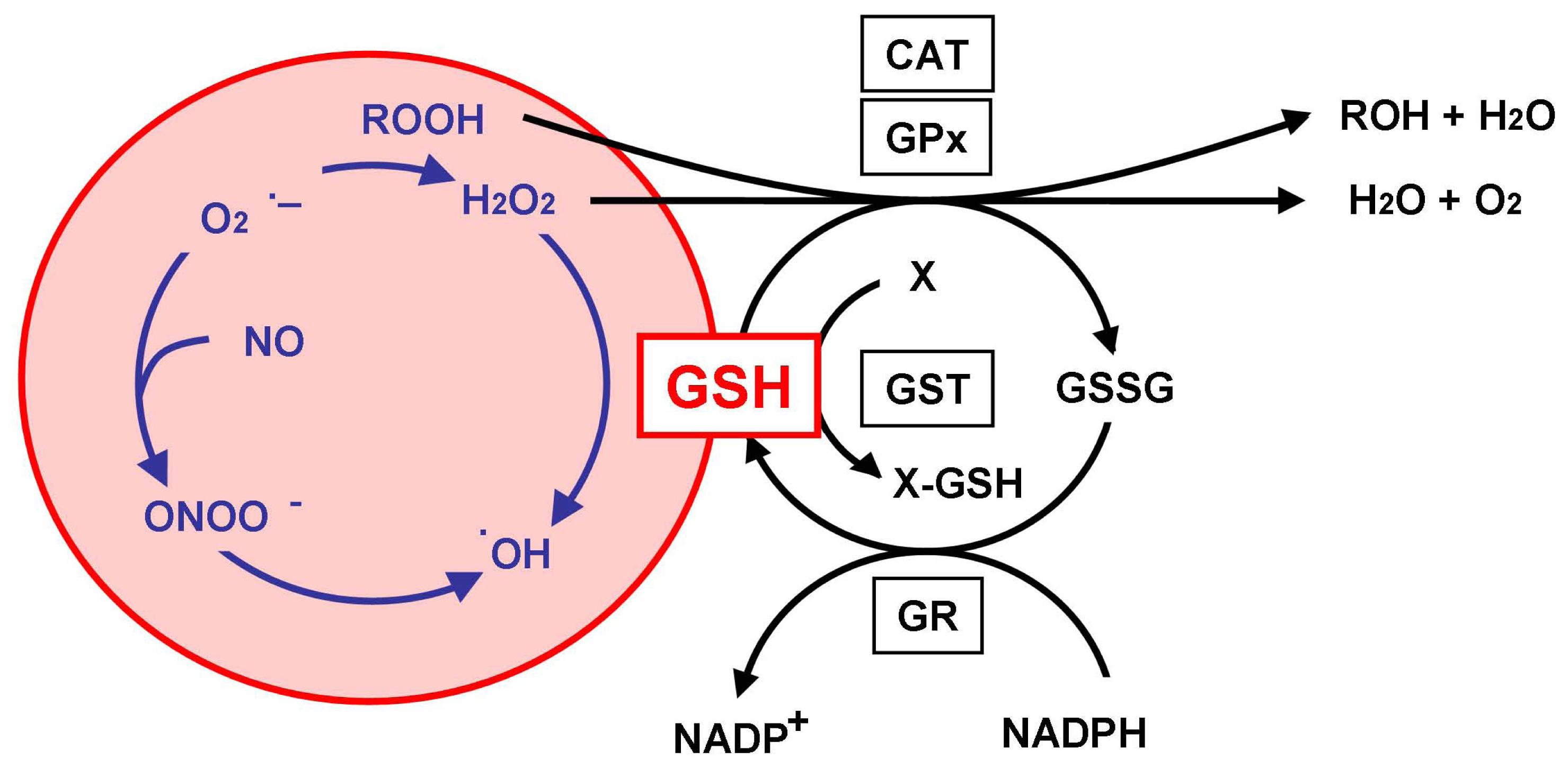
2. Glutathione as an Antioxidant
3. Glutathione Depletion and Neurodegeneration
4. Approaches to Increase the GSH Levels in the Brain
4.1. Cysteine Uptake
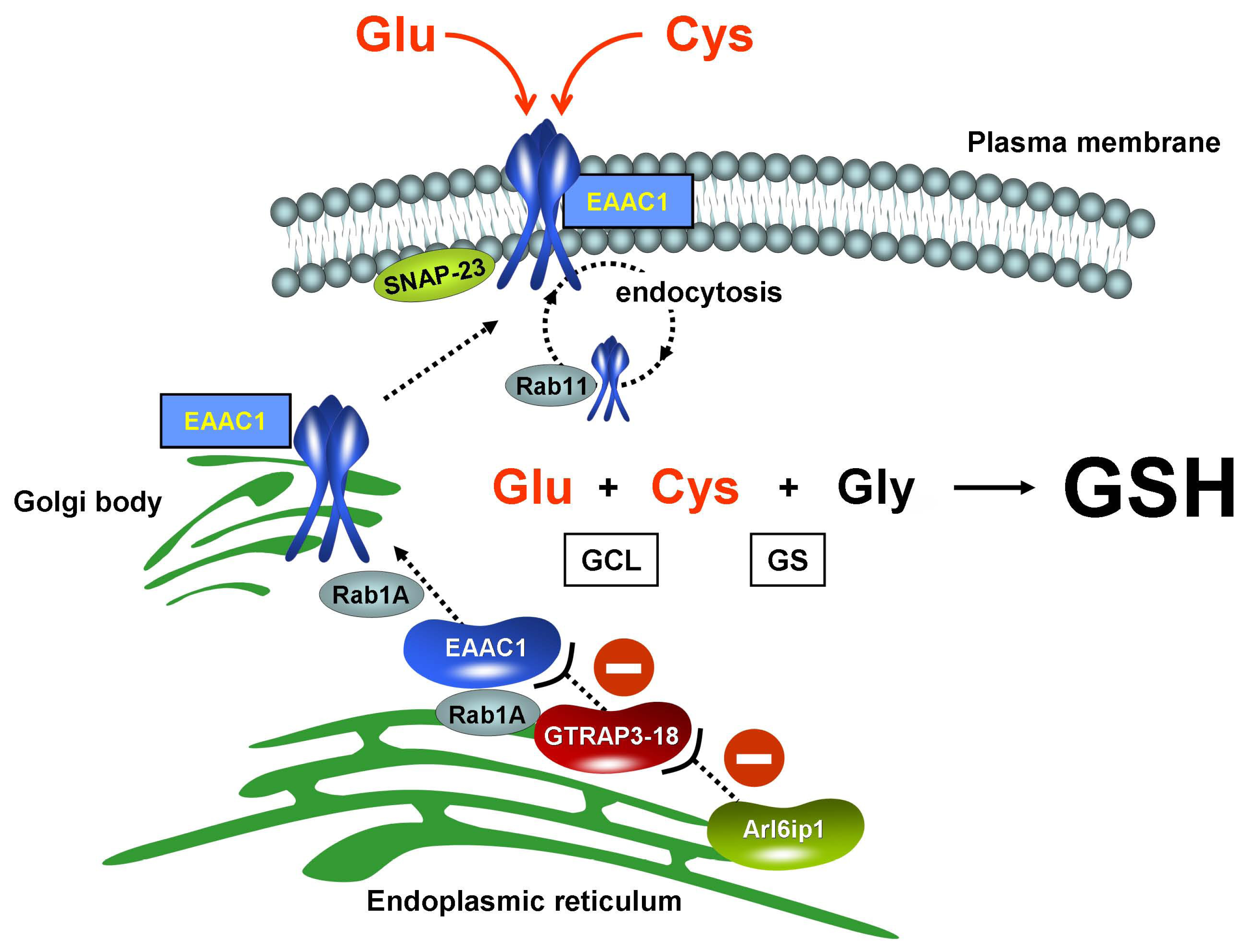
4.2. EAAC1 for Neuronal Cysteine Uptake
4.3. Regulation of EAAC1 Translocation to the Plasma Membrane
4.4. GTRAP3-18
4.5. Possible Regulation of ER-Golgi Transport by GTRAP3-18
4.6. Physiological Roles of GTRAP3-18
4.7. Induction of GTRAP3-18 by Methyl-β-cyclodextrin
4.8. Regulation of GSH Synthesis by GTRAP3-18
4.9. GTRAP3-18-Deficient Mouse
5. Antioxidant Supplementation
5.1. l-cysteine and Glutathione
5.2. Vitamins
5.3. Uric Acid
5.4. N-acetylcysteine
6. Conclusions
References
- Ho, Y.S.; Magnenat, J.L.; Bronson, R.T.; Cao, J.; Gargano, M.; Sugawara, M.; Funk, C.D. Mice deficient in cellular glutathione peroxidase develop normally and show no increased sensitivity to hyperoxia. J. Biol. Chem 1997, 272, 16644–16651. [Google Scholar]
- Dringen, R. Metabolism and functions of glutathione in brain. Prog. Neurobiol 2000, 62, 649–671. [Google Scholar]
- Coyle, J.T.; Puttfarcken, P. Oxidative stress, glutamate, and neurodegenerative disorders. Science 1993, 262, 689–695. [Google Scholar]
- Bains, J.S.; Shaw, C.A. Neurodegenerative disorders in humans: The role of glutathione in oxidative stress-mediated neuronal death. Brain Res. Brain Res. Rev 1997, 25, 335–358. [Google Scholar]
- Reed, T.T. Lipid peroxidation and neurodegenerative disease. Free Radic. Biol. Med 2011, 51, 1302–1319. [Google Scholar]
- Aoyama, K.; Watabe, M.; Nakaki, T. Regulation of neuronal glutathione synthesis. J. Pharmacol. Sci 2008, 108, 227–238. [Google Scholar]
- Pacher, P.; Beckman, J.S.; Liaudet, L. Nitric oxide and peroxynitrite in health and disease. Physiol. Rev 2007, 87, 315–424. [Google Scholar]
- Aoyama, K.; Matsubara, K.; Fujikawa, Y.; Nagahiro, Y.; Shimizu, K.; Umegae, N.; Hayase, N.; Shiono, H.; Kobayashi, S. Nitration of manganese superoxide dismutase in cerebrospinal fluids is a marker for peroxynitrite-mediated oxidative stress in neurodegenerative diseases. Ann. Neurol 2000, 47, 524–527. [Google Scholar]
- Szabo, C.; Ischiropoulos, H.; Radi, R. Peroxynitrite: Biochemistry, pathophysiology and development of therapeutics. Nat. Rev. Drug Discov 2007, 6, 662–680. [Google Scholar]
- Beckman, J.S. Peroxynitrite versus hydroxyl radical: The role of nitric oxide in superoxide-dependent cerebral injury. Ann. N. Y. Acad. Sci 1994, 738, 69–75. [Google Scholar]
- Kuhn, H.; Borchert, A. Regulation of enzymatic lipid peroxidation: The interplay of peroxidizing and peroxide reducing enzymes. Free Radic. Biol. Med 2002, 33, 154–172. [Google Scholar]
- Dringen, R.; Gutterer, J.M. Glutathione reductase from bovine brain. Methods Enzymol 2002, 348, 281–288. [Google Scholar]
- Dringen, R.; Kussmaul, L.; Gutterer, J.M.; Hirrlinger, J.; Hamprecht, B. The glutathione system of peroxide detoxification is less efficient in neurons than in astroglial cells. J. Neurochem 1999, 72, 2523–2530. [Google Scholar]
- Meister, A.; Anderson, M.E. Glutathione. Annu. Rev. Biochem 1983, 52, 711–760. [Google Scholar]
- Maher, P. The effects of stress and aging on glutathione metabolism. Ageing Res. Rev 2005, 4, 288–314. [Google Scholar]
- Giustarini, D.; Rossi, R.; Milzani, A.; Colombo, R.; Dalle-Donne, I. S-glutathionylation: From redox regulation of protein functions to human diseases. J. Cell. Mol. Med 2004, 8, 201–212. [Google Scholar]
- Cooper, A.J.; Kristal, B.S. Multiple roles of glutathione in the central nervous system. Biol. Chem 1997, 378, 793–802. [Google Scholar]
- Kang, Y.; Viswanath, V.; Jha, N.; Qiao, X.; Mo, J.Q.; Andersen, J.K. Brain gamma-glutamyl cysteine synthetase (GCS) mRNA expression patterns correlate with regional-specific enzyme activities and glutathione levels. J. Neurosci. Res 1999, 58, 436–441. [Google Scholar]
- Dringen, R.; Pawlowski, P.G.; Hirrlinger, J. Peroxide detoxification by brain cells. J. Neurosci. Res 2005, 79, 157–165. [Google Scholar]
- Desagher, S.; Glowinski, J.; Premont, J. Astrocytes protect neurons from hydrogen peroxide toxicity. J. Neurosci 1996, 16, 2553–2562. [Google Scholar]
- Aebi, H. Catalase in vitro. Methods Enzymol 1984, 105, 121–126. [Google Scholar]
- Jain, A.; Martensson, J.; Stole, E.; Auld, P.A.; Meister, A. Glutathione deficiency leads to mitochondrial damage in brain. Proc. Natl. Acad. Sci. USA 1991, 88, 1913–1917. [Google Scholar]
- Dringen, R.; Gutterer, J.M.; Hirrlinger, J. Glutathione metabolism in brain metabolic interaction between astrocytes and neurons in the defense against reactive oxygen species. Eur. J. Biochem 2000, 267, 4912–4916. [Google Scholar]
- Schulz, J.B.; Lindenau, J.; Seyfried, J.; Dichgans, J. Glutathione, oxidative stress and neurodegeneration. Eur. J. Biochem 2000, 267, 4904–4911. [Google Scholar]
- Sian, J.; Dexter, D.T.; Lees, A.J.; Daniel, S.; Agid, Y.; Javoy-Agid, F.; Jenner, P.; Marsden, C.D. Alterations in glutathione levels in Parkinson’s disease and other neurodegenerative disorders affecting basal ganglia. Ann. Neurol 1994, 36, 348–355. [Google Scholar]
- Ramassamy, C.; Averill, D.; Beffert, U.; Theroux, L.; Lussier-Cacan, S.; Cohn, J.S.; Christen, Y.; Schoofs, A.; Davignon, J.; Poirier, J. Oxidative insults are associated with apolipoprotein E genotype in Alzheimer’s disease brain. Neurobiol. Dis 2000, 7, 23–37. [Google Scholar]
- Riederer, P.; Sofic, E.; Rausch, W.D.; Schmidt, B.; Reynolds, G.P.; Jellinger, K.; Youdim, M.B. Transition metals, ferritin, glutathione, and ascorbic acid in parkinsonian brains. J. Neurochem 1989, 52, 515–520. [Google Scholar]
- Dexter, D.T.; Ward, R.J.; Wells, F.R.; Daniel, S.E.; Lees, A.J.; Peters, T.J.; Jenner, P.; Marsden, C.D. Alpha-tocopherol levels in brain are not altered in Parkinson’s disease. Ann. Neurol 1992, 32, 591–593. [Google Scholar]
- Dexter, D.T.; Sian, J.; Rose, S.; Hindmarsh, J.G.; Mann, V.M.; Cooper, J.M.; Wells, F.R.; Daniel, S.E.; Lees, A.J.; Schapira, A.H.; et al. Indices of oxidative stress and mitochondrial function in individuals with incidental Lewy body disease. Ann. Neurol 1994, 35, 38–44. [Google Scholar]
- Jenner, P. Oxidative damage in neurodegenerative disease. Lancet 1994, 344, 796–798. [Google Scholar]
- Jenner, P. Oxidative stress in Parkinson’s disease. Ann. Neurol 2003, 53, S26–36, discussion S36–28. [Google Scholar]
- Dringen, R.; Pfeiffer, B.; Hamprecht, B. Synthesis of the antioxidant glutathione in neurons: Supply by astrocytes of CysGly as precursor for neuronal glutathione. J. Neurosci 1999, 19, 562–569. [Google Scholar]
- Puka-Sundvall, M.; Eriksson, P.; Nilsson, M.; Sandberg, M.; Lehmann, A. Neurotoxicity of cysteine: Interaction with glutamate. Brain Res 1995, 705, 65–70. [Google Scholar]
- Janaky, R.; Varga, V.; Hermann, A.; Saransaari, P.; Oja, S.S. Mechanisms of l-cysteine neurotoxicity. Neurochem. Res 2000, 25, 1397–1405. [Google Scholar]
- Gazit, V.; Ben-Abraham, R.; Pick, C.G.; Ben-Shlomo, I.; Katz, Y. Long-term neurobehavioral and histological damage in brain of mice induced by l-cysteine. Pharmacol. Biochem. Behav 2003, 75, 795–799. [Google Scholar]
- Gazit, V.; Ben-Abraham, R.; Coleman, R.; Weizman, A.; Katz, Y. Cysteine-induced hypoglycemic brain damage: An alternative mechanism to excitotoxicity. Amino Acids 2004, 26, 163–168. [Google Scholar]
- Richman, P.G.; Meister, A. Regulation of gamma-glutamyl-cysteine synthetase by nonallosteric feedback inhibition by glutathione. J. Biol. Chem 1975, 250, 1422–1426. [Google Scholar]
- Holmseth, S.; Dehnes, Y.; Huang, Y.H.; Follin-Arbelet, V.V.; Grutle, N.J.; Mylonakou, M.N.; Plachez, C.; Zhou, Y.; Furness, D.N.; Bergles, D.E.; et al. The density of EAAC1 (EAAT3) glutamate transporters expressed by neurons in the mammalian CNS. J. Neurosci 2012, 32, 6000–6013. [Google Scholar]
- Volterra, A.; Trotti, D.; Tromba, C.; Floridi, S.; Racagni, G. Glutamate uptake inhibition by oxygen free radicals in rat cortical astrocytes. J. Neurosci 1994, 14, 2924–2932. [Google Scholar]
- Brennan, A.M.; Suh, S.W.; Won, S.J.; Narasimhan, P.; Kauppinen, T.M.; Lee, H.; Edling, Y.; Chan, P.H.; Swanson, R.A. NADPH oxidase is the primary source of superoxide induced by NMDA receptor activation. Nat. Neurosci 2009, 12, 857–863. [Google Scholar]
- Lehre, K.P.; Levy, L.M.; Ottersen, O.P.; Storm-Mathisen, J.; Danbolt, N.C. Differential expression of two glial glutamate transporters in the rat brain: Quantitative and immunocytochemical observations. J. Neurosci 1995, 15, 1835–1853. [Google Scholar]
- Haugeto, O.; Ullensvang, K.; Levy, L.M.; Chaudhry, F.A.; Honore, T.; Nielsen, M.; Lehre, K.P.; Danbolt, N.C. Brain glutamate transporter proteins form homomultimers. J. Biol. Chem 1996, 271, 27715–27722. [Google Scholar]
- Tanaka, K.; Watase, K.; Manabe, T.; Yamada, K.; Watanabe, M.; Takahashi, K.; Iwama, H.; Nishikawa, T.; Ichihara, N.; Kikuchi, T.; et al. Epilepsy and exacerbation of brain injury in mice lacking the glutamate transporter GLT-1. Science 1997, 276, 1699–1702. [Google Scholar]
- Zerangue, N.; Kavanaugh, M.P. Interaction of l-cysteine with a human excitatory amino acid transporter. J. Physiol 1996, 493, 419–423. [Google Scholar]
- Bendahan, A.; Armon, A.; Madani, N.; Kavanaugh, M.P.; Kanner, B.I. Arginine 447 plays a pivotal role in substrate interactions in a neuronal glutamate transporter. J. Biol. Chem 2000, 275, 37436–37442. [Google Scholar]
- Rothstein, J.D.; Martin, L.; Levey, A.I.; Dykes-Hoberg, M.; Jin, L.; Wu, D.; Nash, N.; Kuncl, R.W. Localization of neuronal and glial glutamate transporters. Neuron 1994, 13, 713–725. [Google Scholar]
- Shashidharan, P.; Huntley, G.W.; Murray, J.M.; Buku, A.; Moran, T.; Walsh, M.J.; Morrison, J.H.; Plaitakis, A. Immunohistochemical localization of the neuron-specific glutamate transporter EAAC1 (EAAT3) in rat brain and spinal cord revealed by a novel monoclonal antibody. Brain Res 1997, 773, 139–148. [Google Scholar]
- Fournier, K.M.; Gonzalez, M.I.; Robinson, M.B. Rapid trafficking of the neuronal glutamate transporter, EAAC1: Evidence for distinct trafficking pathways differentially regulated by protein kinase C and platelet-derived growth factor. J. Biol. Chem 2004, 279, 34505–34513. [Google Scholar]
- Escartin, C.; Won, S.J.; Malgorn, C.; Auregan, G.; Berman, A.E.; Chen, P.C.; Deglon, N.; Johnson, J.A.; Suh, S.W.; Swanson, R.A. Nuclear factor erythroid 2-related factor 2 facilitates neuronal glutathione synthesis by upregulating neuronal excitatory amino acid transporter 3 expression. J. Neurosci 2011, 31, 7392–7401. [Google Scholar]
- Davis, K.E.; Straff, D.J.; Weinstein, E.A.; Bannerman, P.G.; Correale, D.M.; Rothstein, J.D.; Robinson, M.B. Multiple signaling pathways regulate cell surface expression and activity of the excitatory amino acid carrier 1 subtype of Glu transporter in C6 glioma. J. Neurosci 1998, 18, 2475–2485. [Google Scholar]
- Fournier, K.M.; Robinson, M.B. A dominant-negative variant of SNAP-23 decreases the cell surface expression of the neuronal glutamate transporter EAAC1 by slowing constitutive delivery. Neurochem. Int 2006, 48, 596–603. [Google Scholar]
- Zerial, M.; McBride, H. Rab proteins as membrane organizers. Nat. Rev. Mol. Cell Biol 2001, 2, 107–117. [Google Scholar]
- Fukuda, M. Regulation of secretory vesicle traffic by Rab small GTPases. Cell. Mol. Life Sci 2008, 65, 2801–2813. [Google Scholar]
- Gonzalez, M.I.; Susarla, B.T.; Fournier, K.M.; Sheldon, A.L.; Robinson, M.B. Constitutive endocytosis and recycling of the neuronal glutamate transporter, excitatory amino acid carrier 1. J. Neurochem 2007, 103, 1917–1931. [Google Scholar]
- Li, X.; Valencia, A.; Sapp, E.; Masso, N.; Alexander, J.; Reeves, P.; Kegel, K.B.; Aronin, N.; Difiglia, M. Aberrant Rab11-dependent trafficking of the neuronal glutamate transporter EAAC1 causes oxidative stress and cell death in Huntington’s disease. J. Neurosci 2010, 30, 4552–4561. [Google Scholar]
- Lin, C.I.; Orlov, I.; Ruggiero, A.M.; Dykes-Hoberg, M.; Lee, A.; Jackson, M.; Rothstein, J.D. Modulation of the neuronal glutamate transporter EAAC1 by the interacting protein GTRAP3-18. Nature 2001, 410, 84–88. [Google Scholar]
- Abdul-Ghani, M.; Gougeon, P.Y.; Prosser, D.C.; Da-Silva, L.F.; Ngsee, J.K. PRA isoforms are targeted to distinct membrane compartments. J. Biol. Chem 2001, 276, 6225–6233. [Google Scholar]
- Butchbach, M.E.; Lai, L.; Lin, C.L. Molecular cloning, gene structure, expression profile and functional characterization of the mouse glutamate transporter (EAAT3) interacting protein GTRAP3-18. Gene 2002, 292, 81–90. [Google Scholar]
- Ruggiero, A.M.; Liu, Y.; Vidensky, S.; Maier, S.; Jung, E.; Farhan, H.; Robinson, M.B.; Sitte, H.H.; Rothstein, J.D. The endoplasmic reticulum exit of glutamate transporter is regulated by the inducible mammalian Yip6b/GTRAP3-18 protein. J. Biol. Chem 2008, 283, 6175–6183. [Google Scholar]
- Pfeffer, S.; Aivazian, D. Targeting Rab GTPases to distinct membrane compartments. Nat. Rev. Mol. Cell Biol 2004, 5, 886–896. [Google Scholar]
- Akiduki, S.; Ikemoto, M.J. Modulation of the neural glutamate transporter EAAC1 by the addicsin-interacting protein ARL6IP1. J. Biol. Chem 2008, 283, 31323–31332. [Google Scholar]
- Takai, Y.; Sasaki, T.; Matozaki, T. Small GTP-binding proteins. Physiol. Rev 2001, 81, 153–208. [Google Scholar]
- Marshall, C.J. Protein prenylation: A mediator of protein-protein interactions. Science 1993, 259, 1865–1866. [Google Scholar]
- Gonzalez, L., Jr; Scheller, R.H. Regulation of membrane trafficking: Structural insights from a Rab/effector complex. Cell 1999, 96, 755–758. [Google Scholar]
- Maier, S.; Reiterer, V.; Ruggiero, A.M.; Rothstein, J.D.; Thomas, S.; Dahm, R.; Sitte, H.H.; Farhan, H. GTRAP3-18 serves as a negative regulator of Rab1 in protein transport and neuronal differentiation. J. Cell. Mol. Med 2009, 13, 114–124. [Google Scholar]
- Takai, Y.; Sasaki, T.; Shirataki, H.; Nakanishi, H. Rab3A small GTP-binding protein in Ca(2+)-dependent exocytosis. Genes Cells 1996, 1, 615–632. [Google Scholar]
- Watabe, M.; Aoyama, K.; Nakaki, T. Regulation of glutathione synthesis via interaction between glutamate transport-associated protein 3-18 (GTRAP3-18) and excitatory amino acid carrier-1 (EAAC1) at plasma membrane. Mol. Pharmacol 2007, 72, 1103–1110. [Google Scholar]
- Wang, N.P.; Zhou, J.W.; Li, A.P.; Cao, H.X.; Wang, X.R. The mechanism of JWA gene involved in oxidative stress of cells (in Chinese). Zhonghua Lao Dong Wei Sheng Zhi Ye Bing Za Zhi 2003, 21, 212–215. [Google Scholar]
- Mao, W.G.; Li, A.P.; Ye, J.; Huang, S.; Li, A.Q.; Zhou, J.W. Expressions of JWA protein and heat stress protein 70 induced by cell differentiation inducers combined with heat stress in K562 cells (in Chinese). Zhonghua Lao Dong Wei Sheng Zhi Ye Bing Za Zhi 2004, 22, 60–63. [Google Scholar]
- Chen, R.; Qiu, W.; Liu, Z.; Cao, X.; Zhu, T.; Li, A.; Wei, Q.; Zhou, J. Identification of JWA as a novel functional gene responsive to environmental oxidative stress induced by benzo[a]pyrene and hydrogen peroxide. Free Radic. Biol. Med 2007, 42, 1704–1714. [Google Scholar]
- Ikemoto, M.J.; Inoue, K.; Akiduki, S.; Osugi, T.; Imamura, T.; Ishida, N.; Ohtomi, M. Identification of addicsin/GTRAP3-18 as a chronic morphine-augmented gene in amygdala. Neuroreport 2002, 13, 2079–2084. [Google Scholar]
- Wu, Y.; Chen, R.; Zhao, X.; Li, A.; Li, G.; Zhou, J. JWA regulates chronic morphine dependence via the delta opioid receptor. Biochem. Biophys. Res. Commun 2011, 409, 520–525. [Google Scholar]
- Furuta, A.; Martin, L.J.; Lin, C.L.; Dykes-Hoberg, M.; Rothstein, J.D. Cellular and synaptic localization of the neuronal glutamate transporters excitatory amino acid transporter 3 and 4. Neuroscience 1997, 81, 1031–1042. [Google Scholar]
- Hediger, M.A.; Welbourne, T.C. Introduction: Glutamate transport, metabolism, and physiological responses. Am. J. Physiol 1999, 277, F477–F480. [Google Scholar]
- Hediger, M.A. Glutamate transporters in kidney and brain. Am. J. Physiol 1999, 277, F487–492. [Google Scholar]
- Butchbach, M.E.; Guo, H.; Lin, C.L. Methyl-beta-cyclodextrin but not retinoic acid reduces EAAT3-mediated glutamate uptake and increases GTRAP3-18 expression. J. Neurochem 2003, 84, 891–894. [Google Scholar]
- Shouffani, A.; Kanner, B.I. Cholesterol is required for the reconstruction of the sodium- and chloride-coupled, gamma-aminobutyric acid transporter from rat brain. J. Biol. Chem 1990, 265, 6002–6008. [Google Scholar]
- Kilsdonk, E.P.; Yancey, P.G.; Stoudt, G.W.; Bangerter, F.W.; Johnson, W.J.; Phillips, M.C.; Rothblat, G.H. Cellular cholesterol efflux mediated by cyclodextrins. J. Biol. Chem 1995, 270, 17250–17256. [Google Scholar]
- Middleton, A.; Nury, D.; Willington, S.J.; Latif, L.; Hill, S.J.; Middleton, B. Modulation by cellular cholesterol of gene transcription via the cyclic AMP response element. Biochem. Pharmacol 2001, 62, 171–181. [Google Scholar]
- Furuchi, T.; Anderson, R.G. Cholesterol depletion of caveolae causes hyperactivation of extracellular signal-related kinase (ERK). J. Biol. Chem 1998, 273, 21099–21104. [Google Scholar]
- Watabe, M.; Aoyama, K.; Nakaki, T. A dominant role of GTRAP3-18 in neuronal glutathione synthesis. J. Neurosci 2008, 28, 9404–9413. [Google Scholar]
- Aoyama, K.; Wang, F.; Matsumura, N.; Kiyonari, H.; Shioi, G.; Tanaka, K.; Kinoshita, C.; Kikuchi-Utsumi, K.; Watabe, M.; Nakaki, T. Increased neuronal glutathione and neuroprotection in GTRAP3-18-deficient mice. Neurobiol. Dis 2012, 45, 973–982. [Google Scholar]
- Fraticelli, A.I.; Thompson, K.J. EAAC1 protein expression is increased in the rat hippocampus during water maze learning. Soc. Neurosci. Abstr 2011, 37, 342.313/E342. [Google Scholar]
- Levenson, J.; Weeber, E.; Selcher, J.C.; Kategaya, L.S.; Sweatt, J.D.; Eskin, A. Long-term potentiation and contextual fear conditioning increase neuronal glutamate uptake. Nat. Neurosci 2002, 5, 155–161. [Google Scholar]
- Aoyama, K.; Suh, S.W.; Hamby, A.M.; Liu, J.; Chan, W.Y.; Chen, Y.; Swanson, R.A. Neuronal glutathione deficiency and age-dependent neurodegeneration in the EAAC1 deficient mouse. Nat. Neurosci 2006, 9, 119–126. [Google Scholar]
- Lee, S.; Park, S.H.; Zuo, Z. Effects of isoflurane on learning and memory functions of wild-type and glutamate transporter type 3 knockout mice. J. Pharm. Pharmacol 2012, 64, 302–307. [Google Scholar]
- Ng, K.T.; O’Dowd, B.S.; Rickard, N.S.; Robinson, S.R.; Gibbs, M.E.; Rainey, C.; Zhao, W.Q.; Sedman, G.L.; Hertz, L. Complex roles of glutamate in the Gibbs-Ng model of one-trial aversive learning in the new-born chick. Neurosci. Biobehav. Rev 1997, 21, 45–54. [Google Scholar]
- Levenson, J.; Endo, S.; Kategaya, L.S.; Fernandez, R.I.; Brabham, D.G.; Chin, J.; Byrne, J.H.; Eskin, A. Long-term regulation of neuronal high-affinity glutamate and glutamine uptake in Aplysia. Proc. Natl. Acad. Sci. USA 2000, 97, 12858–12863. [Google Scholar]
- Maleszka, R.; Helliwell, P.; Kucharski, R. Pharmacological interference with glutamate re-uptake impairs long-term memory in the honeybee, apis mellifera. Behav. Brain Res 2000, 115, 49–53. [Google Scholar]
- Mennerick, S.; Zorumski, C.F. Glial contributions to excitatory neurotransmission in cultured hippocampal cells. Nature 1994, 368, 59–62. [Google Scholar]
- Cornford, E.M.; Braun, L.D.; Crane, P.D.; Oldendorf, W.H. Blood-brain barrier restriction of peptides and the low uptake of enkephalins. Endocrinology 1978, 103, 1297–1303. [Google Scholar]
- Lash, L.H.; Jones, D.P. Distribution of oxidized and reduced forms of glutathione and cysteine in rat plasma. Arch. Biochem. Biophys 1985, 240, 583–592. [Google Scholar]
- Ammon, H.P.; Melien, M.C.; Verspohl, E.J. Pharmacokinetics of intravenously administered glutathione in the rat. J. Pharm. Pharmacol 1986, 38, 721–725. [Google Scholar]
- Wendel, A.; Cikryt, P. The level and half-life of glutathione in human plasma. FEBS Lett 1980, 120, 209–211. [Google Scholar]
- Wendel, A.; Jaeschke, H. Drug-induced lipid peroxidation in mice—III. Glutathione content of liver, kidney and spleen after intravenous administration of free and liposomally entrapped glutathione. Biochem. Pharmacol 1982, 31, 3607–3611. [Google Scholar]
- Perry, T.L.; Yong, V.W.; Clavier, R.M.; Jones, K.; Wright, J.M.; Foulks, J.G.; Wall, R.A. Partial protection from the dopaminergic neurotoxin N-methyl-4-phenyl-1,2,3,6-tetrahydropyridine by four different antioxidants in the mouse. Neurosci. Lett 1985, 60, 109–114. [Google Scholar]
- Meister, A. Glutathione-ascorbic acid antioxidant system in animals. J. Biol. Chem 1994, 269, 9397–9400. [Google Scholar]
- Gilgun-Sherki, Y.; Melamed, E.; Offen, D. Oxidative stress induced-neurodegenerative diseases: The need for antioxidants that penetrate the blood brain barrier. Neuropharmacology 2001, 40, 959–975. [Google Scholar]
- Grunewald, R.A. Ascorbic acid in the brain. Brain Res. Brain Res. Rev 1993, 18, 123–133. [Google Scholar]
- Agus, D.B.; Gambhir, S.S.; Pardridge, W.M.; Spielholz, C.; Baselga, J.; Vera, J.C.; Golde, D.W. Vitamin C crosses the blood-brain barrier in the oxidized form through the glucose transporters. J. Clin. Invest 1997, 100, 2842–2848. [Google Scholar]
- Metcalfe, T.; Bowen, D.M.; Muller, D.P. Vitamin E concentrations in human brain of patients with Alzheimer’s disease, fetuses with Down’s syndrome, centenarians, and controls. Neurochem. Res 1989, 14, 1209–1212. [Google Scholar]
- Pappert, E.J.; Tangney, C.C.; Goetz, C.G.; Ling, Z.D.; Lipton, J.W.; Stebbins, G.T.; Carvey, P.M. Alpha-tocopherol in the ventricular cerebrospinal fluid of Parkinson’s disease patients: Dose-response study and correlations with plasma levels. Neurology 1996, 47, 1037–1042. [Google Scholar]
- Logroscino, G.; Marder, K.; Cote, L.; Tang, M.X.; Shea, S.; Mayeux, R. Dietary lipids and antioxidants in Parkinson’s disease: A population-based, case-control study. Ann. Neurol 1996, 39, 89–94. [Google Scholar]
- Mendelsohn, A.B.; Belle, S.H.; Stoehr, G.P.; Ganguli, M. Use of antioxidant supplements and its association with cognitive function in a rural elderly cohort: The MoVIES Project. Monongahela Valley Independent Elders Survey. Am. J. Epidemiol 1998, 148, 38–44. [Google Scholar]
- Petersen, R.C.; Thomas, R.G.; Grundman, M.; Bennett, D.; Doody, R.; Ferris, S.; Galasko, D.; Jin, S.; Kaye, J.; Levey, A.; et al. Vitamin E and donepezil for the treatment of mild cognitive impairment. N. Engl. J. Med 2005, 352, 2379–2388. [Google Scholar]
- Isaac, M.G.; Quinn, R.; Tabet, N. Vitamin E for Alzheimer’s disease and mild cognitive impairment. Cochrane Database Syst. Rev 2008, CD002854. [Google Scholar]
- Galasko, D.R.; Peskind, E.; Clark, C.M.; Quinn, J.F.; Ringman, J.M.; Jicha, G.A.; Cotman, C.; Cottrell, B.; Montine, T.J.; Thomas, R.G.; et al. Antioxidants for Alzheimer Disease: A Randomized Clinical Trial With Cerebrospinal Fluid Biomarker Measures. Arch. Neurol 2012, 69, 836–841. [Google Scholar]
- Dexter, D.T.; Nanayakkara, I.; Goss-Sampson, M.A.; Muller, D.P.; Harding, A.E.; Marsden, C.D.; Jenner, P. Nigral dopaminergic cell loss in vitamin E deficient rats. Neuroreport 1994, 5, 1773–1776. [Google Scholar]
- Sano, M.; Ernesto, C.; Thomas, R.G.; Klauber, M.R.; Schafer, K.; Grundman, M.; Woodbury, P.; Growdon, J.; Cotman, C.W.; Pfeiffer, E.; et al. A controlled trial of selegiline, alpha-tocopherol, or both as treatment for Alzheimer’s disease. The Alzheimer’s Disease Cooperative Study. N. Engl. J. Med 1997, 336, 1216–1222. [Google Scholar]
- Cristina Polidori, M.; De Spirt, S.; Stahl, W.; Pientka, L. Conflict of evidence: Carotenoids and other micronutrients in the prevention and treatment of cognitive impairment. Biofactors 2012, 38, 167–171. [Google Scholar]
- Paraskevas, G.P.; Kapaki, E.; Libitaki, G.; Zournas, C.; Segditsa, I.; Papageorgiou, C. Ascorbate in healthy subjects, amyotrophic lateral sclerosis and Alzheimer’s disease. Acta. Neurol. Scand 1997, 96, 88–90. [Google Scholar]
- Chen, H.; Tappel, A.L. Protection by vitamin E selenium, trolox C, ascorbic acid palmitate, acetylcysteine, coenzyme Q, beta-carotene, canthaxanthin, and (+)-catechin against oxidative damage to liver slices measured by oxidized heme proteins. Free Radic. Biol. Med 1994, 16, 437–444. [Google Scholar]
- Ames, B.N.; Cathcart, R.; Schwiers, E.; Hochstein, P. Uric acid provides an antioxidant defense in humans against oxidant- and radical-caused aging and cancer: A hypothesis. Proc. Natl. Acad. Sci. USA 1981, 78, 6858–6862. [Google Scholar]
- Kutzing, M.K.; Firestein, B.L. Altered uric acid levels and disease states. J. Pharmacol. Exp. Ther 2008, 324, 1–7. [Google Scholar]
- Hensley, K.; Maidt, M.L.; Yu, Z.; Sang, H.; Markesbery, W.R.; Floyd, R.A. Electrochemical analysis of protein nitrotyrosine and dityrosine in the Alzheimer brain indicates region-specific accumulation. J. Neurosci 1998, 18, 8126–8132. [Google Scholar]
- Marklund, N.; Ostman, B.; Nalmo, L.; Persson, L.; Hillered, L. Hypoxanthine, uric acid and allantoin as indicators of in vivo free radical reactions. Description of a HPLC method and human brain microdialysis data. Acta Neurochir. (Wien.) 2000, 142, 1135–1141, discussion 1141–1132. [Google Scholar]
- Aoyama, K.; Matsumura, N.; Watabe, M.; Wang, F.; Kikuchi-Utsumi, K.; Nakaki, T. Caffeine and uric acid mediate glutathione synthesis for neuroprotection. Neuroscience 2011, 181, 206–215. [Google Scholar]
- De Flora, S.; Bennicelli, C.; Zanacchi, P.; Camoirano, A.; Morelli, A.; De Flora, A. In vitro effects of N-acetylcysteine on the mutagenicity of direct-acting compounds and procarcinogens. Carcinogenesis 1984, 5, 505–510. [Google Scholar]
- Won, S.J.; Yoo, B.H.; Brennan, A.M.; Shin, B.S.; Kauppinen, T.M.; Berman, A.E.; Swanson, R.A.; Suh, S.W. EAAC1 gene deletion alters zinc homeostasis and exacerbates neuronal injury after transient cerebral ischemia. J. Neurosci 2010, 30, 15409–15418. [Google Scholar]
- Hussain, S.; Slikker, W., Jr; Ali, S.F. Role of metallothionein and other antioxidants in scavenging superoxide radicals and their possible role in neuroprotection. Neurochem. Int. 1996, 29, 145–152. [Google Scholar]
- Banaclocha, M.M.; Hernandez, A.I.; Martinez, N.; Ferrandiz, M.L. N-acetylcysteine protects against age-related increase in oxidized proteins in mouse synaptic mitochondria. Brain Res 1997, 762, 256–258. [Google Scholar]
- Fu, A.L.; Dong, Z.H.; Sun, M.J. Protective effect of N-acetyl-l-cysteine on amyloid beta-peptide-induced learning and memory deficits in mice. Brain Res 2006, 1109, 201–206. [Google Scholar]
- Park, S.W.; Kim, S.H.; Park, K.H.; Kim, S.D.; Kim, J.Y.; Baek, S.Y.; Chung, B.S.; Kang, C.D. Preventive effect of antioxidants in MPTP-induced mouse model of Parkinson’s disease. Neurosci. Lett 2004, 363, 243–246. [Google Scholar]
- Munoz, A.M.; Rey, P.; Soto-Otero, R.; Guerra, M.J.; Labandeira-Garcia, J.L. Systemic administration of N-acetylcysteine protects dopaminergic neurons against 6-hydroxydopamineinduced degeneration. J. Neurosci. Res 2004, 76, 551–562. [Google Scholar]
- Hart, A.M.; Terenghi, G.; Kellerth, J.O.; Wiberg, M. Sensory neuroprotection, mitochondrial preservation, and therapeutic potential of N-acetyl-cysteine after nerve injury. Neuroscience 2004, 125, 91–101. [Google Scholar]
- Sekhon, B.; Sekhon, C.; Khan, M.; Patel, S.J.; Singh, I.; Singh, A.K. N-Acetyl cysteine protects against injury in a rat model of focal cerebral ischemia. Brain Res 2003, 971, 1–8. [Google Scholar]
- Aoyama, K.; Matsumura, N.; Watabe, M.; Nakaki, T. Oxidative stress on EAAC1 is involved in MPTP-induced glutathione depletion and motor dysfunction. Eur. J. Neurosci 2008, 27, 20–30. [Google Scholar]
- Kranich, O.; Dringen, R.; Sandberg, M.; Hamprecht, B. Utilization of cysteine and cysteine precursors for the synthesis of glutathione in astroglial cultures: Preference for cystine. Glia 1998, 22, 11–18. [Google Scholar]

© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Aoyama, K.; Nakaki, T. Inhibition of GTRAP3-18 May Increase Neuroprotective Glutathione (GSH) Synthesis. Int. J. Mol. Sci. 2012, 13, 12017-12035. https://doi.org/10.3390/ijms130912017
Aoyama K, Nakaki T. Inhibition of GTRAP3-18 May Increase Neuroprotective Glutathione (GSH) Synthesis. International Journal of Molecular Sciences. 2012; 13(9):12017-12035. https://doi.org/10.3390/ijms130912017
Chicago/Turabian StyleAoyama, Koji, and Toshio Nakaki. 2012. "Inhibition of GTRAP3-18 May Increase Neuroprotective Glutathione (GSH) Synthesis" International Journal of Molecular Sciences 13, no. 9: 12017-12035. https://doi.org/10.3390/ijms130912017