Modification by Ubiquitin-Like Proteins: Significance in Apoptosis and Autophagy Pathways

Abstract
:1. Introduction
2. Ubiquitin-Like Modifiers as Independent Modules
2.1. Small Ubiquitin-Related Modifier (SUMO)
2.2. Neural Precursor Cell-Expressed Developmentally Down-Regulated (NEDD8)
2.3. Human HLA-F Adjacent Transcript 10 (FAT10)
2.4. Interferon Stimulated Gene 15 (ISG15)
2.5. Autophagy-Related (ATG) Genes
2.6. Ubiquitin-Related Modifier 1 (Urm1)
2.7. Ubiquitin-Fold Modifier 1 (Ufm1)
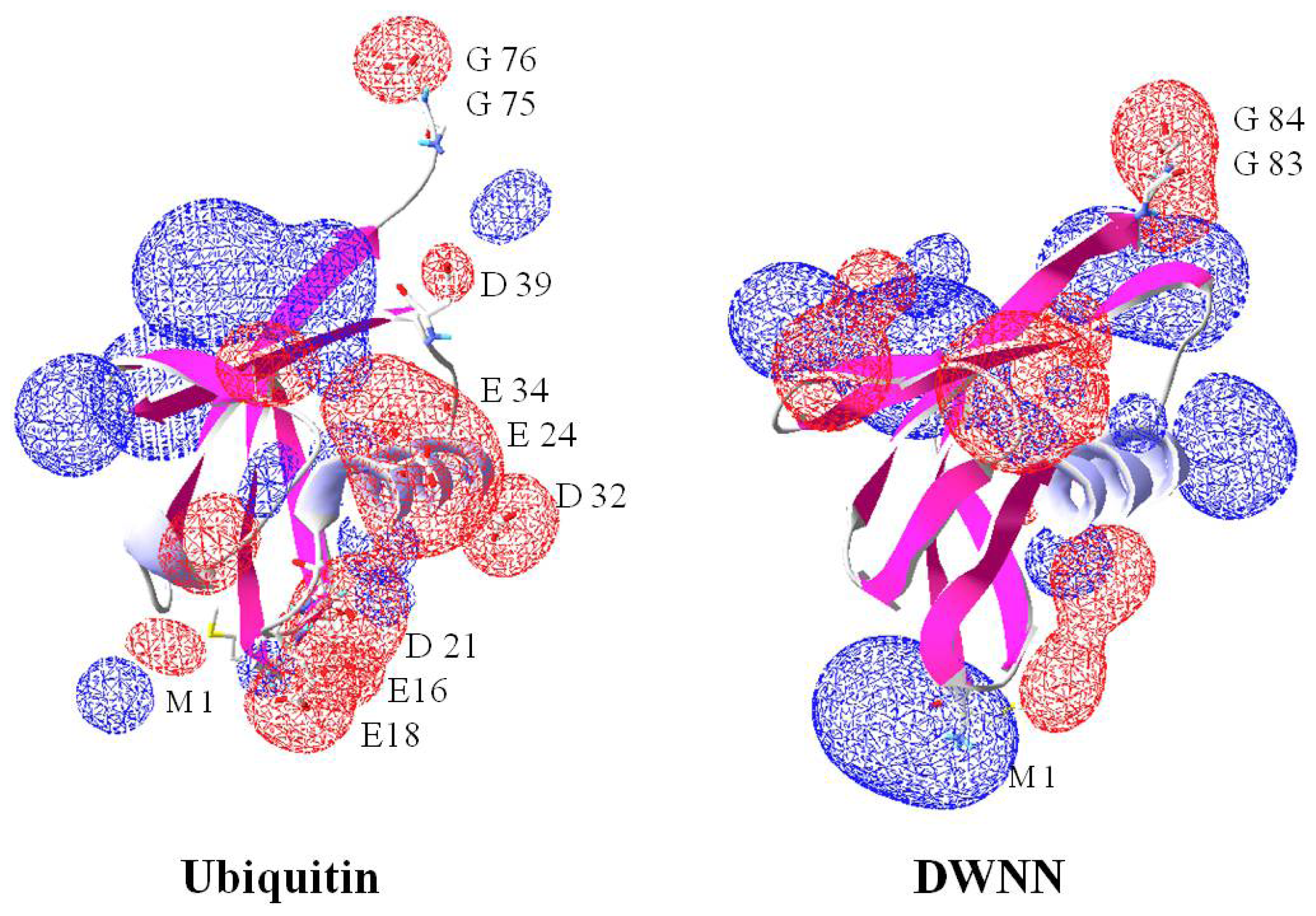
2.8. Domain with no Name (DWNN)
3. Proteins with an Ubiquitin Domain
3.1. Homocysteine-Inducible, Endoplasmic Reticulum Stress-Inducible, Ubiquitin-Like Domain Member 1 (Herpud1)
3.2. Parkin
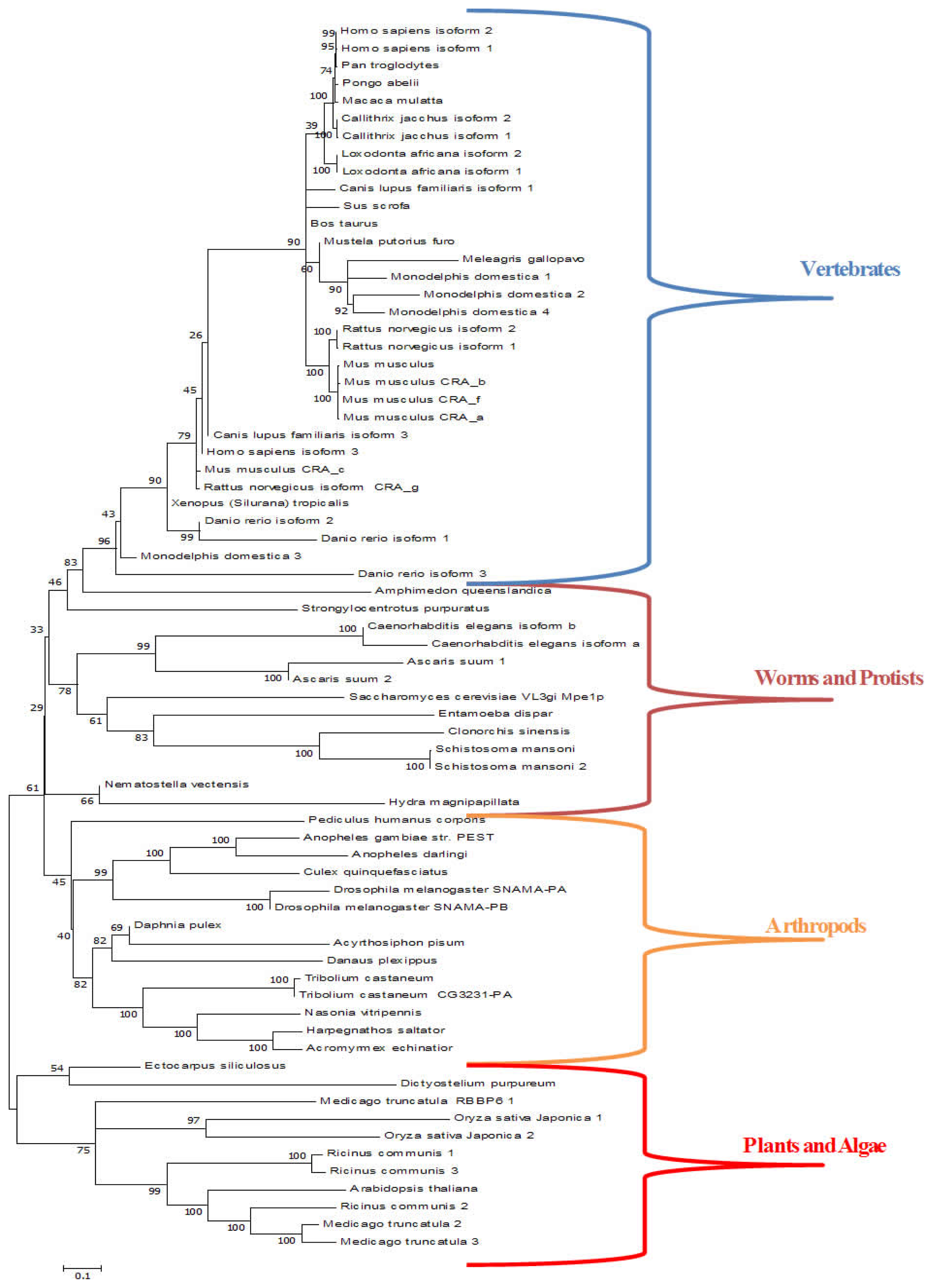
3.3. Retinoblastoma Binding Protein 6 (RBBP6)
3.4. Ubiquilin
4. Potential for Therapeutic Intervention
5. The Ubiquitin and Ubiquitin-Like Protein System as a Drug Target
Acknowledgments
References
- Kerscher, O.; Felberbaum, R.; Hochstrasser, M. Modification of proteins by ubiquitin and ubiquitin-like proteins. Annu. Rev. Cell Dev. Biol 2006, 22, 159–180. [Google Scholar]
- Hershko, A. The ubiquitin system for protein degradation and some of its roles in the control of the cell division cycle. Cell Death Differ 2005, 12, 1191–1197. [Google Scholar]
- Sadeh, R.; Breitschopf, K.; Bercovich, B.; Zoabi, M.; Kravtsova-Ivantsiv, Y.; Kornitzer, D.; Schwartz, A.; Ciechanover, A. The N-terminal domain of MyoD is necessary and sufficient for its nuclear localization-dependent degradation by the ubiquitin system. Proc. Natl. Acad. Sci. USA 2008, 105, 15690–15695. [Google Scholar]
- Ciechanover, A.; Ben-Saadon, R. N-terminal ubiquitination: More protein substrates join in. Trends Cell Biol 2004, 14, 103–106. [Google Scholar]
- Fajerman, I.; Schwartz, A.L.; Ciechanover, A. Degradation of the Id2 developmental regulator: Targeting via N-terminal ubiquitination. Biochem. Biophys. Res.Commun 2004, 314, 505–512. [Google Scholar]
- Hershko, A.; Ciechanover, A. The ubiquitin system. Annu. Rev. Biochem 1998, 67, 425–479. [Google Scholar]
- Jentsch, S.; Pyrowolakis, G. Ubiquitin and its kin: How close are the family ties? Trends Cell Biol 2000, 10, 335–341. [Google Scholar]
- Herrmann, J.; Lerman, L.O.; Lerman, A. Ubiquitin and ubiquitin-like proteins in protein regulation. Circ. Res 2007, 100, 1276–1291. [Google Scholar]
- Bohren, K.M.; Nadkarni, V.; Song, J.H.; Gabbay, K.H.; Owerbach, D. A M55V polymorphism in a novel SUMO gene (SUMO-4) differentially activates heat shock transcription factors and is associated with susceptibility to type I diabetes mellitus. J. Biol. Chem 2004, 279, 27233–27238. [Google Scholar]
- Odagiri, S.; Tanji, K.; Mori, F.; Kakita, A.; Takahashi, H.; Kamitani, T.; Wakabayashi, K. Immunohistochemical analysis of Marinesco bodies, using antibodies against proteins implicated in the ubiquitin-proteasome system, autophagy and aggresome formation. Neuropathology 2012, 32, 261–266. [Google Scholar]
- Zhao, C.; Denison, C.; Huibregtse, J.M.; Gygi, S.; Krug, R.M. Human ISG15 conjugation targets both IFN-induced and constitutively expressed proteins functioning in diverse cellular pathways. Proc. Natl. Acad. Sci. USA 2005, 102, 10200–10205. [Google Scholar]
- French, S.W.; Bardag-Gorce, F.; French, B.A.; Li, J.; Oliva, J. The role of innate immunity in the pathogenesis of preneoplasia in drug-induced chronic hepatitis based on a mouse model. Exp Mol Pathol 2011, 91, 653–659. [Google Scholar]
- McNally, T.; Huang, Q.; Janis, R.S.; Lui, Z.; Olejniczak, E.T.; Reilly, R.M. Structural analysis of UBL5, a novel ubiquitin-like modifier. Protein Sci 2003, 12, 1562–1566. [Google Scholar]
- Allende-Vega, N.; Dayal, S.; Agarwala, U.; Sparks, A.; Bourdon, J.C.; Saville, M.K. p53 is activated in response to disruption of the pre-mRNA splicing machinery. Oncogene 2012. [Google Scholar] [CrossRef]
- Kas, K.; Michiels, L.; Merregaert, J. Genomic structure and expression of the human fau gene: Encoding the ribosomal protein S30 fused to a ubiquitin-like protein. Biochem. Biophys. Res. Commun 1992, 187, 927–933. [Google Scholar]
- Geng, J.; Klionsky, D.J. The Atg8 and Atg12 ubiquitin-like conjugation systems in macroautophagy. EMBO Rep 2008, 9, 859–864. [Google Scholar]
- Sasakawa, H.; Sakata, E.; Yamaguchi, Y.; Komatsu, M.; Tatsumi, K.; Kominami, E.; Tanaka, K.; Kato, K. Solution structure and dynamics of Ufm1, a ubiquitin-fold modifier 1. Biochem. Biophys. Res. Commun 2006, 343, 21–26. [Google Scholar]
- Mather, A.; Rakghotho, M.; Ntwasa, M. SNAMA, a novel protein with a DWNN domain and a RING finger-like motif: A possible role in apoptosis. Biochim. Biophys. Acta 2005, 1727, 169–176. [Google Scholar]
- Schlieker, C.D.; van der Veen, A.G.; Damon, J.R.; Spooner, E.; Ploegh, H.L. A functional proteomics approach links the ubiquitin-related modifier Urm1 to a tRNA modification pathway. Proc. Natl. Acad. Sci. USA 2008, 105, 18255–18260. [Google Scholar]
- Matunis, M.J.; Coutavas, E.; Blobel, G. A novel ubiquitin-like modification modulates the partitioning of the Ran-GTPase-activating protein ranGAP1 between the cytosol and the nuclear pore complex. J. Cell Biol 1996, 135, 1457–1470. [Google Scholar]
- Jürgen Dohmen, R. SUMO protein modification. Biochim. Biophys. Acta (BBA)-Mol. Cell Res 2004, 1695, 113–131. [Google Scholar]
- Bayer, P.; Arndt, A.; Metzger, S.; Mahajan, R.; Melchior, F.; Jaenicke, R.; Becker, J. Structure determination of the small ubiquitin-related modifier SUMO-1. J. Mol. Biol 1998, 280, 275–286. [Google Scholar]
- Meulmeester, E.; Melchior, F. Cell biology: SUMO. Nature 2008, 452, 709–711. [Google Scholar]
- Johnson, E.S. Protein modification by SUMO. Annu. Rev. Biochem 2004, 73, 355–382. [Google Scholar]
- Su, H.-L.; Li, S.S.L. Molecular features of human ubiquitin-like SUMO genes and their encoded proteins. Gene 2002, 296, 65–73. [Google Scholar]
- Hay, R.T. SUMO-specific proteases: A twist in the tail. Trends Cell Biol 2007, 17, 370–376. [Google Scholar]
- Tatham, M.H.; Jaffray, E.; Vaughan, O.A.; Desterro, J.M.P.; Botting, C.H.; Naismith, J.H.; Hay, R.T. Polymeric chains of SUMO-2 and SUMO-3 are conjugated to protein substrates by SAE1/SAE2 and ubc9. J. Biol. Chem 2001, 276, 35368–35374. [Google Scholar]
- Rodriguez, M.S.; Dargemont, C.; Hay, R.T. SUMO-1 conjugation in vivo requires both a consensus modification motif and nuclear targeting. J. Biol. Chem 2001, 276, 12654–12659. [Google Scholar]
- Castillo-Lluva, S.; Tatham, M.H.; Jones, R.C.; Jaffray, E.G.; Edmondson, R.D.; Hay, R.T.; Malliri, A. SUMOylation of the GTPase Rac1 is required for optimal cell migration. Nat. Cell Biol 2010, 12, 1078–1085. [Google Scholar]
- Marx, J. SUMO wrestles its way to prominence in the cell. Science 2005, 307, 836–839. [Google Scholar]
- Hay, R.T.; Vuillard, L.; Desterro, J.M.P.; Rodriguez, M.S. Control of NF–κB transcriptional activation by signal induced proteolysis of IκBα. Phil. Trans. R. Soc. Lond. B 1999, 354, 1601–1609. [Google Scholar]
- Mabb, A.; Miyamoto, S. SUMO and NF–κB ties. Cell Mol. Life Sci 2007, 64, 1979–1996. [Google Scholar]
- Kirsh, O.; Seeler, J.-S.; Pichler, A.; Gast, A.; Muller, S.; Miska, E.; Mathieu, M.; Harel-Bellan, A.; Kouzarides, T.; Melchior, F.; et al. The SUMO E3 ligase RanBP2 promotes modification of the HDAC4 deacetylase. EMBO J 2002, 21, 2682–2691. [Google Scholar]
- Yamamoto, H.; Ihara, M.; Matsuura, Y.; Kikuchi, A. Sumoylation is involved in [beta]-catenin-dependent activation of Tcf-4. EMBO J 2003, 22, 2047–2059. [Google Scholar]
- Ihara, M.; Yamamoto, H.; Kikuchi, A. SUMO-1 modification of PIASy, an E3 ligase, is necessary for PIASy-dependent activation of Tcf-4. Mol. Cell Biol 2005, 25, 3506–3518. [Google Scholar]
- Goodson, M.L.; Hong, Y.; Rogers, R.; Matunis, M.J.; Park-Sarge, O.-K.; Sarge, K.D. SUMO-1 modification regulates the DNA binding activity of heat shock transcription factor 2, a promyelocytic leukemia nuclear body associated transcription factor. J. Biol. Chem 2001, 276, 18513–18518. [Google Scholar]
- Stehmeier, P.; Muller, S. Regulation of p53 family members by the ubiquitin-like SUMO system. DNA Repair 2009, 8, 491–498. [Google Scholar]
- Rodriguez, M.S.; Desterro, J.M.P.; Lain, S.; Midgley, C.A.; Lane, D.P.; Hay, R.T. SUMO-1 modification activates the transcriptional response of p53. EMBO J 1999, 18, 6455–6461. [Google Scholar]
- Hoege, C.; Pfander, B.; Moldovan, G.-L.; Pyrowolakis, G.; Jentsch, S. RAD6-dependent DNA repair is linked to modification of PCNA by ubiquitin and SUMO. Nature 2002, 419, 135–141. [Google Scholar]
- Bergink, S.; Jentsch, S. Principles of ubiquitin and SUMO modifications in DNA repair. Nature 2009, 458, 461–467. [Google Scholar]
- Owerbach, D.; McKay, E.M.; Yeh, E.T.H.; Gabbay, K.H.; Bohren, K.M. A proline-90 residue unique to SUMO-4 prevents maturation and sumoylation. Biochem. Biophys. Res. Commun 2005, 337, 517–520. [Google Scholar]
- Kumar, S.; Yoshida, Y.; Noda, M. Cloning of a cDNA which encodes a novel ubiquitin-like protein. Biochem. Biophys. Res. Commun 1993, 195, 393–399. [Google Scholar]
- Ohki, Y.; Funatsu, N.; Konishi, N.; Chiba, T. The mechanism of poly-NEDD8 chain formation in vitro. Biochem. Biophys. Res. Commun 2009, 381, 443–447. [Google Scholar]
- Liakopoulos, D.; Doenges, G.; Matuschewski, K.; Jentsch, S. A novel protein modification pathway related to the ubiquitin system. EMBO J 1998, 17, 2208–2214. [Google Scholar]
- Hori, T.; Osaka, F.; Chiba, T.; Miyamoto, C.; Okabayashi, K.; Shimbara, N.; Kato, S.; Tanaka, K. Covalent modification of all members of human cullin family proteins by NEDD8. Oncogene 1999, 18, 6829–6834. [Google Scholar]
- Kawakami, T.; Chiba, T.; Suzuki, T.; Iwai, K.; Yamanaka, K.; Minato, N.; Suzuki, H.; Shimbara, N.; Hidaka, Y.; Osaka, F.; et al. NEDD8 recruits E2-ubiquitin to SCF E3 ligase. EMBO J 2001, 20, 4003–4012. [Google Scholar]
- Watson, I.R.; Irwin, M.S.; Ohh, M. NEDD8 pathways in cancer, sine quibus non. Cancer Cell 2011, 19, 168–176. [Google Scholar]
- Deshaies, R.J.; Emberley, E.D.; Saha, A. Control of cullin-ring ubiquitin ligase activity by Nedd8. Subcell. Biochem 2010, 54, 41–56. [Google Scholar]
- Sarikas, A.; Hartmann, T.; Pan, Z.-Q. The cullin protein family. Genome Biol 2011, 12, 220. [Google Scholar]
- Liu, L.; Lee, S.; Zhang, J.; Peters, S.B.; Hannah, J.; Zhang, Y.; Yin, Y.; Koff, A.; Ma, L.; Zhou, P. CUL4A abrogation augments DNA damage response and protection against skin carcinogenesis. Mol. Cell 2009, 34, 451–460. [Google Scholar]
- Du, J.; Zhang, J.; Su, Y.; Liu, M.; Ospina, J.K.; Yang, S.; Zhu, A.J. In vivo RNAi screen reveals Neddylation genes as novel regulators of Hedgehog signaling. PLoS One 2011, 6, e24168. [Google Scholar]
- Gao, F.; Cheng, J.; Shi, T.; Yeh, E.T.H. Neddylation of a breast cancer-associated protein recruits a class III histone deacetylase that represses NFκB-dependent transcription. Nat. Cell Biol 2006, 8, 1171–1177. [Google Scholar]
- Stickle, N.H.; Chung, J.; Klco, J.M.; Hill, R.P.; Kaelin, W.G., Jr; Ohh, M. pVHL modification by NEDD8 is required for fibronectin matrix assembly and suppression of tumor development. Mol. Cell Biol. 2004, 24, 3251–3261. [Google Scholar]
- Xirodimas, D.P.; Sundqvist, A.; Nakamura, A.; Shen, L.; Botting, C.; Hay, R.T. Ribosomal proteins are targets for the NEDD8 pathway. EMBO Rep 2008, 9, 280–286. [Google Scholar]
- Shamay, M.; Greenway, M.; Liao, G.; Ambinder, R.F.; Hayward, D. De novo DNA methyltransferase DNMT3b interacts with NEDD8-modified proteins. J. Biol. Chem 2010, 285, 36377–36386. [Google Scholar]
- Broemer, M.; Tenev, T.; Rigbolt, K.T.G.; Hempel, S.; Blagoev, B.; Silke, J.; Ditzel, M.; Meier, P. Systematic in vivo RNAi analysis identifies IAPs as NEDD8-E3 ligases. Mol. Cell 2010, 40, 810–822. [Google Scholar]
- Fan, W.; Cai, W.; Parimoo, S.; Lennon, G.G.; Weissman, S.M. Identification of seven new human MHC class I region genes around the HLA-F locus. Immunogenetics 1996, 44, 97–103. [Google Scholar]
- Liu, Y.-C.; Pan, J.; Zhang, C.; Fan, W.; Collinge, M.; Bender, J.R.; Weissman, S.M. A MHC-encoded ubiquitin-like protein (FAT10) binds noncovalently to the spindle assembly checkpoint protein MAD2. Proc. Natl. Acad. Sci. USA 1999, 96, 4313–4318. [Google Scholar]
- Lee, C.G.; Ren, J.; Cheong, I.S.; Ban, K.H.; Ooi, L.L.; Tan, S.Y.; Kan, A.; Nuchprayoon, I.; Jin, R.; Lee, K.-H.; et al. Expression of the FAT10 gene is highly upregulated in hepatocellular carcinoma and other gastrointestinal and gynecological cancers. Oncogene 2003, 22, 2592–2603. [Google Scholar]
- Aichem, A.; Pelzer, C.; Lukasiak, S.; Kalveram, B.; Sheppard, P.W.; Rani, N.; Schmidtke, G.; Groettrup, M. USE1 is a bispecific conjugating enzyme for ubiquitin and FAT10, which FAT10ylates itself in cis. Nat. Commun 2010, 1. [Google Scholar] [CrossRef]
- Chiu, Y.H.; Sun, Q.; Chen, Z.J. E1-L2 activates both ubiquitin and FAT10. Mol. Cell Biol 2007, 27, 1014–1023. [Google Scholar]
- Lukasiak, S.; Schiller, C.; Oehlschlaeger, P.; Schmidtke, G.; Krause, P.; Legler, D.F.; Autschbach, F.; Schirmacher, P.; Breuhahn, K.; Groettrup, M. Proinflammatory cytokines cause FAT10 upregulation in cancers of liver and colon. Oncogene 2008, 27, 6068–6074. [Google Scholar]
- Canaan, A.; Yu, X.; Booth, C.J.; Lian, J.; Lazar, I.; Gamfi, S.L.; Castille, K.; Kohya, N.; Nakayama, Y.; Liu, Y.-C.; et al. FAT10/Diubiquitin-like protein-deficient mice exhibit minimal phenotypic differences. Mol. Cell Biol 2006, 26, 5180–5189. [Google Scholar]
- Gong, P.; Canaan, A.; Wang, B.; Leventhal, J.; Snyder, A.; Nair, V.; Cohen, C.D.; Kretzler, M.; D’Agati, V.; Weissman, S.; et al. The ubiquitin-like protein FAT10 mediates NF–κB activation. J. Am. Soc. Nephrol 2010, 21, 316–326. [Google Scholar]
- Li, T.; Santockyte, R.; Yu, S.; Shen, R.-F.; Tekle, E.; Lee, C.G.L.; Yang, D.C.H.; Chock, P.B. FAT10 modifies p53 and upregulates its transcriptional activity. Arch. Biochem. Biophys 2011, 509, 164–169. [Google Scholar]
- Ren, J.; Wang, Y.; Gao, Y.; Mehta, S.B.K.; Lee, C.G.L. FAT10 mediates the effect of TNF-α in inducing chromosomal instability. J. Cell Sci 2011, 124, 3665–3675. [Google Scholar]
- Raasi, S.; Schmidtke, G.; Groettrup, M. The ubiquitin-like protein FAT10 forms covalent conjugates and induces apoptosis. J. Biol. Chem 2001, 276, 35334–35343. [Google Scholar]
- Buchsbaum, S.; Bercovich, B.; Ciechanover, A. FAT10 is a proteasomal degradation signal that is itself regulated by ubiquitination. Mol. Biol. Cell 2012, 23, 225–232. [Google Scholar]
- Zhang, D.W.; Jeang, K.T.; Lee, C.G.L. p53 negatively regulates the expression of FAT10, a gene upregulated in various cancers. Oncogene 2006, 25, 2318–2327. [Google Scholar]
- Doyle, S.E.; Vaidya, S.A.; O’Connell, R.; Dadgostar, H.; Dempsey, P.W.; Wu, T.-T.; Rao, G.; Sun, R.; Haberland, M.E.; Modlin, R.L.; et al. IRF3 mediates a TLR3/TLR4-specific antiviral gene program. Immunity 2002, 17, 251–263. [Google Scholar]
- Martensen, P.M. Small ISGs coming forward. J. Interferon Cytokine Res 2004, 24, 1–19. [Google Scholar]
- Reich, N.; Evans, B.; Levy, D.; Fahey, D.; Knight, E., Jr; Darnell, J.E., Jr. Interferon-induced transcription of a gene encoding a 15-kDa protein depends on an upstream enhancer element (promoter/transcription regulation). Proc. Natl. Acad. Sci. USA 1987, 84, 6394–6398. [Google Scholar]
- Hummer, B.T.; Li, X.L.; Hassel, B.A. Role for p53 in gene induction by double-stranded RNA. J. Virol 2001, 75, 7774–7777. [Google Scholar]
- Jeon, Y.J.; Yoo, H.M.; Chung, C.H. ISG15 and immune diseases. Biochim. Biophys. Acta Mol. Basis Dis 2010, 1802, 485–496. [Google Scholar]
- Zou, W.; Zhang, D.-E. The interferon-inducible ubiquitin-protein isopeptide ligase (E3) EFP also functions as an ISG15 E3 ligase. J. Biol. Chem 2006, 281, 3989–3994. [Google Scholar]
- Wong, J.J.Y.; Pung, Y.F.; Sze, N.S.-K.; Chin, K.-C. HERC5 is an IFN-induced HECT-type E3 protein ligase that mediates type I IFN-induced ISGylation of protein targets. Proc. Natl. Acad. Sci. USA 2006, 103, 10735–10740. [Google Scholar]
- Van der Veen, A.G.; Ploegh, H.L. Ubiquitin-like proteins. Annu. Rev. Biochem 2012, 81, 323–357. [Google Scholar]
- Skaug, B.; Chen, Z.J. Emerging role of ISG15 in antiviral immunity. Cell 2010, 143, 187–190. [Google Scholar]
- Durfee, L.A.; Lyon, N.; Seo, K.; Huibregtse, J.M. The ISG15 conjugation system broadly targets newly synthesized proteins: Implications for the antiviral function of ISG15. Mol. Cell 2010, 38, 722–732. [Google Scholar]
- Nisole, S.; Stoye, J.P.; Saïb, A. Trim family proteins: Retroviral restriction and antiviral defense. Nat. Rev. Microbiol 2005, 3, 799–808. [Google Scholar]
- Nakasato, N.; Ikeda, K.; Urano, T.; Horie-Inoue, K.; Takeda, S.; Inoue, S. A ubiquitin E3 ligase Efp is up-regulated by interferons and conjugated with ISG15. Biochem. Biophys. Res. Commun 2006, 351, 540–546. [Google Scholar]
- Lenschow, D.J.; Giannakopoulos, N.V.; Gunn, L.J.; Johnston, C.; O’Guin, A.K.; Schmidt, R.E.; Levine, B.; Virgin, H.W., IV. J. Virol. 2005, 79, 13974–13983.
- Lenschow, D.J.; Lai, C.; Frias-Staheli, N.; Giannakopoulos, N.V.; Lutz, A.; Wolff, T.; Osiak, A.; Levine, B.; Schmidt, R.E.; Garcia-Sastre, A.; et al. IFN-stimulated gene 15 functions as a critical antiviral molecule against influenza, herpes, and Sindbis viruses. Proc. Natl. Acad. Sci. USA 2007, 104, 1371–1376. [Google Scholar]
- Okumura, A.; Lu, G.; Pitha-Rowe, I.; Pitha, P.M. Innate antiviral response targets HIV-1 release by the induction of ubiquitin-like protein ISG15. Proc. Natl. Acad. Sci. USA 2006, 103, 1440–1445. [Google Scholar]
- Okumura, A.; Pitha, P.M.; Harty, R.N. ISG15 inhibits Ebola VP40 VLP budding in an L-domain-dependent manner by blocking Nedd4 ligase activity. Proc. Natl. Acad. Sci. USA 2008, 105, 3974–3979. [Google Scholar]
- Yuan, W.; Krug, R.M. Influenza B virus NS1 protein inhibits conjugation of the interferon (IFN)-induced ubiquitin-like ISG15 protein. EMBO J 2001, 20, 362–371. [Google Scholar]
- Tang, Y.; Zhong, G.; Zhu, L.; Liu, X.; Shan, Y.; Feng, H.; Bu, Z.; Chen, H.; Wang, C. Herc5 attenuates influenza A virus by catalyzing ISGylation of viral NS1 protein. J. Immunol 2010, 184, 5777–5790. [Google Scholar]
- Akutsu, M.; Ye, Y.; Virdee, S.; Chin, J.W.; Komander, D. Molecular basis for ubiquitin and ISG15 cross-reactivity in viral ovarian tumor domains. Proc. Natl. Acad. Sci. USA 2011, 108, 2228–2233. [Google Scholar]
- Malakhova, O.A.; Yan, M.; Malakhov, M.P.; Yuan, Y.; Ritchie, K.J.; Kim, K.I.; Peterson, L.F.; Shuai, K.; Zhang, D.-E. Protein ISGylation modulates the JAK-STAT signaling pathway. Genes Dev 2003, 17, 455–460. [Google Scholar]
- Jeon, Y.J.; Choi, J.S.; Lee, J.Y.; Yu, K.R.; Kim, S.; Ka, S.H.; Oh, K.H.; Kim, K.I.; Zhang, D.-E.; Bang, O.S.; et al. ISG15 modification of filamin B negatively regulates the type I interferon-induced JNK signalling pathway. EMBO Rep 2009, 10, 374–380. [Google Scholar]
- He, C.; Klionsky, D.J. Regulation mechanisms and signaling pathways of autophagy. Annu. Rev. Genet 2009, 3, 67–93. [Google Scholar]
- Hanada, T.; Ohsumi, Y. Structure-function relationship of Atg12, a ubiquitin-like modifier essential for autophagy. Autophagy 2005, 1, 110–118. [Google Scholar]
- Ichimura, Y.; Kirisako, T.; Takao, T.; Satomi, Y.; Shimonishi, Y.; Ishihara, N.; Mizushima, N.; Tanida, I.; Kominami, E.; Ohsumi, M.; et al. A ubiquitin-like system mediates protein lipidation. Nature 2000, 408, 488–492. [Google Scholar]
- Kirisakoa, T.; Ichimura, Y.; Okadac, H.; Kabeyaa, Y.; Mizushimaa, N.; Yoshimoria, T.; Ohsumic, M.; Takaoe, T.; Nodaa, T.; Ohsumia, Y. The reversible modification regulates the membrane-binding state of APG8/AUT7 essential for autophagy and the cytoplasm to vacuole targeting pathway. J. Cell Biol 2000, 151, 263–275. [Google Scholar]
- Tanida, I.; Tanida-Miyake, E.; T.U.; Kominami, E. The human homolog of saccharomyces cerevisiae APG7p is a protein-activating enzyme for multiple substrates including human APG12p, GATE-16, GABARAP, and MAP-LC3. J. Biol. Chem 2001, 276, 1701–1706. [Google Scholar]
- Shintani, T.; Mizushima, N.; Ogawa, Y.; Matsuura, A.; Noda, T.; Ohsum, Y. Apg10p, a novel protein-conjugating enzyme essential for autophagy in yeast. EMBO J 1999, 18, 5234–5241. [Google Scholar]
- Mizushima, N.; Noda, T.; Ohsumi, Y. Apg16p is required for the function of the Apg12p-Apg5p conjugate in the yeast autophagy pathway. EMBO J 1999, 18, 3888–3896. [Google Scholar]
- Radoshevich, L.; Murrow, L.; Chen, N.; Fernandez, E.; Roy, S.; Fung, C.; Debnath, J. ATG12 conjugation to ATG3 regulates mitochondrial homeostasis and cell death. Cell 2010, 142, 590–600. [Google Scholar]
- Furukawa, K.; Mizushima, N.; Noda, T.; Ohsumi, Y. A protein conjugation system in yeast with homology to biosynthetic enzyme reaction of prokaryotes. J. Biol. Chem 2000, 275, 7462–7465. [Google Scholar]
- Singh, S.; Tonelli, M.; Tyler, R.C.; Bahrami, A.; Lee, M.S.; Markley, J.L. Three-dimensional structure of the AAH26994.1 protein from Mus musculus, a putative eukaryotic Urm1. Protein Sci 2005, 14, 2095–2102. [Google Scholar]
- Komatsu, M.; Chiba, T.; Tatsumi, K.; Iemura, S.; Tanida, I.; Okazaki, N.; Ueno, T.; Kominami, E.; Natsume, T.; Tanaka, K. A novel protein-conjugating system for Ufm1, a ubiquitin-fold modifier. EMBO J 2004, 23, 1977–1986. [Google Scholar]
- Tatsumi, K.; Sou, Y.-S.; Tada, N.; Nakamura, E.; Iemura, S.-I.; Natsume, T.; Kang, S.H.; Chung, C.H.; Kasahara, M.; Kominami, E.; et al. A novel type of E3 ligase for the Ufm1 conjugation system. J. Biol. Chem 2010, 285, 5417–5427. [Google Scholar]
- Tatsumi, K.; Yamamoto-Mukai, H.; Shimizu, R.; Waguri, S.; Sou, Y.S.; Sakamoto, A.; Taya, C.; Shitara, H.; Hara, T.; Chung, C.H.; et al. The Ufm1-activating enzyme Uba5 is indispensable for erythroid differentiation in mice. Nat. Commun 2011, 2, 181. [Google Scholar]
- Pugh, D.; Eiso, A.B.; Faro, A.; Lutya, P.; Hoffmann, E.; Rees, D.J. DWNN, a novel ubiquitin-like domain, implicates RBBP6 in mRNA processing and ubiquitin-like pathways. BMC Struct. Biol 2006, 6. [Google Scholar] [CrossRef]
- Ntwasa, M. The retinoblastoma binding protein 6 is a potential target for therapeutic drugs. Biotechnol. Mol. Biol. Rev 2008, 3, 24–31. [Google Scholar]
- Li, L.; Deng, B.; Xing, G.; Teng, Y.; Tian, C.; Cheng, X.; Yin, X.; Yang, J.; Gao, X.; Zhu, Y.; et al. PACT is a negative regulator of p53 and essential for cell growth and embryonic development. Proc. Natl. Acad. Sci. USA 2007, 104, 7951–7956. [Google Scholar]
- Jones, S.N.; Roe, A.E.; Donehower, L.A.; Bradley, A. Rescue of embryonic lethality in Mdm2-deficient mice by absence of p53. Nature 1995, 378, 206–208. [Google Scholar]
- Shimizu, N.; Minoshima, S.; Asakawa, S. Cloning of Parkin gene and its function. No To Shinkei 1999, 51, 487–491. [Google Scholar]
- Bronner, C.; Achour, M.; Arima, Y.; Chataigneau, T.; Saya, H.; Schini-Kerth, V.B. The UHRF family: Oncogenes that are drugable targets for cancer therapy in the near future? Pharmacol. Ther 2007, 115, 419–434. [Google Scholar]
- Masutani, C.; Sugasawa, K.; Yanagisawa, J.; Sonoyama, T.; Ui, M.; Enomoto, T.; Takio, K.; Tanaka, K.; van der Spek, P.J.; Bootsma, D.; et al. Purification and cloning of a nucleotide excision repair complex involving the xeroderma pigmentosum group C protein and a human homologue of yeast RAD23. EMBO J 1994, 13, 1831–1843. [Google Scholar]
- Gamerdinger, M.; Hajieva, P.; Kaya, A.M.; Wolfrum, U.; Hartl, F.U.; Behl, C. Cloning and functional analysis of BAG-1: A novel Bcl-2-binding protein with anti-cell death activity. EMBO J 2009, 28, 889–901. [Google Scholar]
- Leznicki, P.; Clancy, A.; Schwappach, B.; High, S. Bat3 promotes the membrane integration of tail-anchored proteins. J. Cell Sci 2010, 123, 2170–2178. [Google Scholar]
- Sirkis, R.; Gerst, J.E.; Fass, D. Ddi1, a eukaryotic protein with the retroviral protease fold. J. Mol. Biol 2006, 364, 376–387. [Google Scholar]
- Hartmann, R.; Olsen, H.S.; Widder, S.; Jørgensen, R.; Justesen, J. p59OASL, a 2′-5′ oligoadenylate synthetase like protein: A novel human gene related to the 2′-5′ oligoadenylate synthetase family. Nucleic Acids Res 1998, 26, 4121–4127. [Google Scholar]
- Kokame, K.; Agarwala, K.L.; Kato, H.; Miyata, T. Herp, a new ubiquitin-like membrane protein induced by endoplasmic reticulum stress. J. Biol. Chem 2000, 275, 32846–32853. [Google Scholar]
- Ozaki, T.; Hishiki, T.; Toyama, Y.; Yuasa, S.; Nakagawara, A.; Sakiyama, S. Identification of a new cellular protein that can interact specifically with DAN. DNA Cell Biol 1997, 16, 985–991. [Google Scholar]
- Saia, X.; Kokame, K.; Shiraishia, H.; Kawamuraa, Y.; Miyata, T.; Yanagisawa, K.; Komano, H. The ubiquitin-like domain of herp is involved in Herp degradation, but not necessary for its enhancement of amyloid β-protein generation. FEBS Lett 2003, 553, 151–156. [Google Scholar]
- Slodzinski, H.; Moran, L.B.; Michael, G.J.; Wang, B.; Novoselov, S.; Cheetham, M.E.; Pearce, R.K.; Graeber, M.B. Homocysteine-induced endoplasmic reticulum protein (Herp) is up-regulated in parkinsonian substantia nigra and present in the core of Lewy bodies. Clin. Neuropathol 2009, 28, 333–343. [Google Scholar]
- Chan, S.L.; Fu, W.; Zhang, P.; Cheng, A.; Lee, J.; Kokame, K.; Mattson, M.P. Herp stabilizes neuronal Ca2+ homeostasis and mitochondrial function during endoplasmic reticulum stress. J. Biol. Chem 2004, 279, 28733–28743. [Google Scholar]
- Hori, O.; Ichinoda, F.; Yamaguchi, A.; Tamatani, T.; Taniguchi, M.; Koyama, Y.; Katayama, T.; Tohyama, M.; Stern, D.M.; Ozawa, K.; et al. Role of Herp in the endoplasmic reticulum stress response. Genes Cells 2004, 9, 457–469. [Google Scholar]
- Kitada, T.; Asakawa, S.; Minoshima, S.; Mizuno, Y.; Shimizu, N. Molecular cloning, gene expression, and identification of a splicing variant of the mouse parkin gene. Mamm. Genome 2000, 11, 417–421. [Google Scholar]
- Kitada, T.; Asakawa, S.; Hattori, N.; Matsumine, H.; Yamamura, Y.; Minoshima, S.; Yokochi, M.; Mizuno, Y.; Shimizu, N. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 1998, 392, 605–608. [Google Scholar]
- Shimura, H.; Hattori, N.; Kubo, S.-I.; Mizuno, Y.; Asakawa, S.; Minoshima, S.; Shimizu, N.; Iwai, K.; Chiba, T.; Tanaka, K.; et al. Familial parkinson disease gene product, parkin, is a ubiquitin-protein ligase. Nat. Genet 2000, 25, 302–305. [Google Scholar]
- Cookson, M.R. Parkin’s substrates and the pathways leading to neuronal damage. NeuroMol. Med 2003, 3, 1–13. [Google Scholar]
- Bae, Y.-J.; Kang, S.-J.; Park, K.S. Drosophila melanogaster Parkin ubiquitinates peanut and septin1 as an E3 ubiquitin–protein ligase. Insect Biochem. Mol. Biol 2007, 37, 430–439. [Google Scholar]
- Witte, M.M.; Scott, R.E. The proliferation potential protein-related (P2P-R) gene with domains encoding heterogeneous nuclear ribonucleoprotein association and Rb1 binding shows repressed expression during terminal differentiation. Proc. Natl. Acad. Sci. USA 1997, 94, 1212–1217. [Google Scholar]
- Simons, A.; Melamed-Bessudo, C.; Wolkowicz, R.; Sperling, J.; Sperling, R.; Eisenbach, L.; Rotter, V. PACT: Cloning and characterization of a cellular p53 binding protein that interacts with Rb. Oncogene 1997, 14, 145–155. [Google Scholar]
- Scott, R.E.; Giannakouros, T.; Gao, S.; Peidis, P. Functional potential of P2P-R: A role in the cell cycle and cell differentiation related to its interactions with proteins that bind to matrix associated regions of DNA? J. Cell Biochem 2003, 90, 6–12. [Google Scholar]
- Conklina, D.; Holdermana, S.; Whitmoreb, T.E.; Maurerb, M.; Feldhaus, A.L. Molecular cloning, chromosome mapping and characterization of UBQLN3 a testis-specific gene that contains an ubiquitin-like domain. Gene 2000, 249, 91–98. [Google Scholar]
- Ko, H.S.; Uehara, T.; Nomura, Y. Role of ubiquilin associated with protein-disulfide isomerase in the endoplasmic reticulum in stress-induced apoptotic cell death. J. Biol. Chem 2002, 277, 35386–35392. [Google Scholar]
- Hoeller, D.; Hecker, C.-M.; Dikic, I. Ubiquitin and ubiquitin-like proteins in cancer pathogenesis. Nat Rev Cancer 2006, 6, 776–788. [Google Scholar]
- Liu, M.; Hummer, T.; Li, X.; Hassel, B.A. Camptothecin induces the ubiquitin-like protein, ISG15, and enhances ISG15 conjugation in response to interferon. J. Interferon Cytokine Res 2004, 24, 647–654. [Google Scholar]
- Ohwada, S.; Kobayashi, I.; Maemura, M.; Satoh, Y.; Ogawa, T.; Iino, Y.; Morishita, Y. Interferon potentiates antiproliferative activity of CPT-11 against human colon cancer xenografts. Cancer Lett 1996, 110, 149–154. [Google Scholar]
- Yang, Y.; Kitagaki, J.; Wang, H.; Hou, D.-X.; Perantoni, A.O. Targeting the ubiquitin-proteasome system for cancer therapy. Cancer Sci 2009, 100, 24–28. [Google Scholar]
- Richardson, P.G.; Barlogie, B.; Berenson, J.; Singhal, S.; Jagannath, S.; Irwin, D.; Rajkumar, S.V.; Srkalovic, G.; Alsina, M.; Alexanian, R.; et al. A Phase 2 study of Bortezomib in relapsed, refractory myeloma. N. Engl. J. Med 2003, 348, 2609–2617. [Google Scholar]
- Jagannath, S.; Barlogie, B.; Berenson, J.; Siegel, D.; Irwin, D.; Richardson, P.G.; Niesvizky, R.; Alexanian, R.; Limentani, S.A.; Alsina, M.; et al. A phase 2 study of two doses of bortezomib in relapsed or refractory myeloma. Br. J. Haematol 2004, 127, 165–172. [Google Scholar]
- Orlowski, R.Z.; Nagler, A.; Sonneveld, P.; Bladé, J.; Hajek, R.; Spencer, A.; San Miguel, J.; Robak, T.; Dmoszynska, A.; Horvath, N.; et al. Randomized Phase III study of Pegylated liposomal Doxorubicin plus Bortezomib compared with Bortezomib alone in relapsed or refractory multiple myeloma: Combination therapy improves time to progression. J. Clin. Oncol 2007, 25, 3892–3901. [Google Scholar]
- National Cancer Institute. Fda approval for Bortezomib. Available online: http://www.cancer.gov/cancertopics/druginfo/fda-bortezomib accessed on 1 June 2012.
- Kane, R.C.; Bross, P.F.; Farrell, A.T.; Pazdur, R. Velcade®: U.S. FDA approval for the treatment of multiple myeloma progressing on prior therapy. Oncologist 2003, 8, 508–513. [Google Scholar]
- Sun, Y. Targeting E3 ubiquitin ligases for cancer therapy. Cancer Biol. Ther 2003, 2, 623–629. [Google Scholar]
- Saha, M.N.; Jiang, H.; Jayakar, J.; Reece, D.; Branch, D.R.; Chang, H. MDM2 antagonist nutlin plus proteasome inhibitor velcade combination displays a synergistic anti-myeloma activity. Cancer Biol. Ther 2010, 9, 936–944. [Google Scholar]
- Bettermann, K.; Benesch, M.; Weis, S.; Haybaeck, J. SUMOylation in carcinogenesis. Cancer Lett 2012, 316, 113–125. [Google Scholar]
- Mo, Y.-Y.; Moschos, S.J. Targeting Ubc9 for cancer therapy. Expert Opin. Ther. Tar 2005, 9, 1203–1216. [Google Scholar]
- Weisshaar, S.R.; Keusekotten, K.; Krause, A.; Horst, C.; Springer, H.M.; Göttsche, K.; Dohmen, R.J.; Praefcke, G.J.K. Arsenic trioxide stimulates SUMO-2/3 modification leading to RNF4-dependent proteolytic targeting of PML. FEBS Lett 2008, 582, 3174–3178. [Google Scholar]
- Miller, W.H.; Schipper, H.M.; Lee, J.S.; Singer, J.; Waxman, S. Mechanisms of action of arsenic trioxide. Cancer Res 2002, 62, 3893–3903. [Google Scholar]
- Hoeller, D.; Dikic, I. Targeting the ubiquitin system in cancer therapy. Nature 2009, 458, 438–444. [Google Scholar]
- Soucy, T.A.; Smith, P.G.; Milhollen, M.A.; Berger, A.J.; Gavin, J.M.; Adhikari, S.; Brownell, J.E.; Burke, K.E.; Cardin, D.P.; Critchley, S.; et al. An inhibitor of NEDD8-activating enzyme as a new approach to treat cancer. Nature 2009, 458, 732–736. [Google Scholar]
- Cohen, P.; Tcherpakov, M. Will the ubiquitin system furnish as many drug targets as protein kinases? Cell 2010, 143, 686–693. [Google Scholar]
- Colland, F.; Formstecher, E.; Jacq, X.; Reverdy, C.; Planquette, C.; Conrath, S.; Trouplin, V.; Bianchi, J.; Aushev, V.N.; Camonis, J.; et al. Small-molecule inhibitor of USP7/HAUSP ubiquitin protease stabilizes and activates p53 in cells. Mol. Cancer Ther 2009, 8, 2286–2295. [Google Scholar]
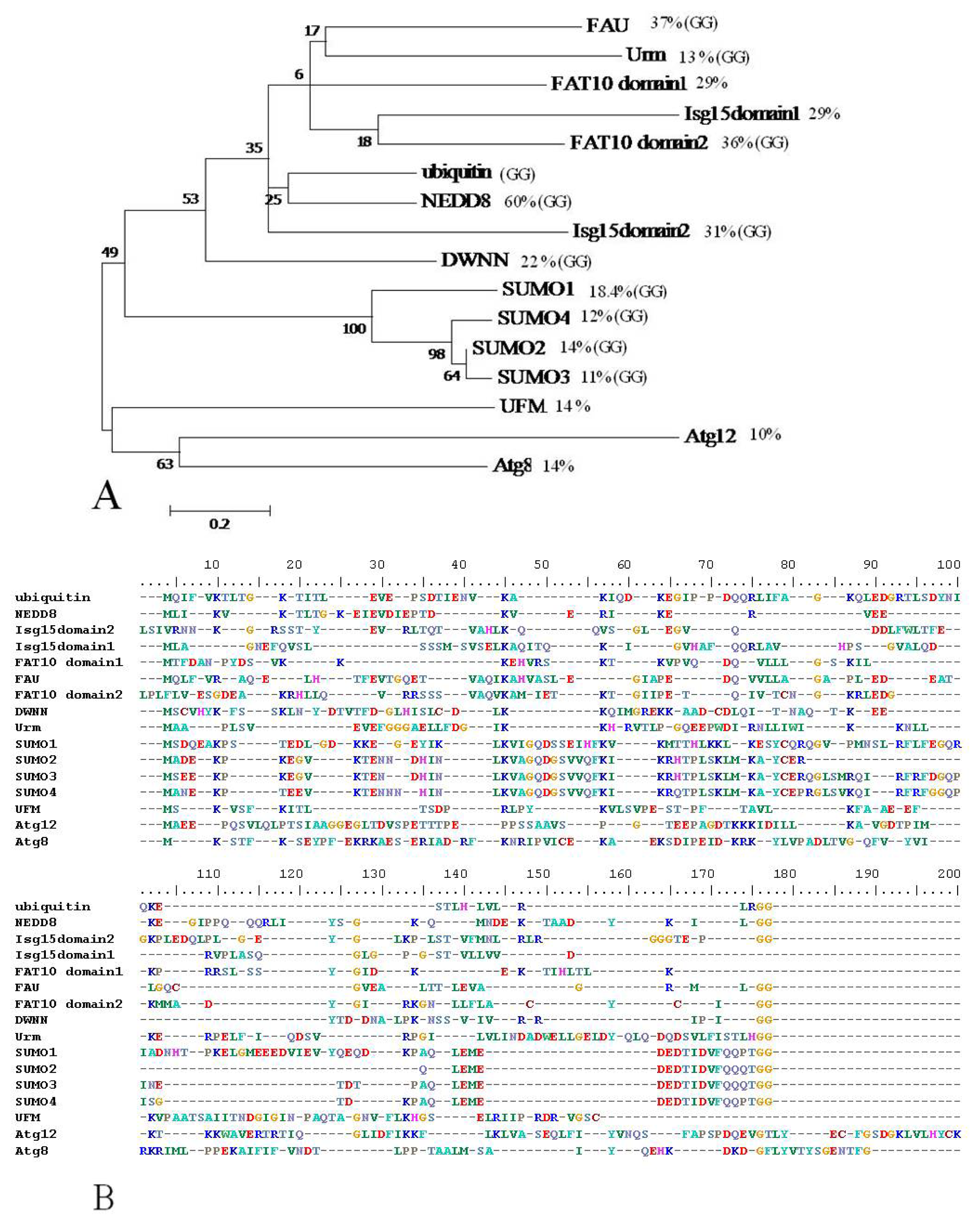
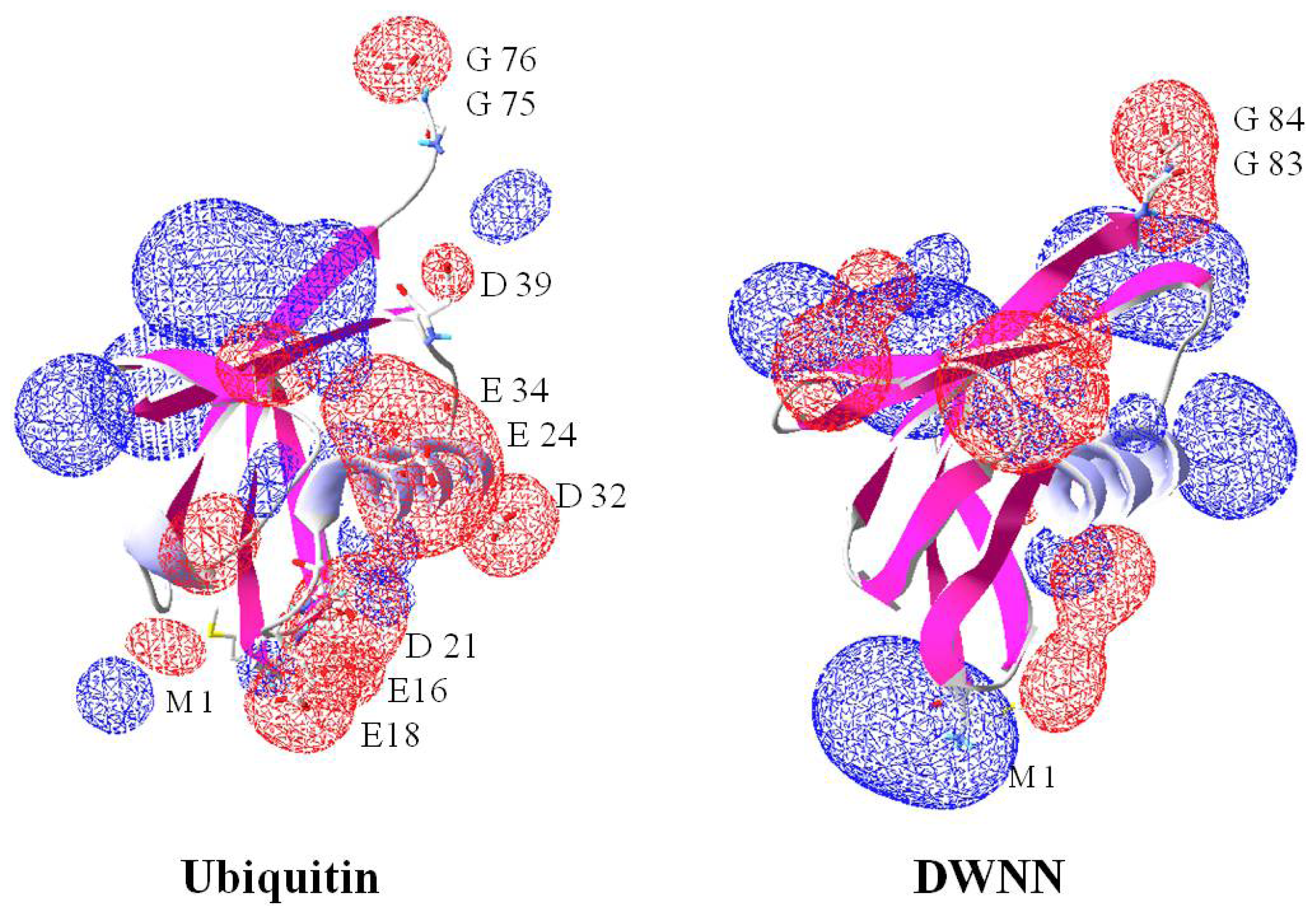
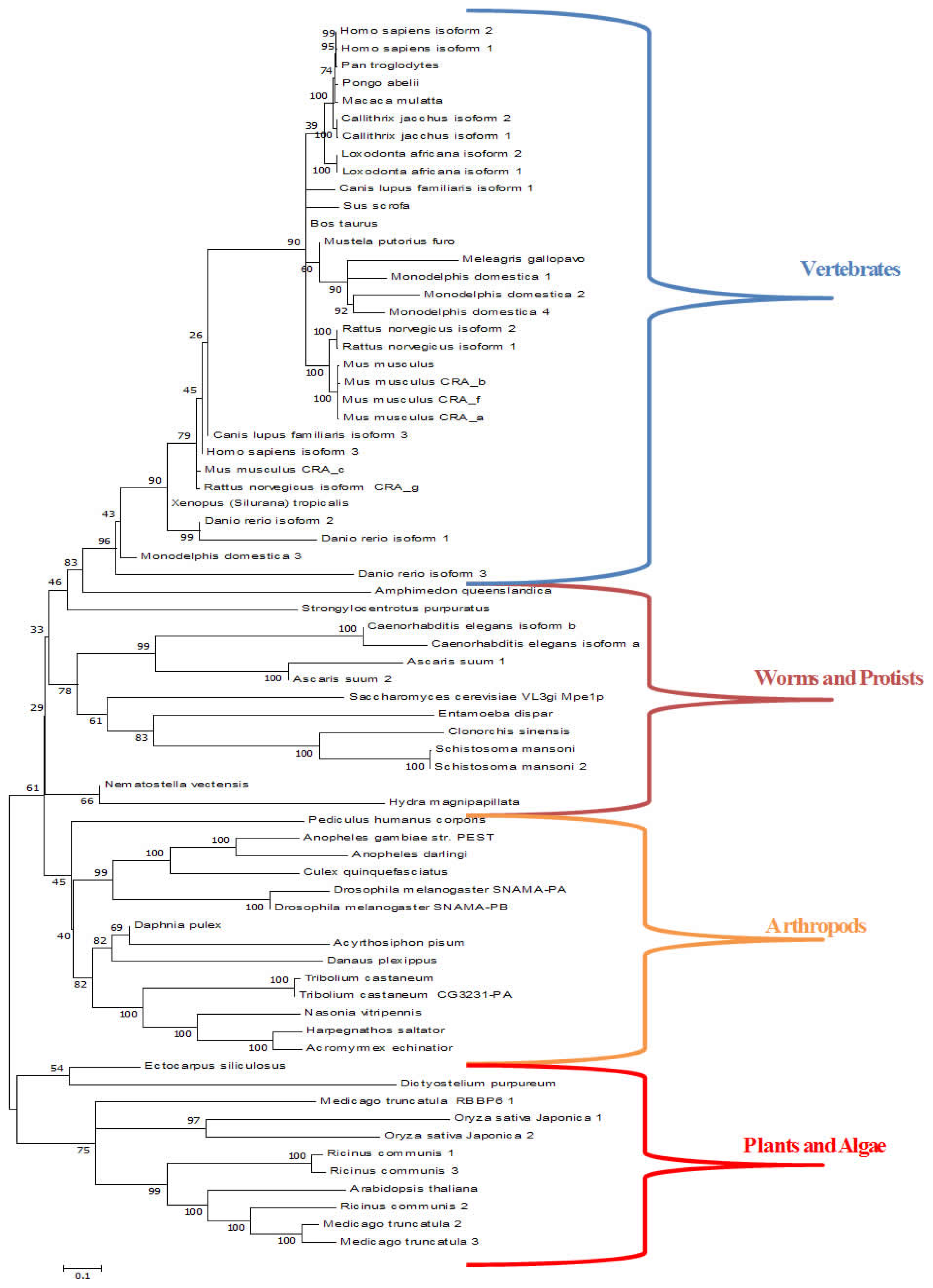

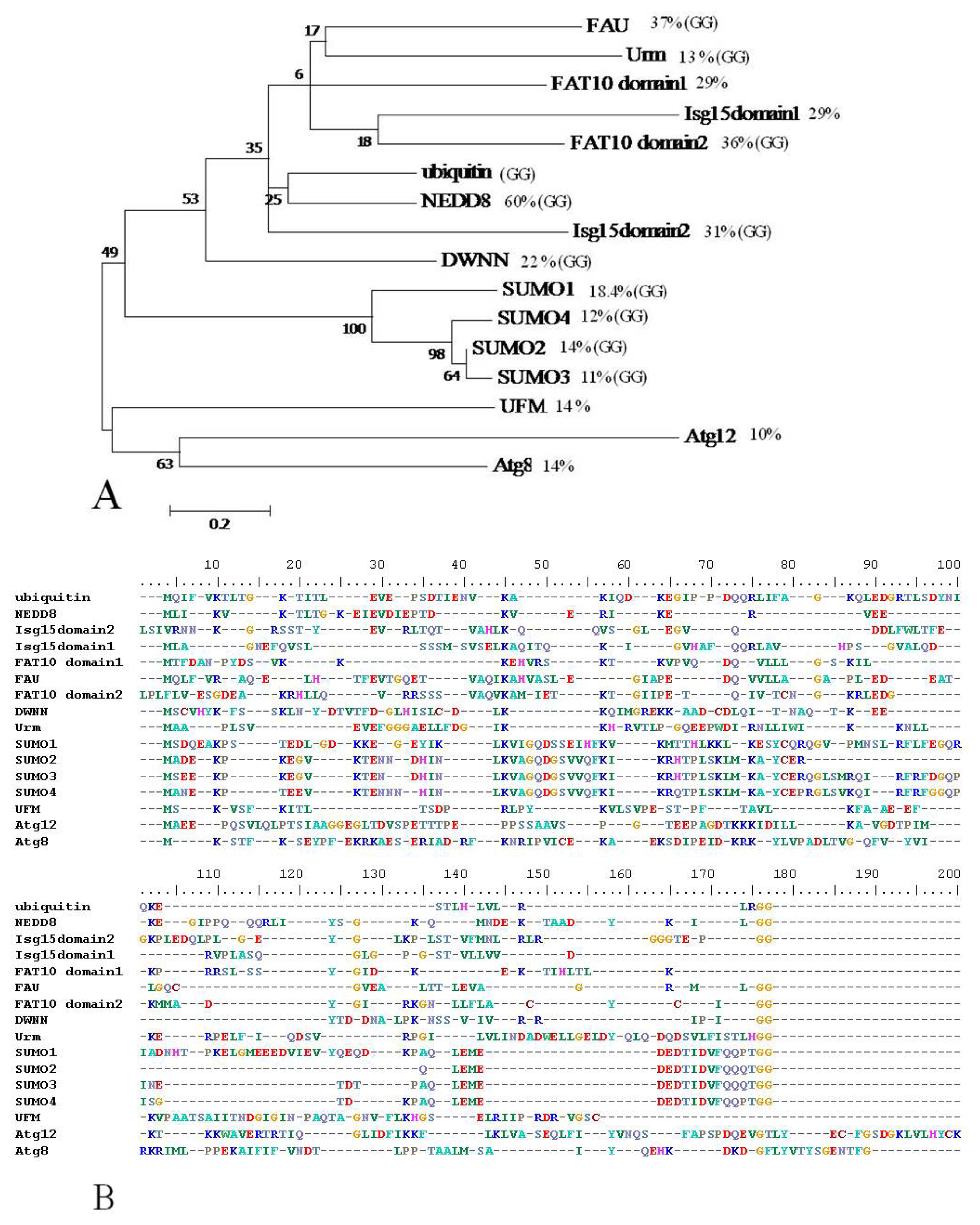{kind=link}
{kind=link}
{kind=link}
| Modifier | Functions | Accession | Reference |
|---|---|---|---|
| SUMO-1 | Nuclear transport, DNA replication and repair, mitosis and signal transduction | NP_001005781 | [7,8] |
| SUMO-2 | Nuclear transport, DNA replication and repair, mitosis and signal transduction | NP_001005849 | [7] |
| SUMO-3 | Nuclear transport, DNA replication and repair, mitosis and signal transduction | NP_008867 | [7] |
| SUMO-4 | May modulate protein sub-cellular localization, stability or activity. Upon oxidative stress, conjugates to various stress defense proteins. Negative regulation of NF-kappa-B-dependent transcription | NP_001002255 | [9] |
| NEDD8 | Cell cycle control and embryogenesis | NP_006147 | [7] |
| May be involved in the formation of aggresomes | [10] | ||
| ISG15 | Modifies STAT1, SERPINA3G/SPI2A, JAK1, MAPK3/ERK1, PLCG1, EIF2AK2/PKR, MX1/MxA, and RIG-1. May serve as a trans-acting binding factor directing the association of ligated target proteins to intermediate filaments. May also be involved in autocrine, paracrine and endocrine mechanisms. Displays antiviral activity | NP_005092 | [7,11] |
| FAT10 | Protein degradation. Regulates TNF-alpha-induced and LPS-mediated activation of innate immunity. Mediates mitotic non-disjunction and chromosome instability, in cancers. May be involved in the formation of aggresomes when proteasome is saturated or impaired. Mediates apoptosis in a caspase-dependent manner | NP_006389 | [1,12] |
| Ubl5 | Negatively regulates p53 | NP_001041706 | [13,14] |
| FAU | Translation Gene expression. Viral infectious cycle. Endocrine pancreas development. Cellular protein metabolic process | NP_001988 | [15] |
| Apg12/Atg12 | Autophagic vacuole assembly. Negative regulation of type I interferon production | NP_004698 | [16] |
| Putative Atg8 homologs | Increases cell-surface expression of kappa-type opioid receptor. Intra-Golgi traffic and transport. Intracellular transport of GABA (A) receptors. Apoptosis. Formation of autophagosomes Formation of autophagosomes. Formation of autophagosomes. Formation of autophagosomes | CAG38511 | |
| GABARAPL1 (Atg8L) | NP_009266.7 | ||
| GABARAPL2 (Atg8C) | NP_00209.1 | ||
| GABARAP (Atg8A) | NP_07379.1 | ||
| MAP1LC3C (Atg8E) | NP_1159031 | ||
| MAP1LC3A (Atg8F) | NP_001078950 | ||
| MAP1LC3B | |||
| MAP1LC3B2 | |||
| Ufm1, UFM1 | Protein Ufmylation | NP_057701 | [17] |
| DWNN | Unknown but may be involved in protein ubiquitination involved in ubiquitin-dependent protein catabolic process | 2C7H_A | [18] |
| URM | Unknown | CAI13492 | [19] |
| Protein | Function | Accession Number | Reference |
|---|---|---|---|
| Parkin | Acts as a positive regulator of autophagy. Promotes the autophagic degradation of dysfunctional depolarized mitochondria. Regulates neuron death. Limits the production of reactive oxygen species. Regulates cyclin-E during neuronal apoptosis. May represent a tumor suppressor gene | NP_004553 | [108] |
| UHRF1 | Important for G1/S transition. May be involved in DNA repair and chromosomal stability | NP_037414.3 | [109] |
| UHRF2 | Important for G1/S transition. May be involved in DNA repair and chromosomal stability | CAI13295.1 | [109] |
| RAD23 | Involved in nucleotide excision repair | NP_005044.1 | [110] |
| BAG1 | Regulation of proteasomal and lysosomal protein | NP_001165886 | [111] |
| BAT3 | Involved in DNA damage-induced apoptosis. Immuno-proteasomes to generate antigenic peptides via targeted degradation, Post-translational delivery of tail-anchored (TA) membrane proteins to the endoplasmic reticulum membrane | BAB63390.1 | [112] |
| DDI1 | Protein degradation | NP_001001711.1 | [113] |
| DDI2 | Protein degradation | NP_115717.3 | [113] |
| OASL | Immune response cytokine-mediated signaling pathway interferon-gamma-mediated signaling pathway type I interferon-mediated signaling pathway | NP_003724 | [114] |
| HERPUD1 | Cellular calcium ion homeostasis. Response to unfolded protein. Endoplasmic reticulum unfolded protein response. Negative regulation of caspase activity | NP_001010989 | [115] |
| RBBP6 | Unknown but may be involved in protein ubiquitination involved in ubiquitin-dependent protein catabolic process | NP_008841.2 | [104] |
| UBQLN1 (Ubiquilin) | Response to Hypoxia. Apoptosis. Regulation of ubiquitination | NP_038466 | [116] |
| Drug | Mode of Action | Target | Disease | State of Development | Reference |
|---|---|---|---|---|---|
| Bortezomib/Velcade® | Selective proteasome inhibitor | Proteasome | Multiple myeloma | In clinical use | [135–137] |
| Nutlin | Mdm2 inhibitor | Mdm2 | Multiple myeloma | Preclinical | [141] |
| Multiple approaches to target SUMO pathway | Inhibition of the active site Inhibiting association with E1 Blocking association with target protein | The E2 enzyme in the SUMO pathway UBC9 | Cancer | Experimental phases | [146] |
| Arsenic trioxide | Targets SUMOylation | Degradation of PML–RAR-α | Leukemia | In use clinically | [146] |
| All-trans retinoic acid | See above | See above | See above | See above | [146] |
| MLN-4924 | Inhibits NEDD8 E1-activating enzyme (NAE) | NEDD8 activating enzyme (NAE) | Cancer, Multiple myeloma and Hodgkin’s lymphoma | Phase II clinical trials | [147] [148] |
| HBX 41108 | Inhibits USP7 deubiquitinating activity | Ubiquitin-specific proteases (USP) | Cancer | Phase I | [149] |
| JNJ26854165 | Inhibitor | E3-Hdm2 | Multiple myeloma and solid tumors | Phase I | [148] |
| GDC-0152 | Inhibitor | E3-IAP | Metastatic malignancies | Phase I | [148] |
| LCL161 | Inhibitor | E3-IAP | Solid tumors | Phase I | [148] |
| AT-406 | Inhibitor | E3-IAP | Solid tumors and lymphoma | Phase I | [148] |
| AEG 35156 | Inhibitor | E3-IAP | AML and liver cancer | Phase II | [148] |
| AEG 40826 | Inhibitor | E3-IAP | Lymphoid tumors | Phase I | [148] |
| TL 32711 | Inhibitor | E3-IAP | Solid tumors and lymphoma | Phase I | [148] |
| YM155 | Inhibitor | E3-IAP | Lung cancer | Phase II | [148] |
© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Cajee, U.-F.; Hull, R.; Ntwasa, M. Modification by Ubiquitin-Like Proteins: Significance in Apoptosis and Autophagy Pathways. Int. J. Mol. Sci. 2012, 13, 11804-11831. https://doi.org/10.3390/ijms130911804
Cajee U-F, Hull R, Ntwasa M. Modification by Ubiquitin-Like Proteins: Significance in Apoptosis and Autophagy Pathways. International Journal of Molecular Sciences. 2012; 13(9):11804-11831. https://doi.org/10.3390/ijms130911804
Chicago/Turabian StyleCajee, Umar-Faruq, Rodney Hull, and Monde Ntwasa. 2012. "Modification by Ubiquitin-Like Proteins: Significance in Apoptosis and Autophagy Pathways" International Journal of Molecular Sciences 13, no. 9: 11804-11831. https://doi.org/10.3390/ijms130911804