Chromatin Dynamics during Nucleotide Excision Repair: Histones on the Move

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
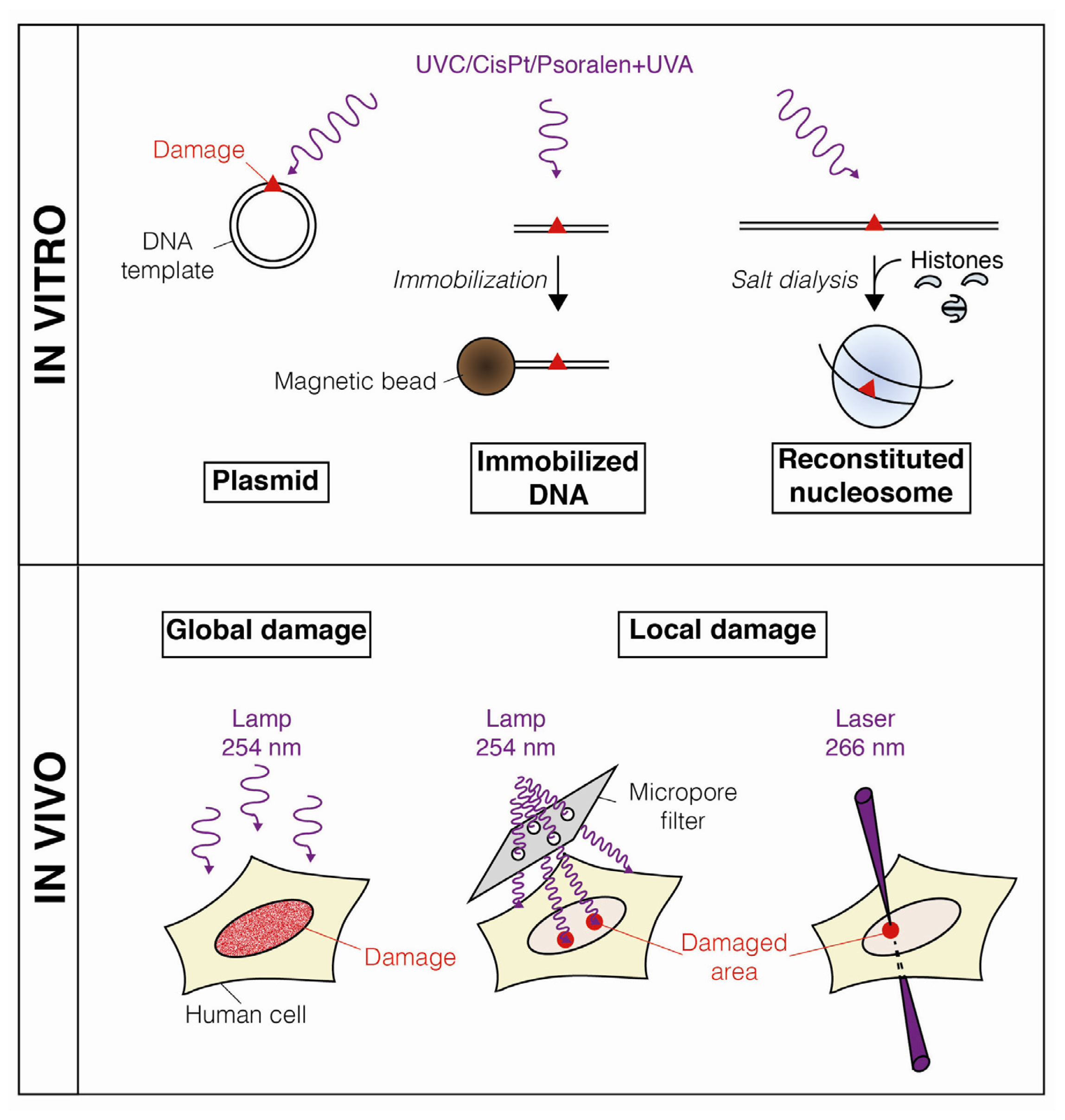
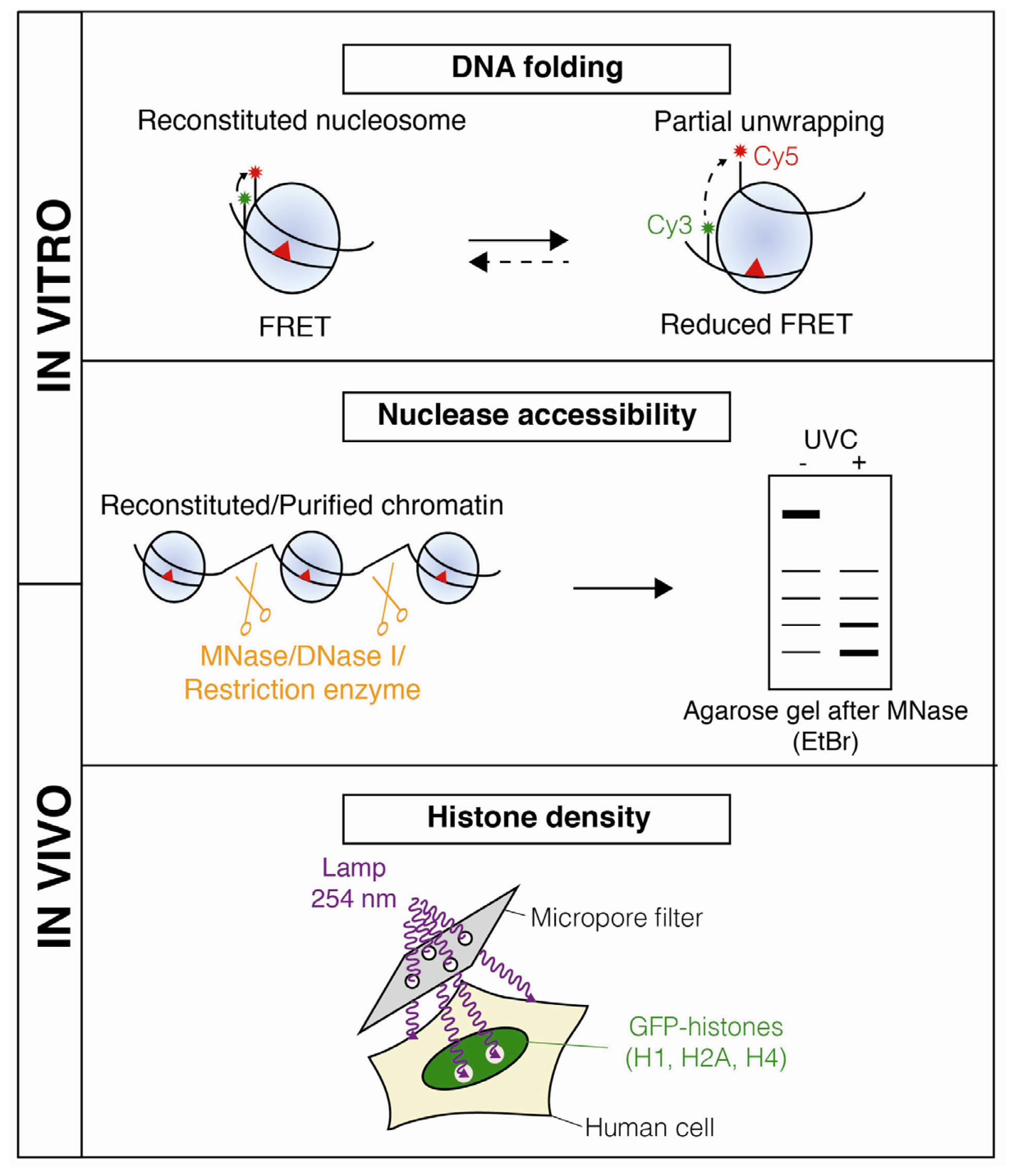
2. Dynamic Changes in Chromatin Structure during Nucleotide Excision Repair
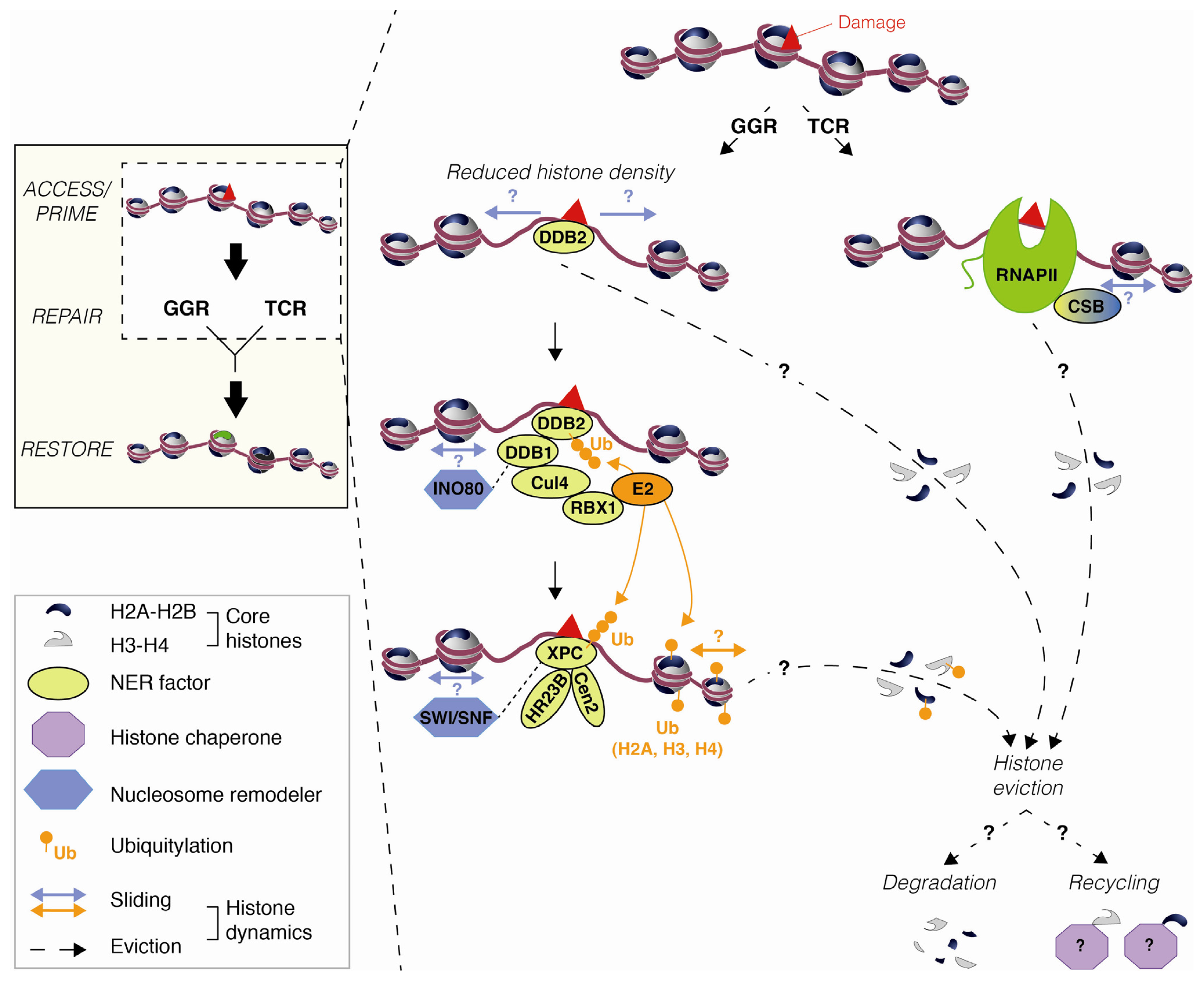
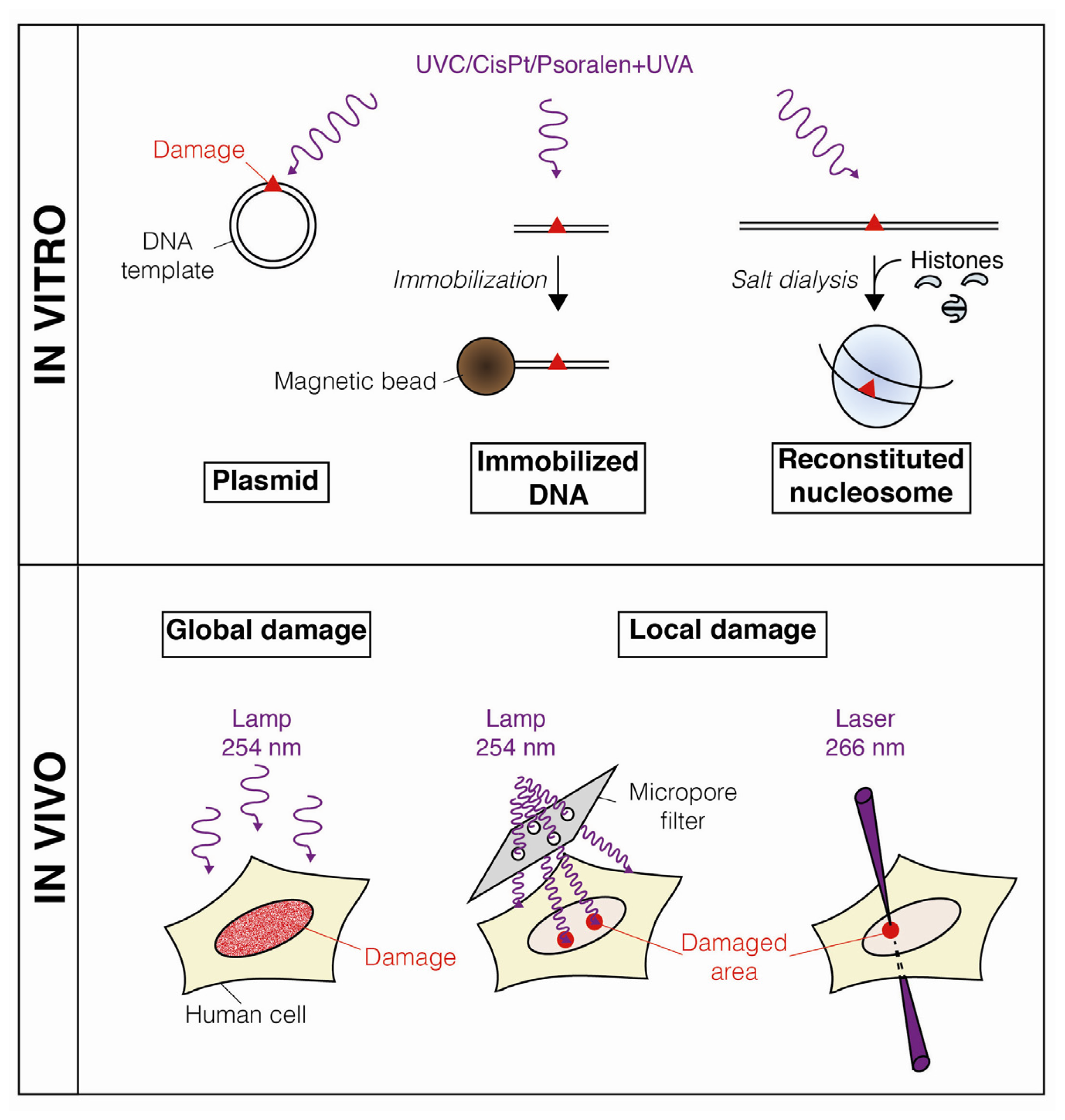
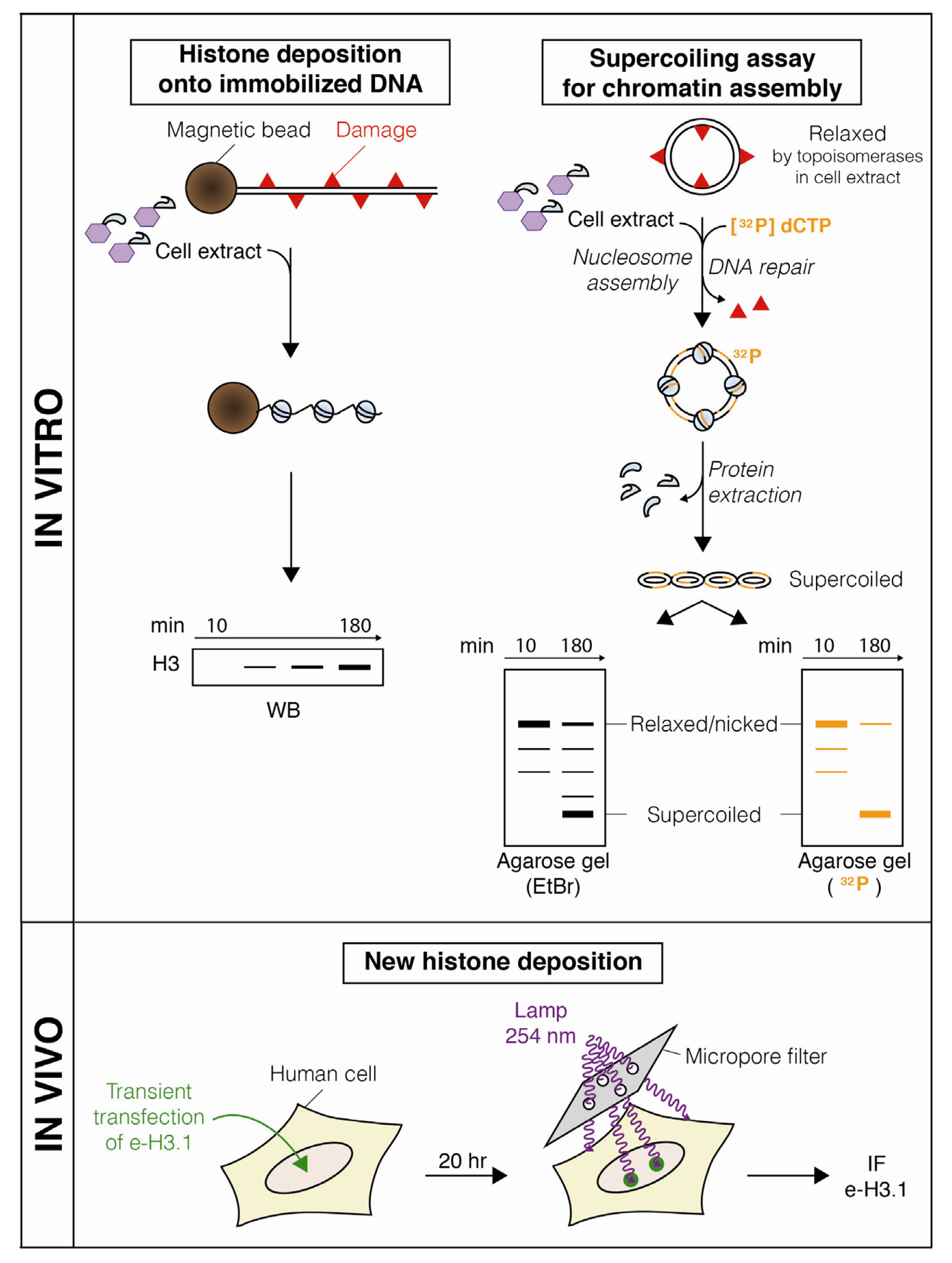
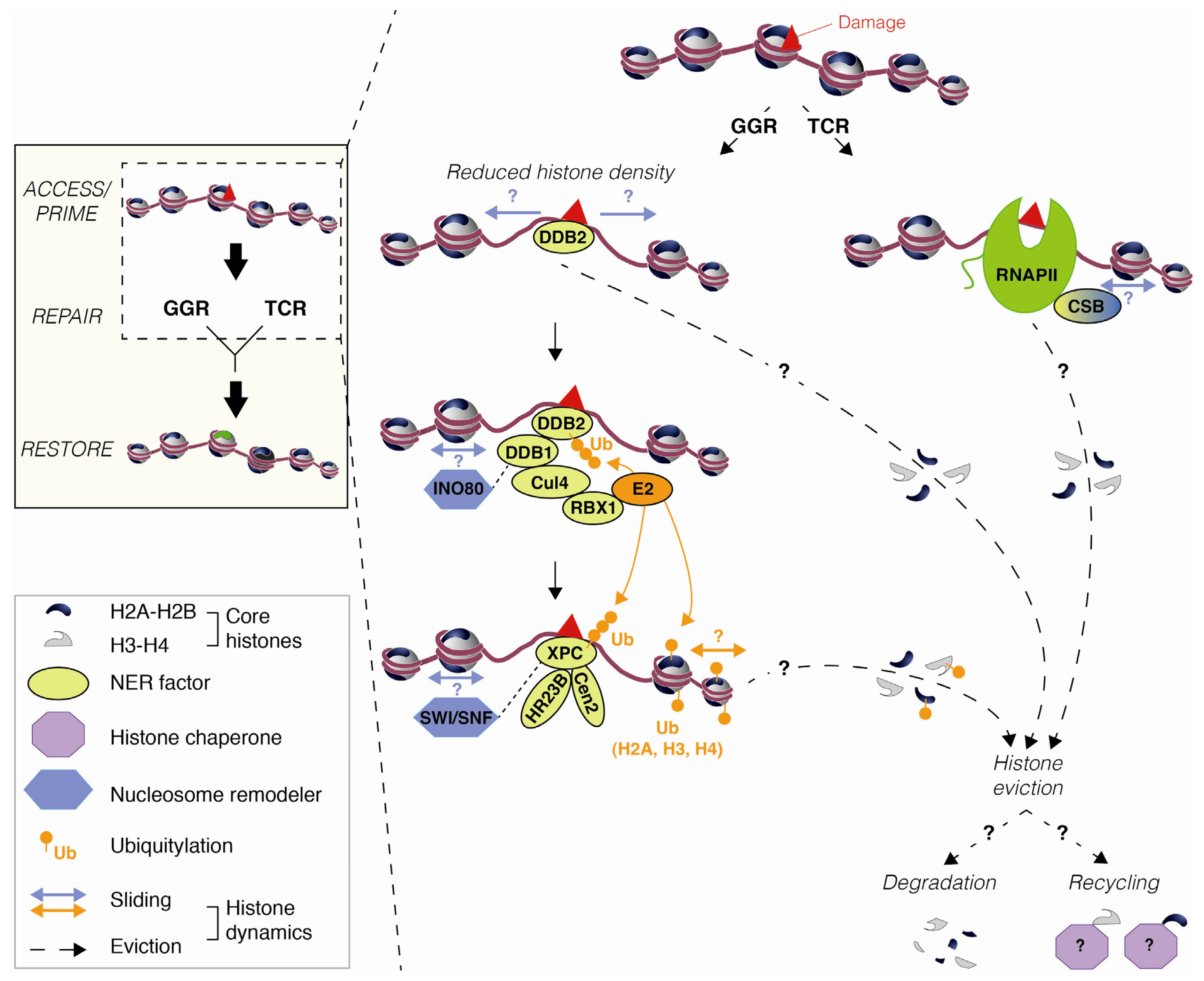
2.1. DNA Damage-Induced Nucleosome Destabilization and Histone Mobilization
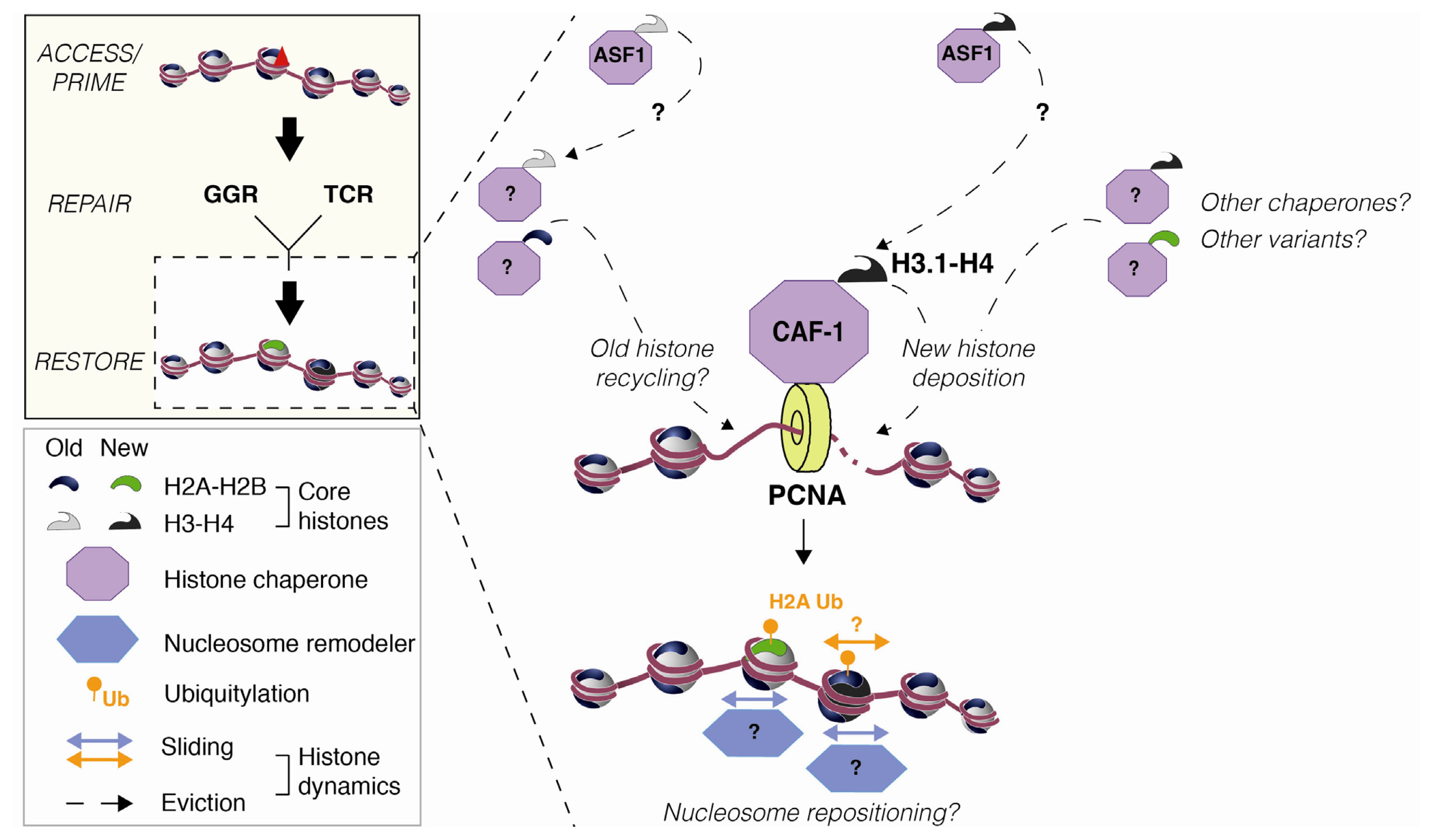
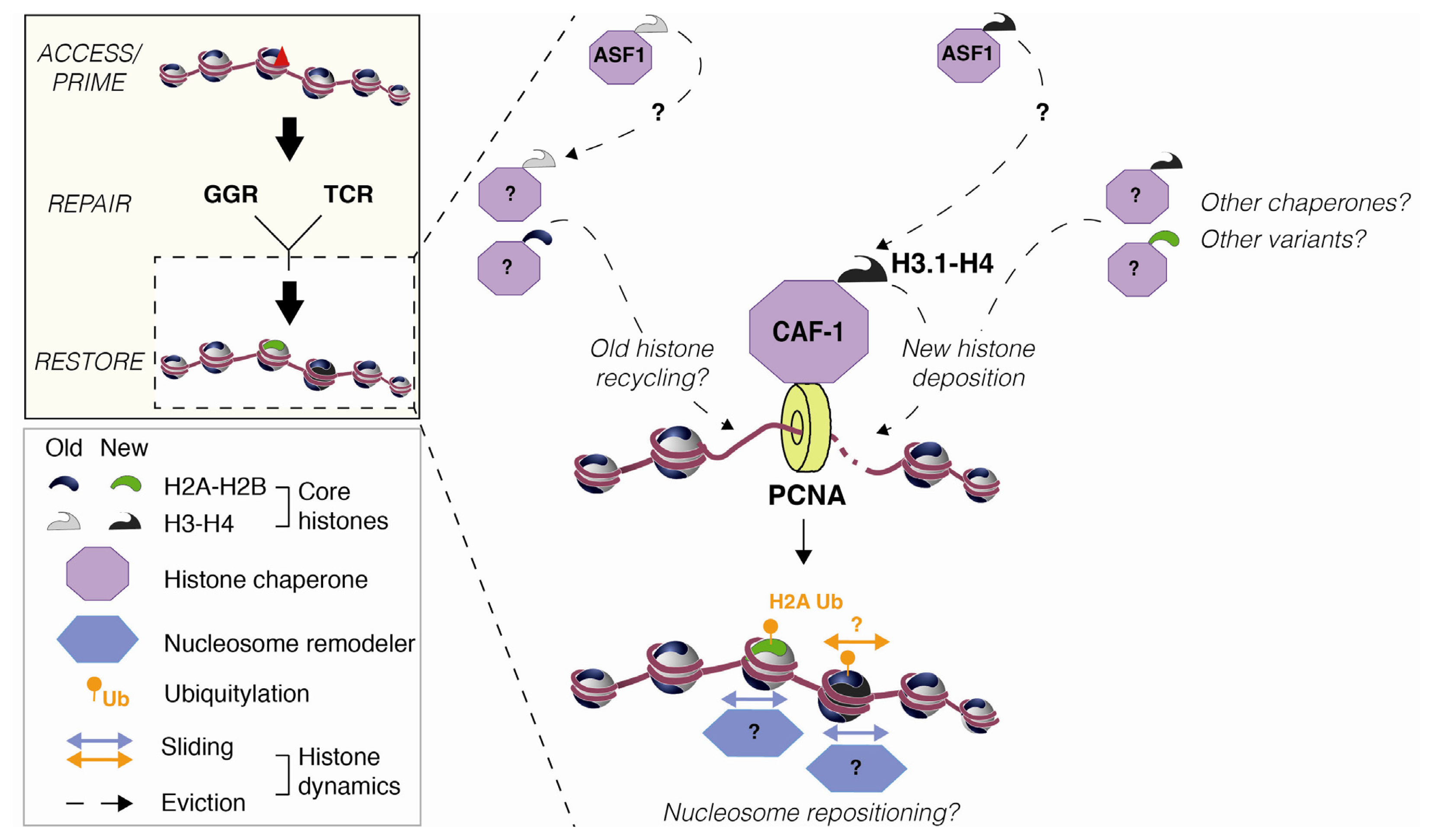
2.2. Nucleosome Restoration and New Histone Deposition Coupled to NER
3. Mechanisms Underlying Histone Dynamics during Nucleotide Excision Repair
3.1. Chromatin Accessibility and Histone Post-Translational Modifications
3.2. Nucleosome Mobilization by Chromatin Remodeling Factors
3.3. Histone Mobilization by Histone Chaperones
4. Conclusions and Open Issues
Acknowledgments
- Conflict of InterestThe authors declare no conflict of interest.
References
- Kornberg, R.D. Structure of chromatin. Annu. Rev. Biochem 1977, 46, 931–954. [Google Scholar]
- Luger, K.; Mäder, A.W.; Richmond, R.K.; Sargent, D.F.; Richmond, T.J. Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature 1997, 389, 251–260. [Google Scholar]
- Probst, A.V.; Dunleavy, E.; Almouzni, G. Epigenetic inheritance during the cell cycle. Nat. Rev. Mol. Cell Biol 2009, 10, 192–206. [Google Scholar]
- Li, G.; Reinberg, D. Chromatin higher-order structures and gene regulation. Curr. Opin. Genet. Dev 2011, 21, 175–186. [Google Scholar]
- Talbert, P.B.; Henikoff, S. Histone variants—Ancient wrap artists of the epigenome. Nat. Rev. Mol. Cell Biol 2010, 11, 264–275. [Google Scholar]
- Talbert, P.B.; Ahmad, K.; Almouzni, G.; Ausió, J.; Berger, F.; Bhalla, P.L.; Bonner, W.M.; Cande, W.Z.; Chadwick, B.P.; Chan, S.W.L.; et al. A unified phylogeny-based nomenclature for histone variants. Epigenetics Chromatin 2012, 5. [Google Scholar] [CrossRef]
- Bannister, A.J.; Kouzarides, T. Regulation of chromatin by histone modifications. Cell Res 2011, 21, 381–395. [Google Scholar]
- Luger, K.; Dechassa, M.L.; Tremethick, D.J. New insights into nucleosome and chromatin structure: An ordered state or a disordered affair? Nat. Rev. Mol. Cell Biol 2012, 13, 436–447. [Google Scholar]
- Flaus, A.; Owen-Hughes, T. Mechanisms for ATP-dependent chromatin remodelling: The means to the end. FEBS J 2011, 278, 3579–3595. [Google Scholar]
- Hargreaves, D.C.; Crabtree, G.R. ATP-dependent chromatin remodeling: Genetics, genomics and mechanisms. Cell Res 2011, 21, 396–420. [Google Scholar]
- De Koning, L.; Corpet, A.; Haber, J.E.; Almouzni, G. Histone chaperones: An escort network regulating histone traffic. Nat. Struct. Mol. Biol 2007, 14, 997–1007. [Google Scholar]
- Avvakumov, N.; Nourani, A.; Côté, J. Histone chaperones: Modulators of chromatin marks. Mol. Cell 2011, 41, 502–514. [Google Scholar]
- Smerdon, M.J. DNA repair and the role of chromatin structure. Curr. Opin. Cell Biol 1991, 3, 422–428. [Google Scholar]
- Green, C.M.; Almouzni, G. When repair meets chromatin. First in series on chromatin dynamics. EMBO Rep 2002, 3, 28–33. [Google Scholar]
- Groth, A.; Rocha, W.; Verreault, A.; Almouzni, G. Chromatin challenges during DNA replication and repair. Cell 2007, 128, 721–733. [Google Scholar]
- Soria, G.; Polo, S.E.; Almouzni, G. Prime, repair, restore: The active role of chromatin in the DNA damage response. Mol. Cell 2012, 46, 722–734. [Google Scholar]
- Nouspikel, T. DNA repair in mammalian cells: Nucleotide excision repair: Variations on versatility. Cell. Mol. Life Sci 2009, 66, 994–1009. [Google Scholar]
- Taylor, J.S.; Brockie, I.R.; O’Day, C.L. A building block for the sequence-specific introduction of cis-syn thymine dimers into oligonucleotides. Solid-phase synthesis of TpT[c,s]pTpT. J. Am. Chem. Soc 1987, 109, 6735–6742. [Google Scholar]
- Mello, J.A.; Moggs, J.G.; Almouzni, G. Analysis of DNA repair and chromatin assembly in vitro using immobilized damaged DNA substrates. Methods Mol. Biol 2006, 314, 477–487. [Google Scholar]
- Ura, K.; Araki, M.; Saeki, H.; Masutani, C.; Ito, T.; Iwai, S.; Mizukoshi, T.; Kaneda, Y.; Hanaoka, F. ATP-dependent chromatin remodeling facilitates nucleotide excision repair of UV-induced DNA lesions in synthetic dinucleosomes. EMBO J 2001, 20, 2004–2014. [Google Scholar]
- Duan, M.-R.; Smerdon, M.J. UV damage in DNA promotes nucleosome unwrapping. J. Biol. Chem 2010, 285, 26295–26303. [Google Scholar]
- Katsumi, S.; Kobayashi, N.; Imoto, K.; Nakagawa, A.; Yamashina, Y.; Muramatsu, T.; Shirai, T.; Miyagawa, S.; Sugiura, S.; Hanaoka, F.; et al. In situ visualization of ultraviolet-light-induced DNA damage repair in locally irradiated human fibroblasts. J. Invest. Dermatol 2001, 117, 1156–1161. [Google Scholar]
- Moné, M.J.; Volker, M.; Nikaido, O.; Mullenders, L.H.; van Zeeland, A.A.; Verschure, P.J.; Manders, E.M.; van Driel, R. Local UV-induced DNA damage in cell nuclei results in local transcription inhibition. EMBO Rep 2001, 2, 1013–1017. [Google Scholar]
- Guerrero-Santoro, J.; Levine, A.S.; Rapić-Otrin, V. Co-localization of DNA repair proteins with UV-induced DNA damage in locally irradiated cells. Methods Mol. Biol 2011, 682, 149–161. [Google Scholar]
- Dinant, C.; de Jager, M.; Essers, J.; van Cappellen, W.A.; Kanaar, R.; Houtsmuller, A.B.; Vermeulen, W. Activation of multiple DNA repair pathways by sub-nuclear damage induction methods. J. Cell Sci 2007, 120, 2731–2740. [Google Scholar]
- Edenberg, H.; Hanawalt, P. Size of repair patches in the DNA of ultraviolet-irradiated HeLa cells. Biochim. Biophys. Acta 1972, 272, 361–372. [Google Scholar]
- Huang, J.C.; Sancar, A. Determination of minimum substrate size for human excinuclease. J. Biol. Chem 1994, 269, 19034–19040. [Google Scholar]
- Smerdon, M.J.; Lieberman, M.W. Nucleosome rearrangement in human chromatin during UV-induced DNA-repair synthesis. Proc. Natl. Acad. Sci. USA 1978, 75, 4238–4241. [Google Scholar]
- Smerdon, M.J.; Lieberman, M.W. Distribution within chromatin of deoxyribonucleic acid repair synthesis occurring at different times after ultraviolet radiation. Biochemistry 1980, 19, 2992–3000. [Google Scholar]
- Gong, F.; Fahy, D.; Smerdon, M.J. Rad4-Rad23 interaction with SWI/SNF links ATP-dependent chromatin remodeling with nucleotide excision repair. Nat. Struct. Mol. Biol 2006, 13, 902–907. [Google Scholar]
- Gérard, A.; Polo, S.E.; Roche, D.; Almouzni, G. Methods for studying chromatin assembly coupled to DNA repair. Meth. Enzymol 2006, 409, 358–374. [Google Scholar]
- Luijsterburg, M.S.; Lindh, M.; Acs, K.; Vrouwe, M.G.; Pines, A.; van Attikum, H.; Mullenders, L.H.; Dantuma, N.P. DDB2 promotes chromatin decondensation at UV-induced DNA damage. J. Cell Biol 2012, 197, 267–281. [Google Scholar]
- Pattison, D.I.; Davies, M.J. Actions of ultraviolet light on cellular structures. Experientia Supplementum 2006, 96, 131–157. [Google Scholar]
- Bader, N.; Grune, T. Protein oxidation and proteolysis. Biol. Chem 2006, 387, 1351–1355. [Google Scholar]
- Mathis, G.A.; Althaus, F.R. Isolation of 8-methoxypsoralen accessible DNA domains from chromatin of intact cells. Cell Biol. Toxicol 1990, 6, 35–45. [Google Scholar]
- Rubbi, C.P.; Milner, J. p53 is a chromatin accessibility factor for nucleotide excision repair of DNA damage. EMBO J 2003, 22, 975–986. [Google Scholar]
- Smerdon, M.J.; Watkins, J.F.; Lieberman, M.W. Effect of histone H1 removal on the distribution of ultraviolet-induced deoxyribonucleic acid repair synthesis within chromatin. Biochemistry 1982, 21, 3879–3885. [Google Scholar]
- Gaillard, P.H.; Martini, E.M.; Kaufman, P.D.; Stillman, B.; Moustacchi, E.; Almouzni, G. Chromatin assembly coupled to DNA repair: A new role for chromatin assembly factor I. Cell 1996, 86, 887–896. [Google Scholar]
- Gaillard, P.H.; Moggs, J.G.; Roche, D.M.; Quivy, J.P.; Becker, P.B.; Wood, R.D.; Almouzni, G. Initiation and bidirectional propagation of chromatin assembly from a target site for nucleotide excision repair. EMBO J 1997, 16, 6281–6289. [Google Scholar]
- Polo, S.E.; Roche, D.; Almouzni, G. New histone incorporation marks sites of UV repair in human cells. Cell 2006, 127, 481–493. [Google Scholar]
- Tagami, H.; Ray-Gallet, D.; Almouzni, G.; Nakatani, Y. Histone H3.1 and H3.3 complexes mediate nucleosome assembly pathways dependent or independent of DNA synthesis. Cell 2004, 116, 51–61. [Google Scholar]
- Loyola, A.; Bonaldi, T.; Roche, D.; Imhof, A.; Almouzni, G. PTMs on H3 variants before chromatin assembly potentiate their final epigenetic state. Mol. Cell 2006, 24, 309–316. [Google Scholar]
- Jansen, L.E.T.; Black, B.E.; Foltz, D.R.; Cleveland, D.W. Propagation of centromeric chromatin requires exit from mitosis. J. Cell Biol 2007, 176, 795–805. [Google Scholar]
- Dunleavy, E.M.; Almouzni, G.; Karpen, G.H. H3.3 is deposited at centromeres in S phase as a placeholder for newly assembled CENP-A in G1 phase. Nucleus 2011, 2, 146–157. [Google Scholar]
- Ray-Gallet, D.; Woolfe, A.; Vassias, I.; Pellentz, C.; Lacoste, N.; Puri, A.; Schultz, D.C.; Pchelintsev, N.A.; Adams, P.D.; Jansen, L.E.T.; et al. Dynamics of histone h3 deposition in vivo reveal a nucleosome gap-filling mechanism for h3.3 to maintain chromatin integrity. Mol. Cell 2011, 44, 928–941. [Google Scholar]
- Bodor, D.L.; Rodríguez, M.G.; Moreno, N. Analysis of protein turnover by quantitative SNAP-based pulse-chase imaging. Curr. Protoc. Cell Biol 2012, 8.1–8.8.34. [Google Scholar]
- Reed, S.H. Nucleotide excision repair in chromatin: Damage removal at the drop of a HAT. DNA Repair 2011, 10, 734–742. [Google Scholar]
- Kapetanaki, M.G.; Guerrero-Santoro, J.; Bisi, D.C.; Hsieh, C.L.; Rapić-Otrin, V.; Levine, A.S. The DDB1-CUL4ADDB2 ubiquitin ligase is deficient in xeroderma pigmentosum group E and targets histone H2A at UV-damaged DNA sites. Proc. Natl. Acad. Sci. USA 2006, 103, 2588–2593. [Google Scholar]
- Wang, H.; Zhai, L.; Xu, J.; Joo, H.-Y.; Jackson, S.; Erdjument-Bromage, H.; Tempst, P.; Xiong, Y.; Zhang, Y. Histone H3 and H4 ubiquitylation by the CUL4-DDB-ROC1 ubiquitin ligase facilitates cellular response to DNA damage. Mol. Cell 2006, 22, 383–394. [Google Scholar]
- Guerrero-Santoro, J.; Kapetanaki, M.G.; Hsieh, C.L.; Gorbachinsky, I.; Levine, A.S.; Rapić-Otrin, V. The cullin 4B-based UV-damaged DNA-binding protein ligase binds to UV-damaged chromatin and ubiquitinates histone H2A. Cancer Res 2008, 68, 5014–5022. [Google Scholar]
- Bergink, S.; Salomons, F.A.; Hoogstraten, D.; Groothuis, T.A.M.; de Waard, H.; Wu, J.; Yuan, L.; Citterio, E.; Houtsmuller, A.B.; Neefjes, J.; et al. DNA damage triggers nucleotide excision repair-dependent monoubiquitylation of histone H2A. Genes Dev 2006, 20, 1343–1352. [Google Scholar]
- Zhu, Q.; Wani, G.; Arab, H.H.; El-Mahdy, M.A.; Ray, A.; Wani, A.A. Chromatin restoration following nucleotide excision repair involves the incorporation of ubiquitinated H2A at damaged genomic sites. DNA Repair 2009, 8, 262–273. [Google Scholar]
- Marteijn, J.A.; Bekker-Jensen, S.; Mailand, N.; Lans, H.; Schwertman, P.; Gourdin, A.M.; Dantuma, N.P.; Lukas, J.; Vermeulen, W. Nucleotide excision repair-induced H2A ubiquitination is dependent on MDC1 and RNF8 and reveals a universal DNA damage response. J. Cell Biol 2009, 186, 835–847. [Google Scholar]
- Lan, L.; Nakajima, S.; Kapetanaki, M.G.; Hsieh, C.L.; Fagerburg, M.; Thickman, K.; Rodriguez-Collazo, P.; Leuba, S.H.; Levine, A.S.; Rapić-Otrin, V. Monoubiquitinated H2A destabilizes photolesion-containing nucleosomes with the concomitant release of the UV-damaged DNA-binding protein E3 ligase. J. Biol. Chem 2012, 287, 12036–12049. [Google Scholar]
- Jiang, Y.; Wang, X.; Bao, S.; Guo, R.; Johnson, D.G.; Shen, X.; Li, L. INO80 chromatin remodeling complex promotes the removal of UV lesions by the nucleotide excision repair pathway. Proc. Natl. Acad. Sci. USA 2010, 107, 17274–17279. [Google Scholar]
- Gong, F.; Fahy, D.; Liu, H.; Wang, W.; Smerdon, M.J. Role of the mammalian SWI/SNF chromatin remodeling complex in the cellular response to UV damage. Cell Cycle 2008, 7, 1067–1074. [Google Scholar]
- Zhao, Q.; Wang, Q.-E.; Ray, A.; Wani, G.; Han, C.; Milum, K.; Wani, A.A. Modulation of nucleotide excision repair by mammalian SWI/SNF chromatin-remodeling complex. J. Biol. Chem 2009, 284, 30424–30432. [Google Scholar]
- Zhang, L.; Zhang, Q.; Jones, K.; Patel, M.; Gong, F. The chromatin remodeling factor BRG1 stimulates nucleotide excision repair by facilitating recruitment of XPC to sites of DNA damage. Cell Cycle 2009, 8, 3953–3959. [Google Scholar]
- Ray, A.; Mir, S.N.; Wani, G.; Zhao, Q.; Battu, A.; Zhu, Q.; Wang, Q.-E.; Wani, A.A. Human SNF5/INI1, a component of the human SWI/SNF chromatin remodeling complex, promotes nucleotide excision repair by influencing ATM recruitment and downstream H2AX phosphorylation. Mol. Cell. Biol 2009, 29, 6206–6219. [Google Scholar]
- Citterio, E.; van den Boom, V.; Schnitzler, G.; Kanaar, R.; Bonte, E.; Kingston, R.E.; Hoeijmakers, J.H.; Vermeulen, W. ATP-dependent chromatin remodeling by the Cockayne syndrome B DNA repair-transcription-coupling factor. Mol. Cell. Biol 2000, 20, 7643–7653. [Google Scholar]
- Nouspikel, T. Multiple roles of ubiquitination in the control of nucleotide excision repair. Mech. Ageing Dev 2011, 132, 355–365. [Google Scholar]
- Lans, H.; Marteijn, J.A.; Vermeulen, W. ATP-dependent chromatin remodeling in the DNA-damage response. Epigenetics Chromatin 2012, 5, 4. [Google Scholar]
- Rajagopalan, S.; Nepa, J.; Venkatachalam, S. Chromodomain helicase DNA-binding protein 2 affects the repair of X-ray and UV-induced DNA damage. Environ. Mol. Mutagen 2012, 53, 44–50. [Google Scholar]
- Ransom, M.; Dennehey, B.K.; Tyler, J.K. Chaperoning histones during DNA replication and repair. Cell 2010, 140, 183–195. [Google Scholar]
- Smith, S.; Stillman, B. Purification and characterization of CAF-I, a human cell factor required for chromatin assembly during DNA replication in vitro. Cell 1989, 58, 15–25. [Google Scholar]
- Stillman, B. Chromatin assembly during SV40 DNA replication in vitro. Cell 1986, 45, 555–565. [Google Scholar]
- Moggs, J.G.; Grandi, P.; Quivy, J.P.; Jónsson, Z.O.; Hübscher, U.; Becker, P.B.; Almouzni, G. A CAF-1-PCNA-mediated chromatin assembly pathway triggered by sensing DNA damage. Mol. Cell. Biol 2000, 20, 1206–1218. [Google Scholar]
- Martini, E.; Roche, D.M.; Marheineke, K.; Verreault, A.; Almouzni, G. Recruitment of phosphorylated chromatin assembly factor 1 to chromatin after UV irradiation of human cells. J. Cell Biol 1998, 143, 563–575. [Google Scholar]
- Green, C.M.; Almouzni, G. Local action of the chromatin assembly factor CAF-1 at sites of nucleotide excision repair in vivo. EMBO J 2003, 22, 5163–5174. [Google Scholar]
- Shibahara, K.; Stillman, B. Replication-dependent marking of DNA by PCNA facilitates CAF-1-coupled inheritance of chromatin. Cell 1999, 96, 575–585. [Google Scholar]
- Mello, J.A.; Silljé, H.H.W.; Roche, D.M.J.; Kirschner, D.B.; Nigg, E.A.; Almouzni, G. Human Asf1 and CAF-1 interact and synergize in a repair-coupled nucleosome assembly pathway. EMBO Rep 2002, 3, 329–334. [Google Scholar]
- Kim, J.-A.; Haber, J.E. Chromatin assembly factors Asf1 and CAF-1 have overlapping roles in deactivating the DNA damage checkpoint when DNA repair is complete. Proc. Natl. Acad. Sci. USA 2009, 106, 1151–1156. [Google Scholar]
- Battu, A.; Ray, A.; Wani, A.A. ASF1A and ATM regulate H3K56-mediated cell-cycle checkpoint recovery in response to UV irradiation. Nucleic Acids Res 2011, 39, 7931–7945. [Google Scholar]
- Groth, A.; Corpet, A.; Cook, A.J.L.; Roche, D.; Bartek, J.; Lukas, J.; Almouzni, G. Regulation of replication fork progression through histone supply and demand. Science 2007, 318, 1928–1931. [Google Scholar]
- Luijsterburg, M.S.; Dinant, C.; Lans, H.; Stap, J.; Wiernasz, E.; Lagerwerf, S.; Warmerdam, D.O.; Lindh, M.; Brink, M.C.; Dobrucki, J.W.; et al. Heterochromatin protein 1 is recruited to various types of DNA damage. J. Cell Biol 2009, 185, 577–586. [Google Scholar]
- Livingstone-Zatchej, M.; Marcionelli, R.; Möller, K.; de Pril, R.; Thoma, F. Repair of UV lesions in silenced chromatin provides in vivo evidence for a compact chromatin structure. J. Biol. Chem 2003, 278, 37471–37479. [Google Scholar]
- Irizar, A.; Yu, Y.; Reed, S.H.; Louis, E.J.; Waters, R. Silenced yeast chromatin is maintained by Sir2 in preference to permitting histone acetylations for efficient NER. Nucleic Acids Res 2010, 38, 4675–4686. [Google Scholar]

© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Adam, S.; Polo, S.E. Chromatin Dynamics during Nucleotide Excision Repair: Histones on the Move. Int. J. Mol. Sci. 2012, 13, 11895-11911. https://doi.org/10.3390/ijms130911895
Adam S, Polo SE. Chromatin Dynamics during Nucleotide Excision Repair: Histones on the Move. International Journal of Molecular Sciences. 2012; 13(9):11895-11911. https://doi.org/10.3390/ijms130911895
Chicago/Turabian StyleAdam, Salomé, and Sophie E. Polo. 2012. "Chromatin Dynamics during Nucleotide Excision Repair: Histones on the Move" International Journal of Molecular Sciences 13, no. 9: 11895-11911. https://doi.org/10.3390/ijms130911895