A Combination of 3D-QSAR, Molecular Docking and Molecular Dynamics Simulation Studies of Benzimidazole-Quinolinone Derivatives as iNOS Inhibitors

Abstract
:1. Introduction
2. Results and Discussion
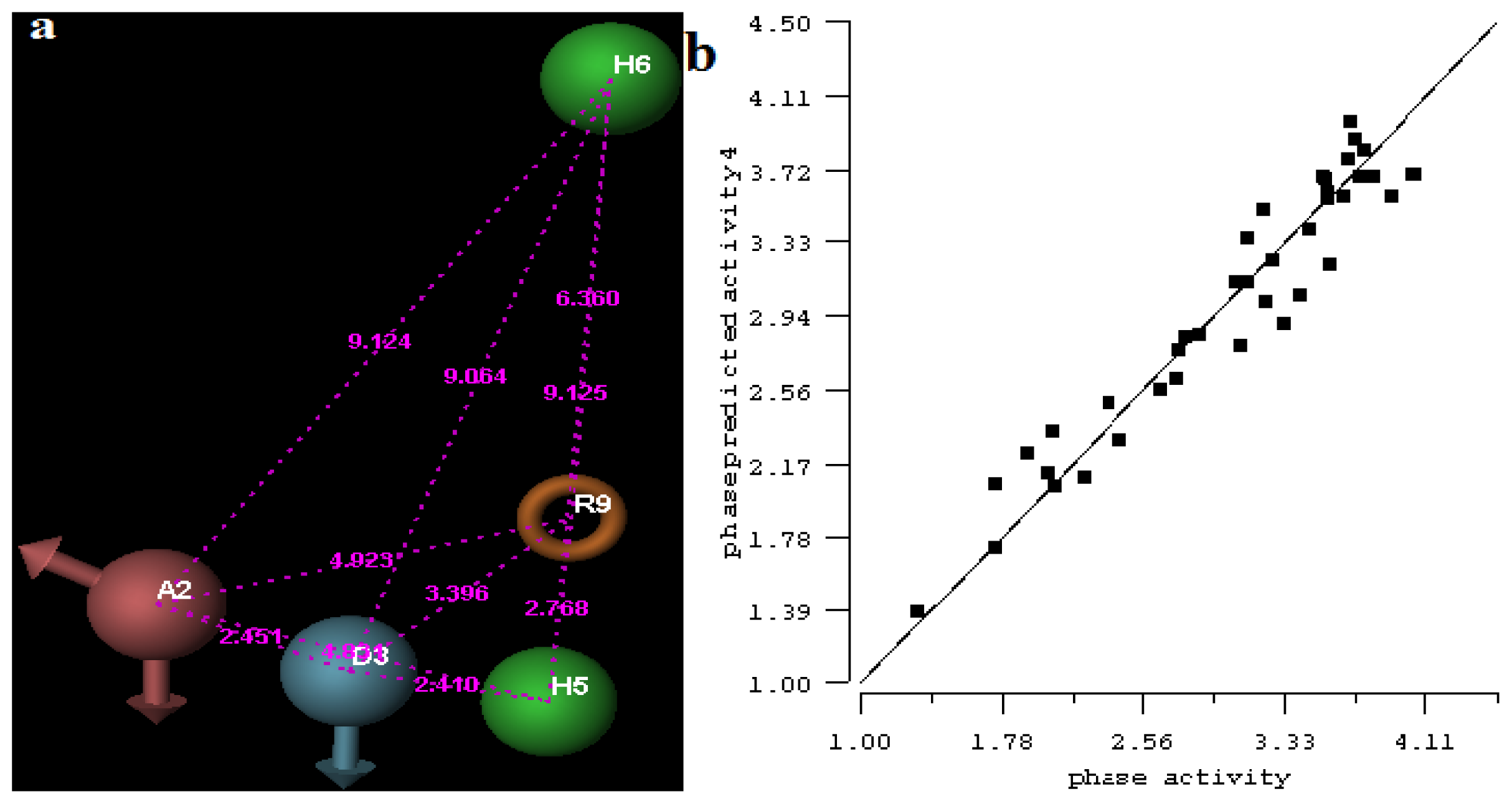
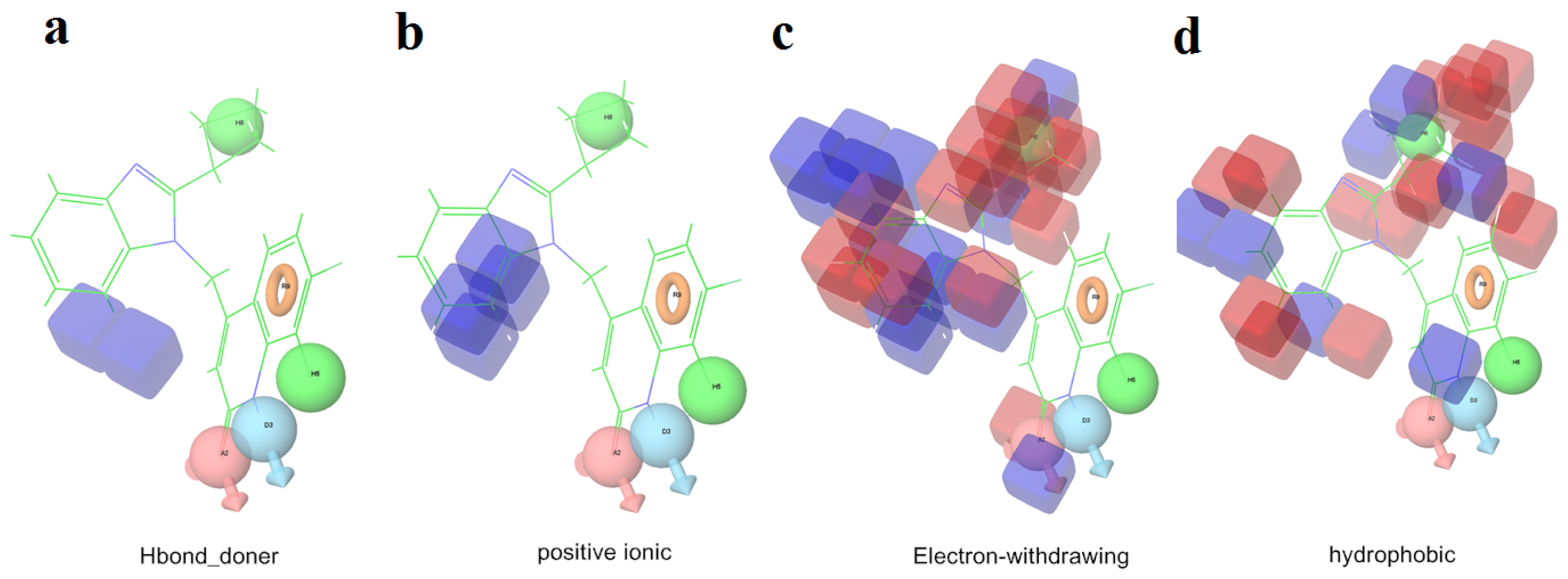
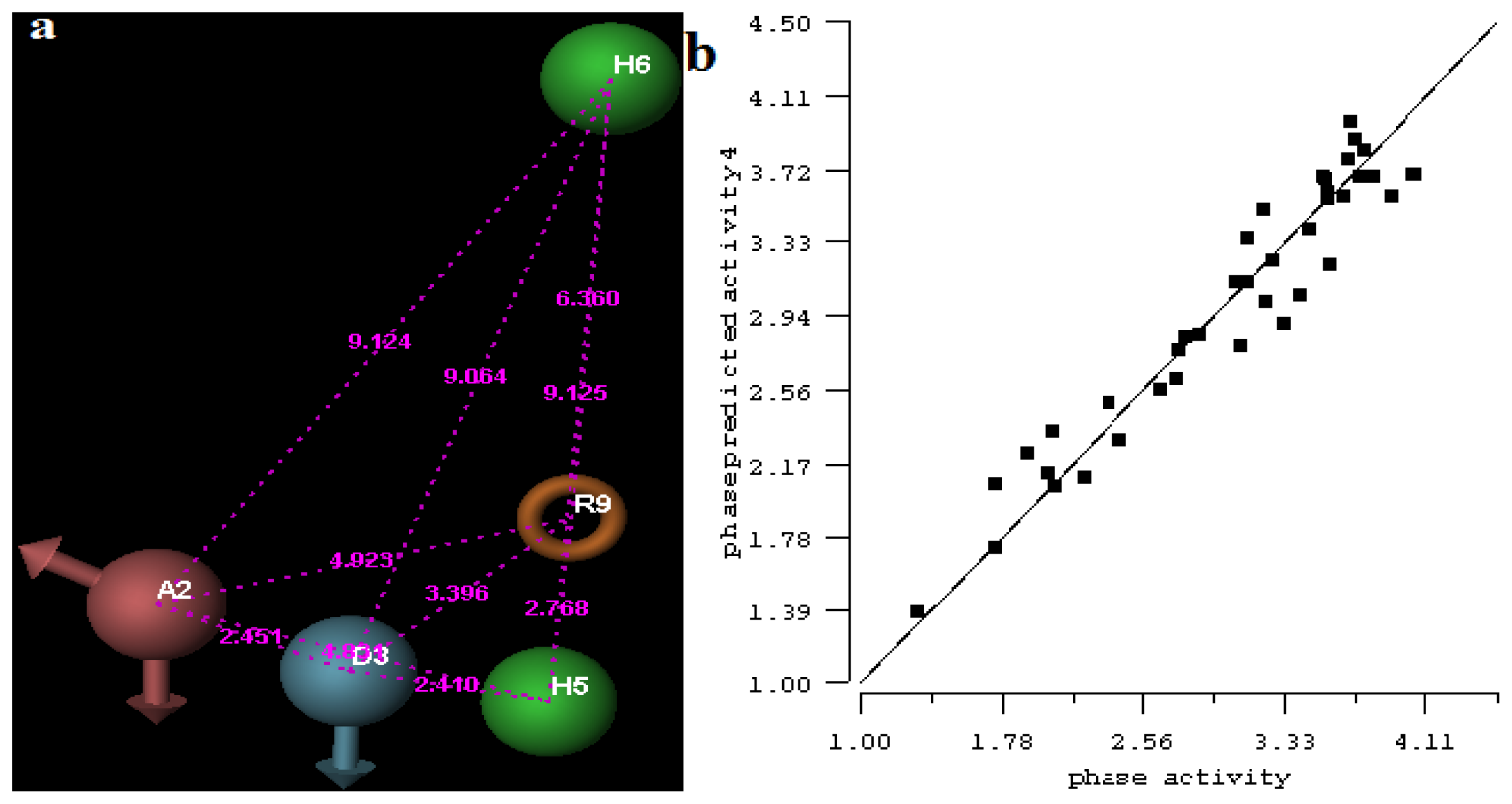
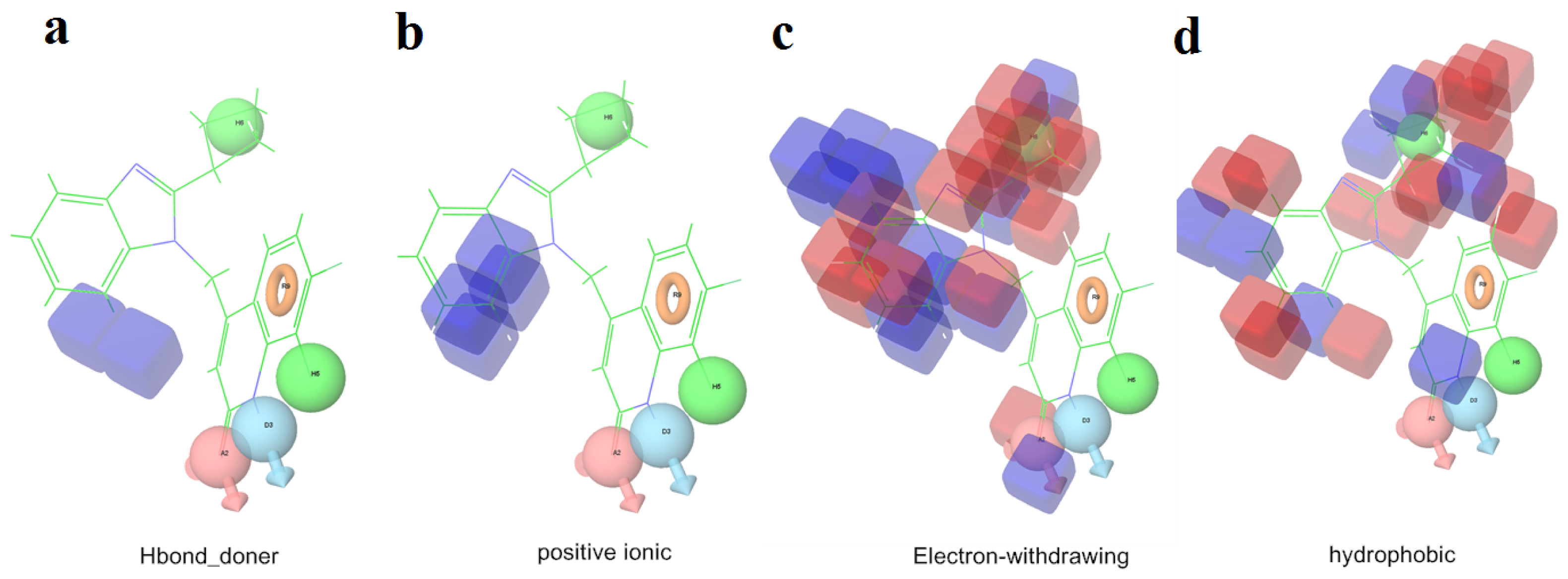
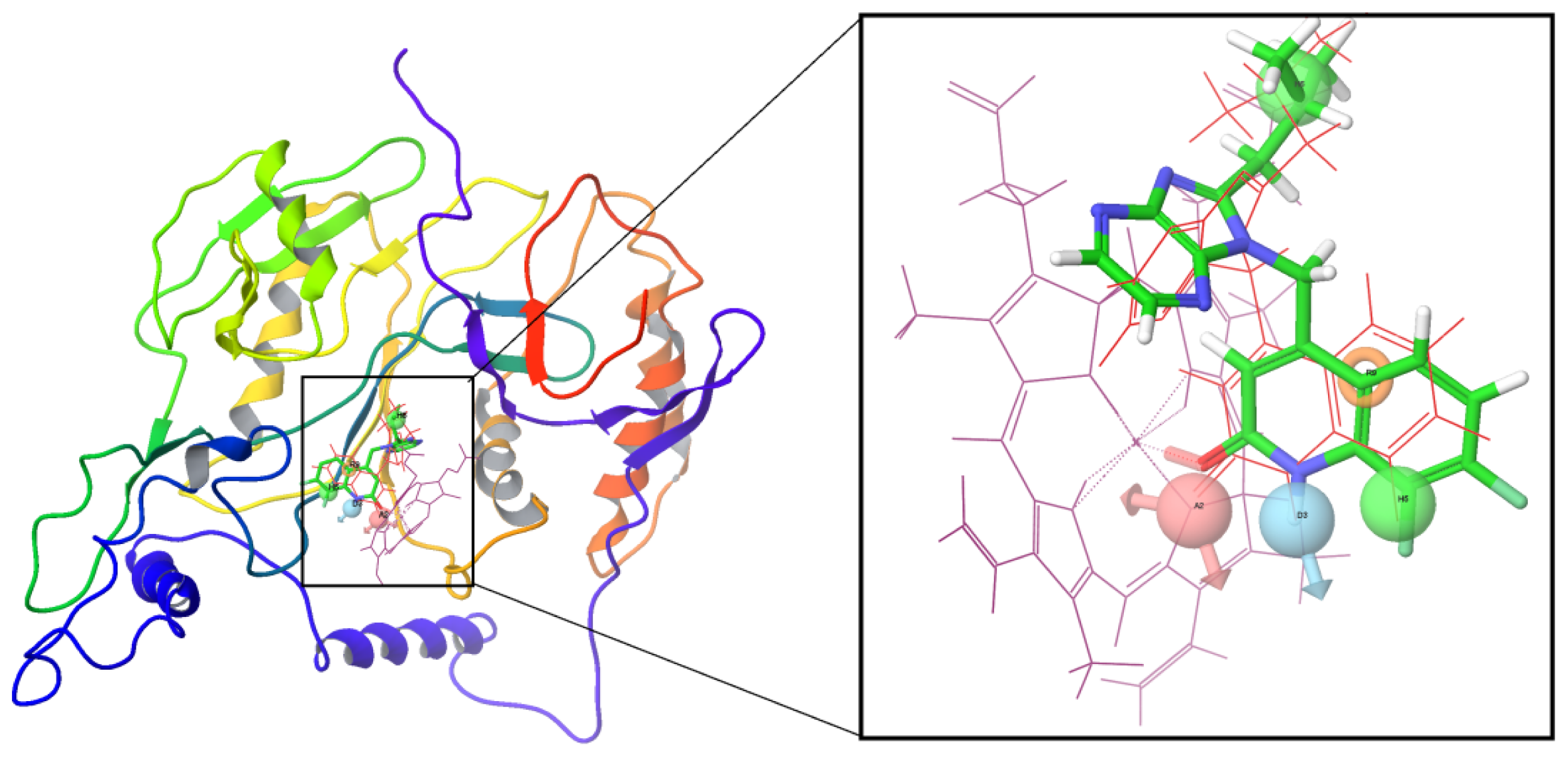
2.1. 3D-QSAR Analyses
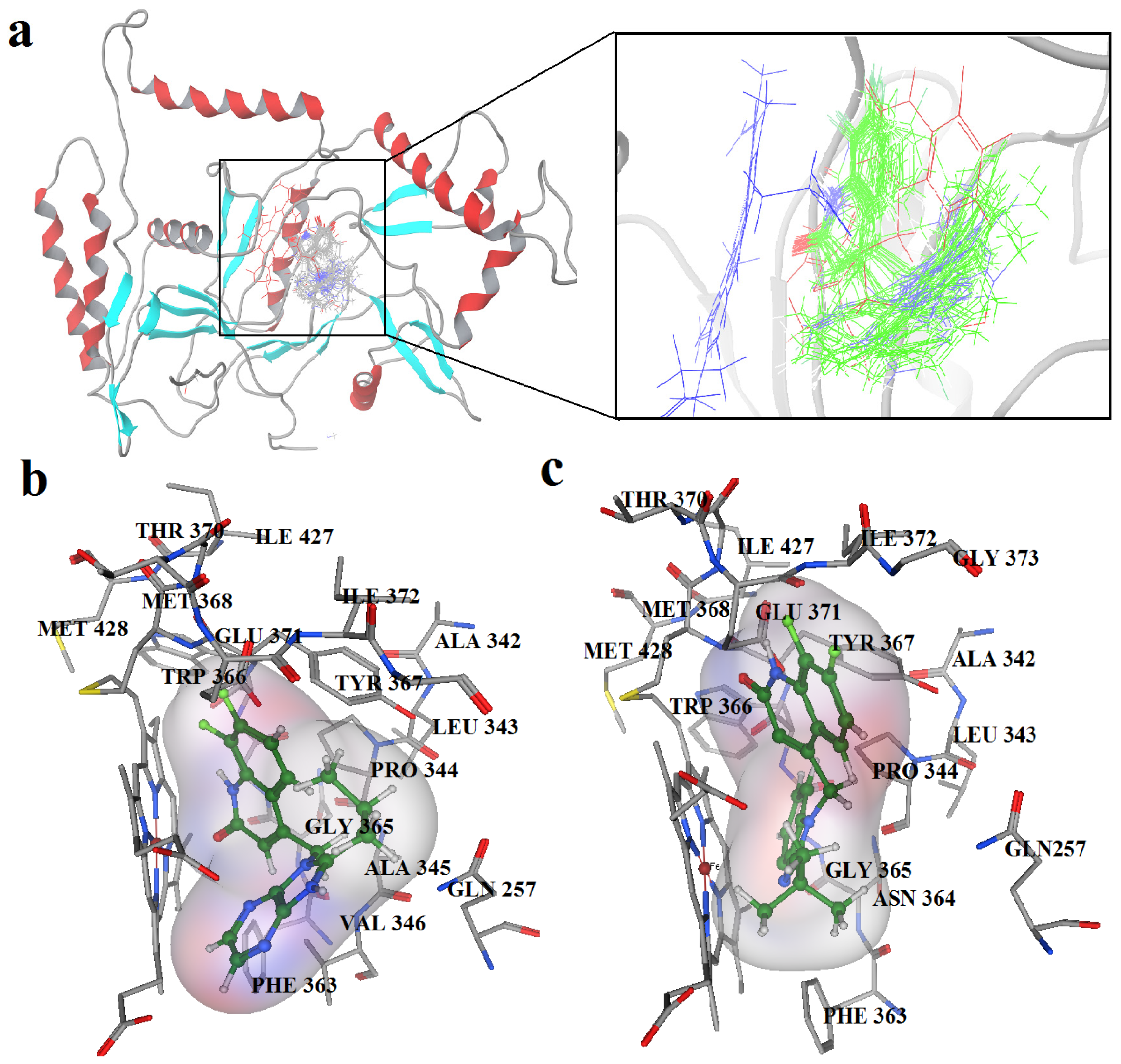
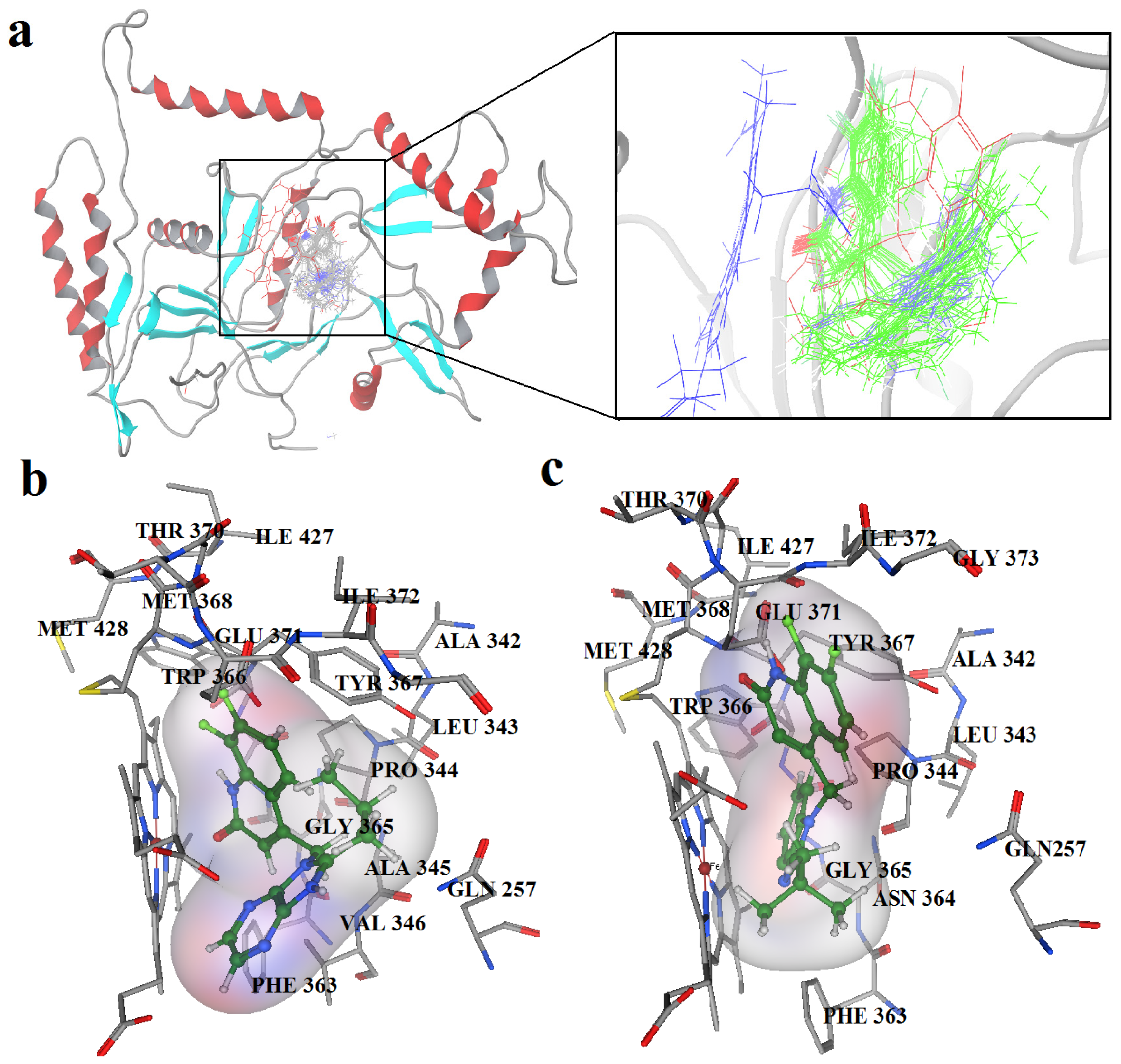
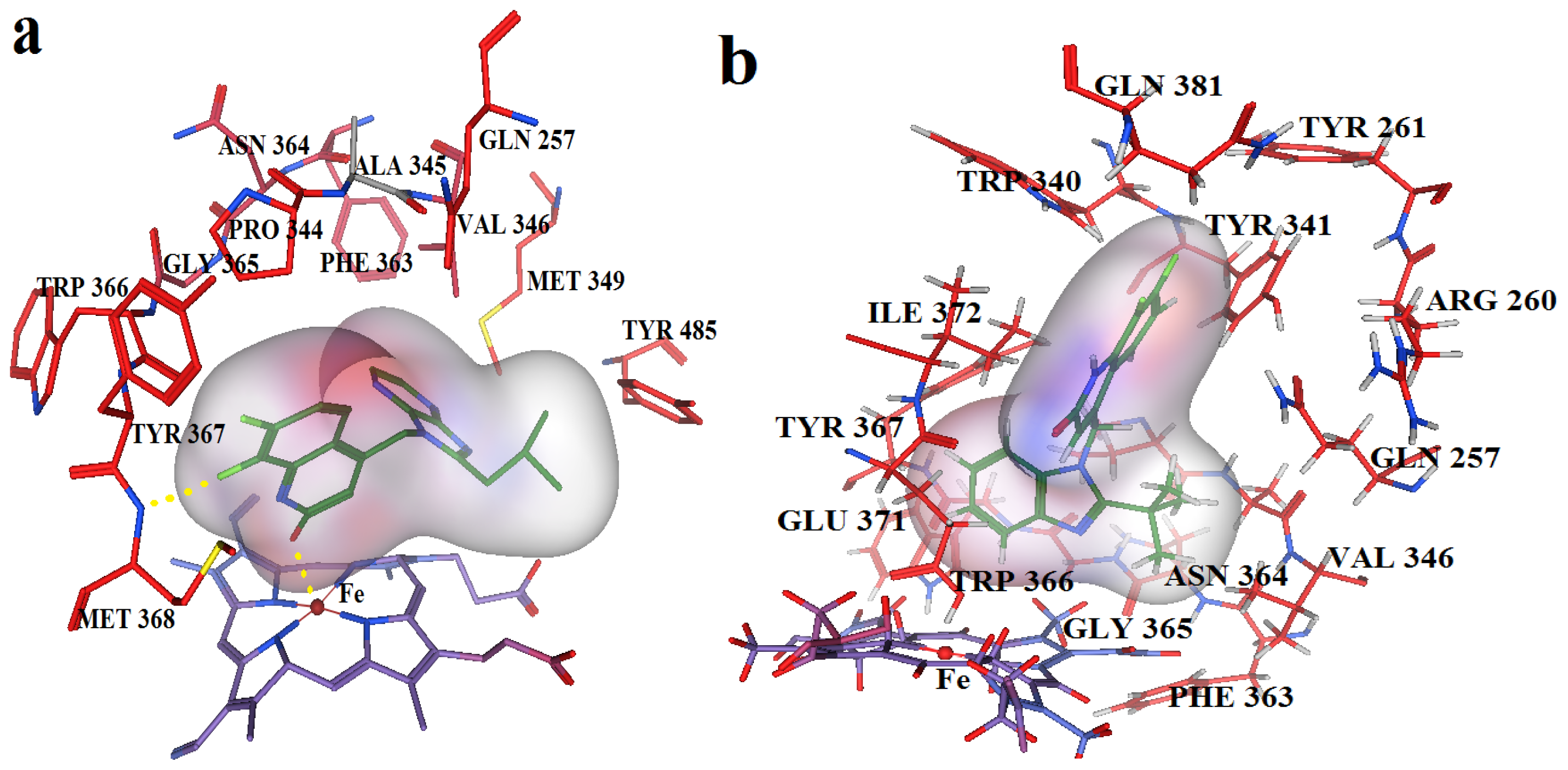
2.2. Docking Results
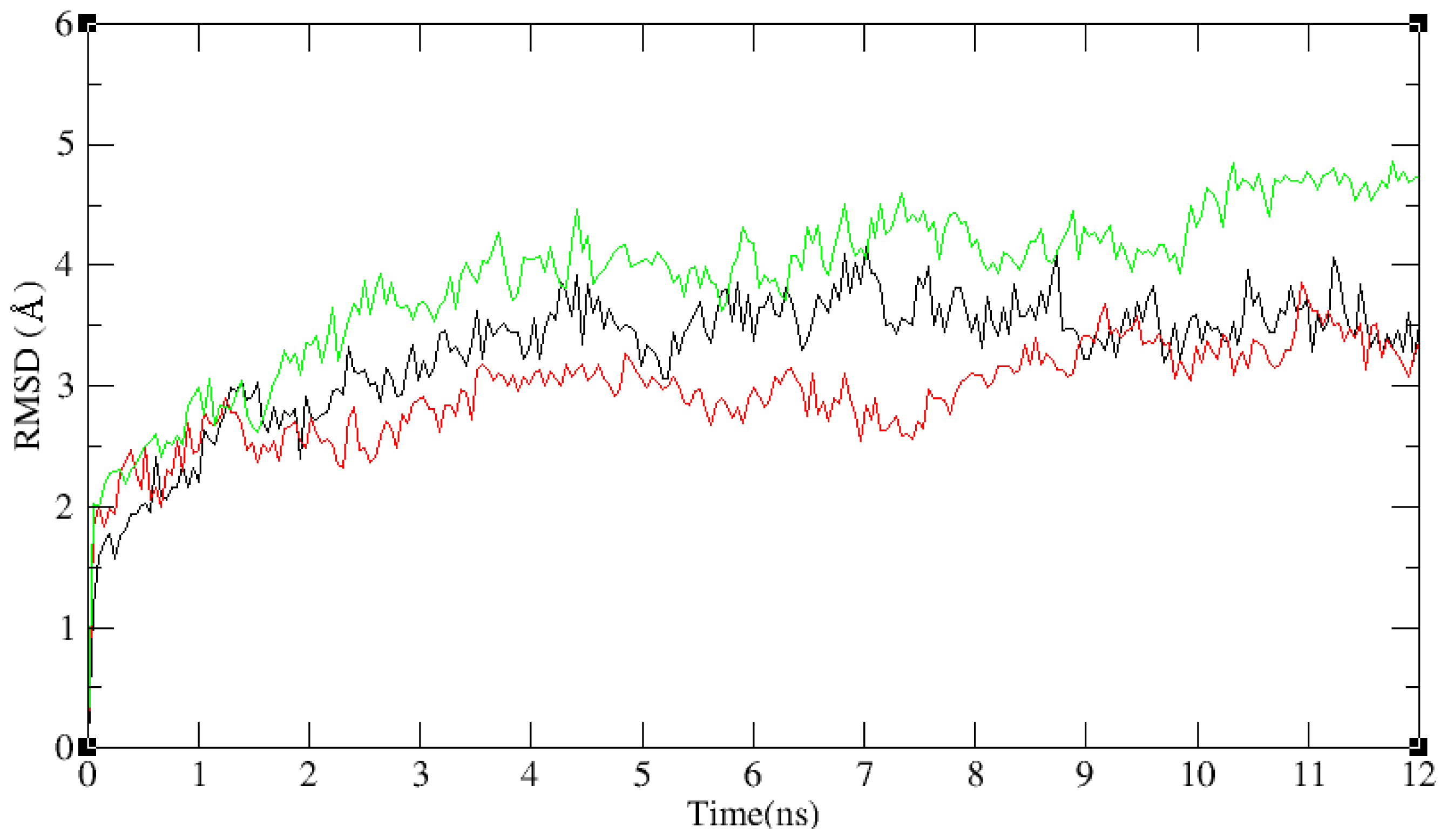
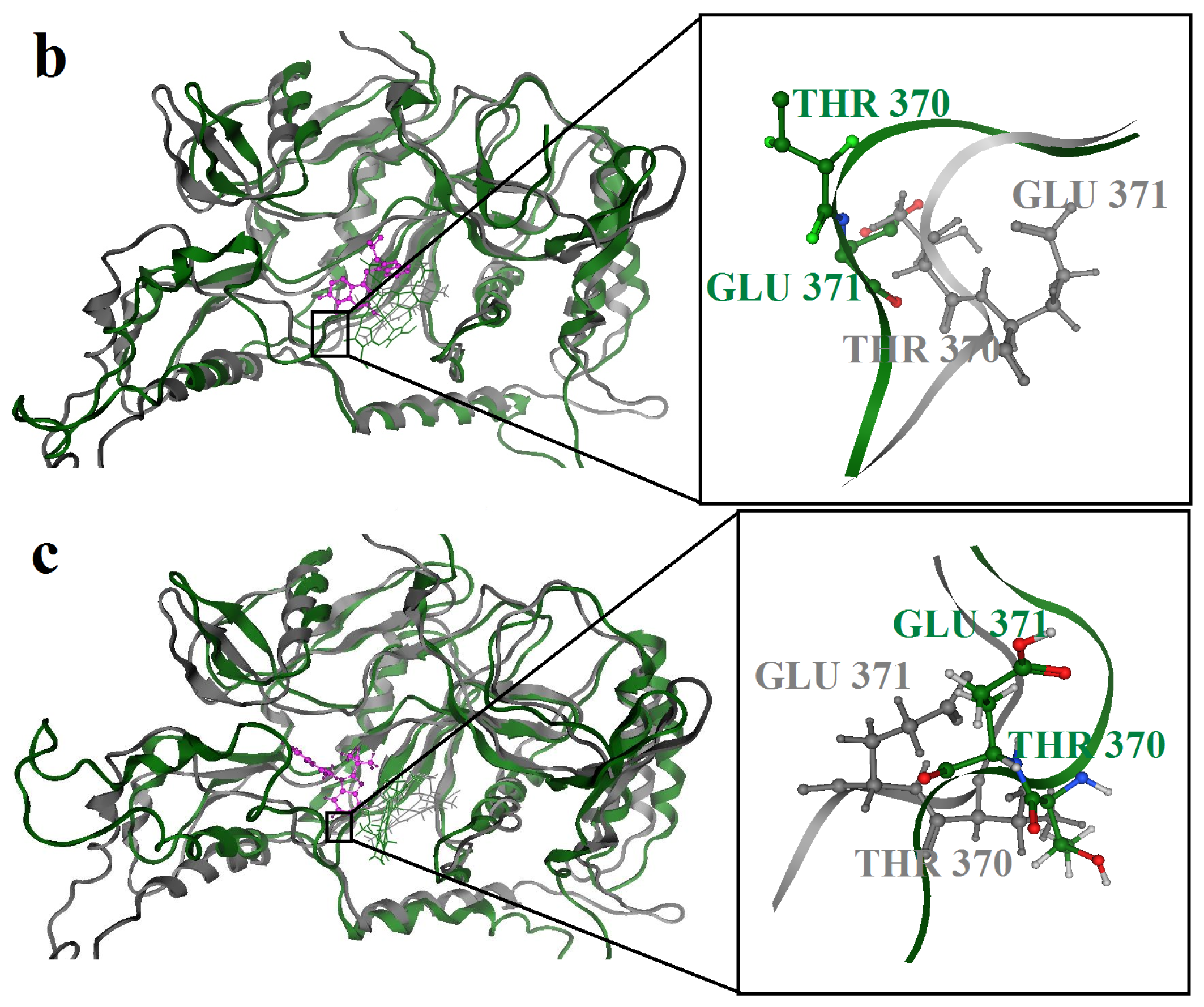
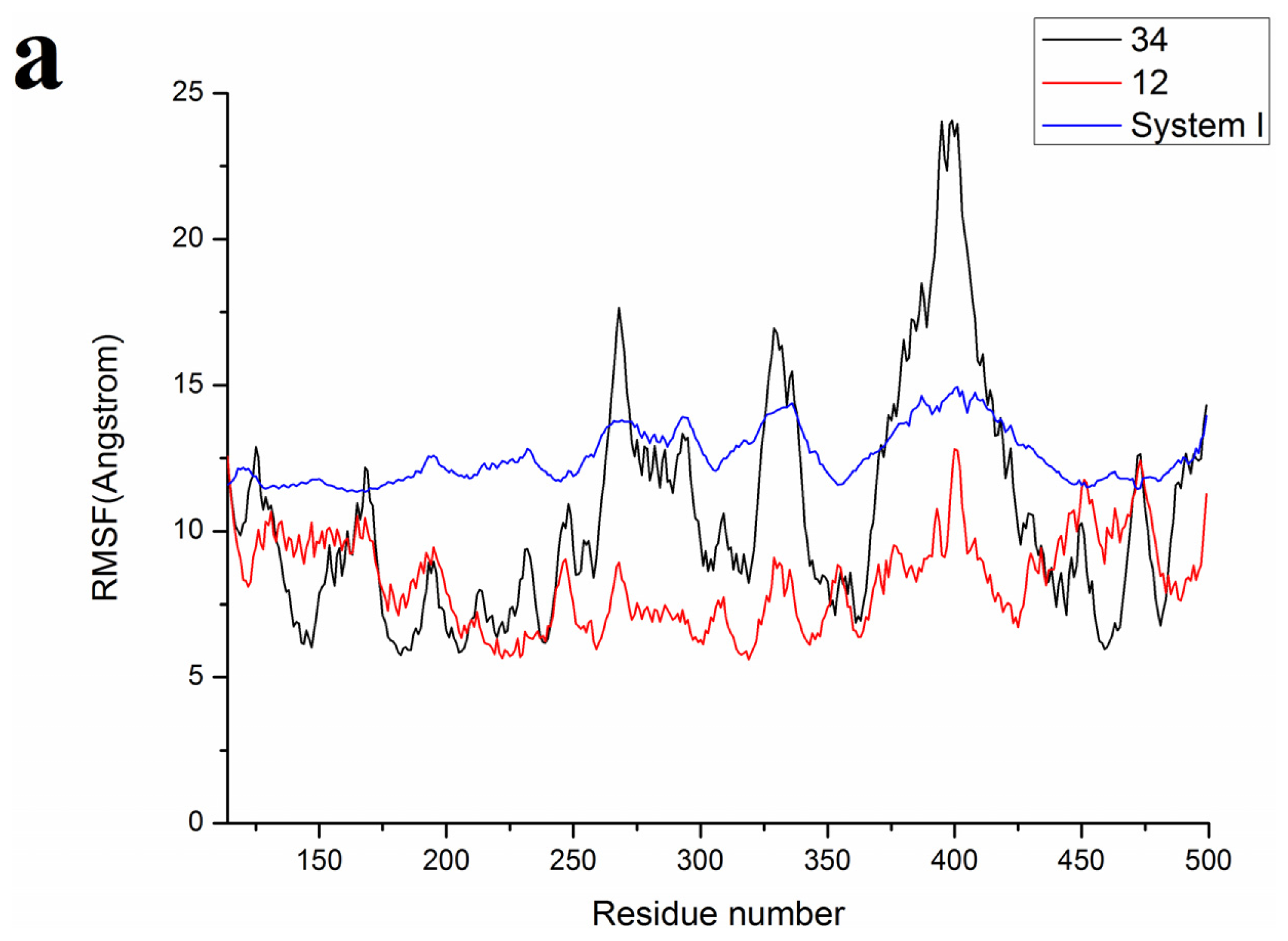
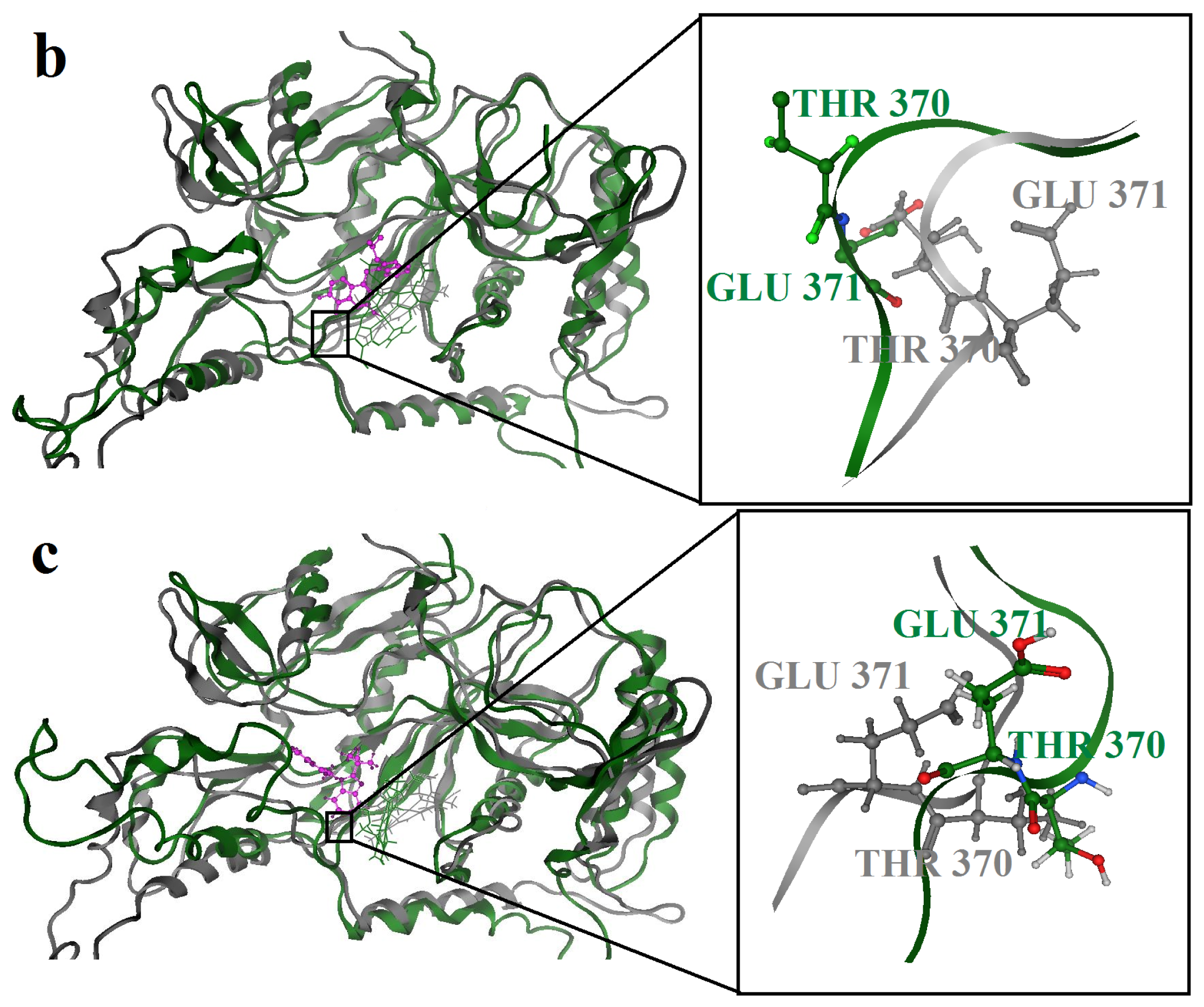
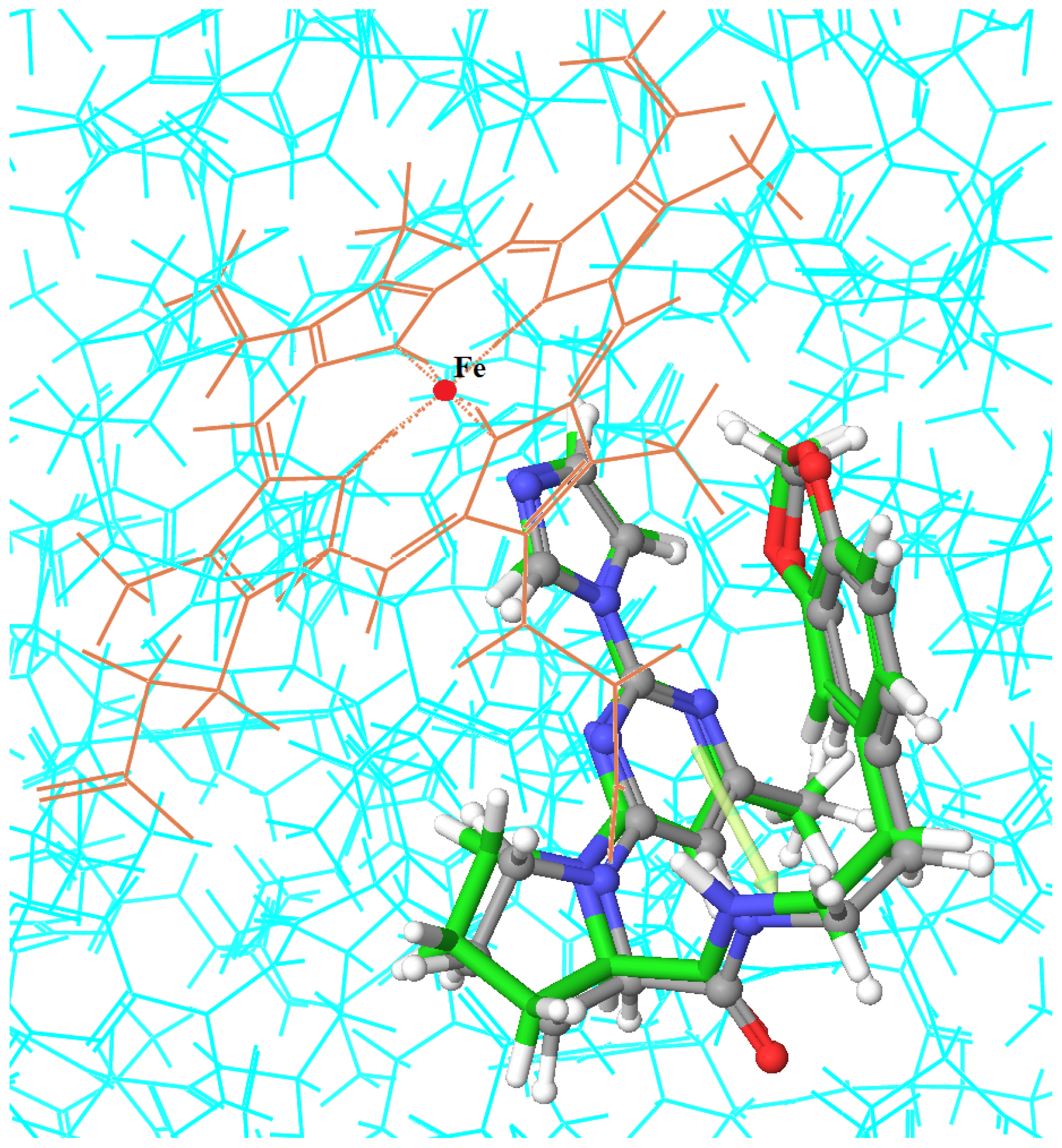
2.3. Molecular Dynamics’ Simulation Studies
3. Materials and Methods
3.1. Data Sets and Tools
3.2. 3D-QSAR Studies
3.3. Molecular Docking and Dynamics Simulations
4. Conclusions
Acknowledgments
References
- Hanafy, K.A.; Krumenacker, J.S.; Murad, F. NO, nitrotyrosine, and cyclic GMP in signal transduction. Med. Sci. Monit 2001, 7, 801–819. [Google Scholar]
- Kobzik, L.; Reid, M.B.; Bredt, D.S.; Stamler, J.S. Nitric oxide in skeletal muscle. Nature 1994, 372, 546–548. [Google Scholar]
- Baranano, D.E.; Snyder, S.H. Neural roles for heme oxygenase: Contrasts to nitric oxide synthase. Proc. Natl. Acad. Sci. USA 2001, 98, 10996–11002. [Google Scholar]
- Rand, M.J. Nitrergic transmission: Nitric oxide as a mediator of non-adrenergic, non-cholinergic neuro-effector transmission. Clin. Exp. Pharm. Physiol 1992, 19, 147–169. [Google Scholar]
- Napoli, C.; Ignarro, L.J. Nitric oxide and atherosclerosis. Nitric Oxide 2001, 5, 88–97. [Google Scholar]
- Hibbs, J.B., Jr; Taintor, R.R.; Vavrin, Z.; Rachlin, E.M. Nitric oxide: A cytotoxic activated macrophage effector molecule. Biochem. Biophys. Res. Commun. 1988, 157, 87–94. [Google Scholar]
- Bogdan, C. Nitric oxide and the immune response. Nat. Immunol 2001, 2, 907–916. [Google Scholar]
- Cirino, G.; Distrutti, E.; Wallace, J.L. Nitric oxide and inflammation. Inflamm. Allergy Drug Targets 2006, 5, 115–119. [Google Scholar]
- Marletta, M.A. Nitric oxide synthase: Aspects concerning structure and catalysis. Cell 1994, 78, 927–930. [Google Scholar]
- Alderton, W.K.; Cooper, C.E.; Knowles, R.G. Nitric oxide synthases: Structure, function and inhibition. Biochem. J 2001, 357, 593–615. [Google Scholar]
- Nagy, G.; Clark, J.M.; Buzas, E.I.; Gorman, C.L.; Cope, A.P. Nitric oxide, chronic inflammation and autoimmunity. Immunol Lett 2007, 111, 1–5. [Google Scholar]
- Dare, A.J.; Phillips, A.R.; Hickey, A.J.; Mittal, A.; Loveday, B.; Thompson, N.; Windsor, J.A. A systematic review of experimental treatments for mitochondrial dysfunction in sepsis and multiple organ dysfunction syndrome. Free Radic. Biol. Med 2009, 47, 1517–1525. [Google Scholar]
- Alderton, W.K.; Angell, A.D.; Craig, C.; Dawson, J.; Garvey, E.; Moncada, S.; Monkhouse, J.; Rees, D.; Russell, L.J.; Russell, R.J.; et al. GW274150 and GW273629 are potent and highly selective inhibitors of inducible nitric oxide synthase in vitro and in vivo. Br. J. Pharm 2005, 145, 301–312. [Google Scholar]
- LaBuda, C.J.; Koblish, M.; Tuthill, P.; Dolle, R.E.; Little, P.J. Antinociceptive activity of the selective iNOS inhibitor AR-C102222 in rodent models of inflammatory, neuropathic and post-operative pain. Eur. J. Pain 2006, 10, 505–512. [Google Scholar]
- Davey, D.D.; Adler, M.; Arnaiz, D.; Eagen, K.; Erickson, S.; Guilford, W.; Kenrick, M.; Morrissey, M.M.; Ohlmeyer, M.; Pan, G.; et al. Design, synthesis, and activity of 2-imidazol-1-ylpyrimidine derived inducible nitric oxide synthase dimerization inhibitors. J. Med. Chem 2007, 50, 1146–1157. [Google Scholar]
- Kubes, P. Inducible nitric oxide synthase: A little bit of good in all of us. Gut 2000, 47, 6–9. [Google Scholar]
- Proskuryakov, S.Y.; Konoplyannikov, A.G.; Skvortsov, V.G.; Mandrugin, A.A.; Fedoseev, V.M. Structure and activity of NO synthase inhibitors specific to the l-arginine binding site. Biochemistry 2005, 70, 8–23. [Google Scholar]
- McMillan, K.; Adler, M.; Auld, D.S.; Baldwin, J.J.; Blasko, E.; Browne, L.J.; Chelsky, D.; Davey, D.; Dolle, R.E.; Eagen, K.A.; et al. Allosteric inhibitors of inducible nitric oxide synthase dimerization discovered via combinatorial chemistry. Proc. Natl. Acad. Sci. USA 2000, 97, 1506–1511. [Google Scholar]
- Ohtsuka, M.; Konno, F.; Honda, H.; Oikawa, T.; Ishikawa, M.; Iwase, N.; Isomae, K.; Ishii, F.; Hemmi, H.; Sato, S. PPA250 [3-(2,4-difluorophenyl)-6-[2-[4-(1H-imidazol-1-ylmethyl)phenoxy] ethoxy]-2-phenylpyridine], a novel orally effective inhibitor of the dimerization of inducible nitric-oxide synthase, exhibits an anti-inflammatory effect in animal models of chronic arthritis. J. Pharm. Exp. Ther 2002, 303, 52–57. [Google Scholar]
- Chida, N.; Hirasawa, Y.; Ohkawa, T.; Ishii, Y.; Sudo, Y.; Tamura, K.; Mutoh, S. Pharmacological profile of FR260330, a novel orally active inducible nitric oxide synthase inhibitor. Eur. J. Pharm 2005, 509, 71–76. [Google Scholar]
- Payne, J.E.; Bonnefous, C.; Symons, K.T.; Nguyen, P.M.; Sablad, M.; Rozenkrants, N.; Zhang, Y.; Wang, L.; Yazdani, N.; Shiau, A.K.; et al. Discovery of dual inducible/neuronal nitric oxide synthase (iNOS/nNOS) inhibitor development candidate 4-((2-cyclobutyl-1H-imidazo[4,5-b]pyrazin-1-yl)methyl)-7,8-difluoroquinolin-2(1H)-one (KD7332) part 2: Identification of a novel, potent, and selective series of benzimidazole-quinolinone iNOS/nNOS dimerization inhibitors that are orally active in pain models. J. Med. Chem 2010, 53, 7739–7755. [Google Scholar]
- Dixon, S.L.; Smondyrev, A.M.; Knoll, E.H.; Rao, S.N.; Shaw, D.E.; Friesner, R.A. PHASE: A new engine for pharmacophore perception, 3D-QSAR model development, and 3D database screening: 1. Methodology and preliminary results. J. Comput. Aided Mol. Des 2006, 20, 647–671. [Google Scholar]
- Dixon, S.L.; Smondyrev, A.M.; Rao, S.N. PHASE: A novel approach to pharmacophore modeling and 3D database searching. Chem. Biol. Drug Des 2006, 67, 370–372. [Google Scholar]
- Stanton, D.T. QSAR and QSPR model interpretation using partial least squares (PLS) analysis. Curr. Comput. Aided Drug Des 2012, 8, 107–127. [Google Scholar]
- Evans, D.A.; Doman, T.N.; Thorner, D.A.; Bodkin, M.J. 3D-QSAR methods: Phase and catalyst compared. J. Chem. Inf. Model 2007, 47, 1248–1257. [Google Scholar]
- Impact 5.0; Schrödinger, LLC: New York, NY, USA, 2008.
- LigPrep 2.2; Schrödinger, LLC: New York, NY, USA, 2008.
- Phase 3.0; Schrödinger, LLC: New York, NY, USA, 2008.
- Maestro 8.5; Schrödinger, LLC: New York, NY, USA, 2008.
- Glide 5.0; Schrödinger, LLC: New York, NY, USA, 2008.
- Desmond 2.0; D.E. Shaw Research/Schrödinger, LLC: New York, NY, USA, 2008.
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem 2004, 47, 1739–1749. [Google Scholar]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys 1983, 79, 926–935. [Google Scholar]
- MacroModel 9.6; Schrödinger, LLC: New York, NY, USA, 2008.
- Kaminski, G.A.; Friesner, R.A. Evaluation and reparametrization of the OPLS-AA force field for proteins via comparison with accurate quantum chemical calculations on peptides. J. Phys. Chem 2001, 105, 6474–6487. [Google Scholar]
- Andersen, H.C. Rattle: A “velocity” version of the shake algorithm for molecular dynamics calculations. J. Comput. Phys 1983, 52, 24–34. [Google Scholar]
- Martyna, G.J.; Klein, M.L.; Tuckerman, M. Nose-Hoover chains: The canonical ensemble via continuous dynamics. J. Comput. Phys 1992, 97, 3684–3690. [Google Scholar]
- Strahan, G.D.; Keniry, M.A.; Shafer, R.H. NMR structure refinement and dynamics of the K+-[d(G3T4G3)]2 quadruplex via particle mesh Ewald molecular dynamics simulations. Biophys. J 1998, 75, 968–981. [Google Scholar]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph 1996, 14, 33–38. [Google Scholar]
- Knapp, B.; Lederer, N.; Omasits, U.; Schreiner, W. vmdICE: A plug-in for rapid evaluation of molecular dynamics simulations using VMD. J. Comput. Chem 2010, 31, 2868–2673. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
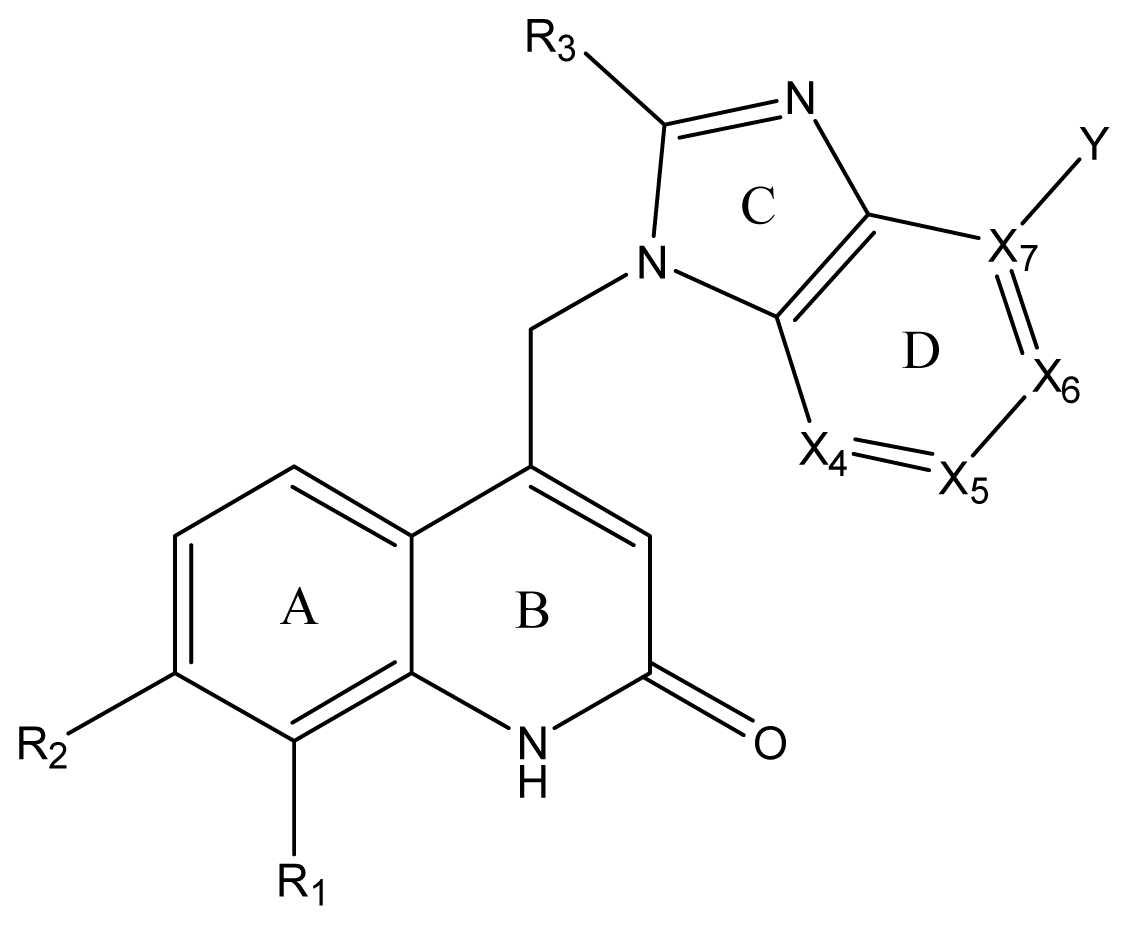
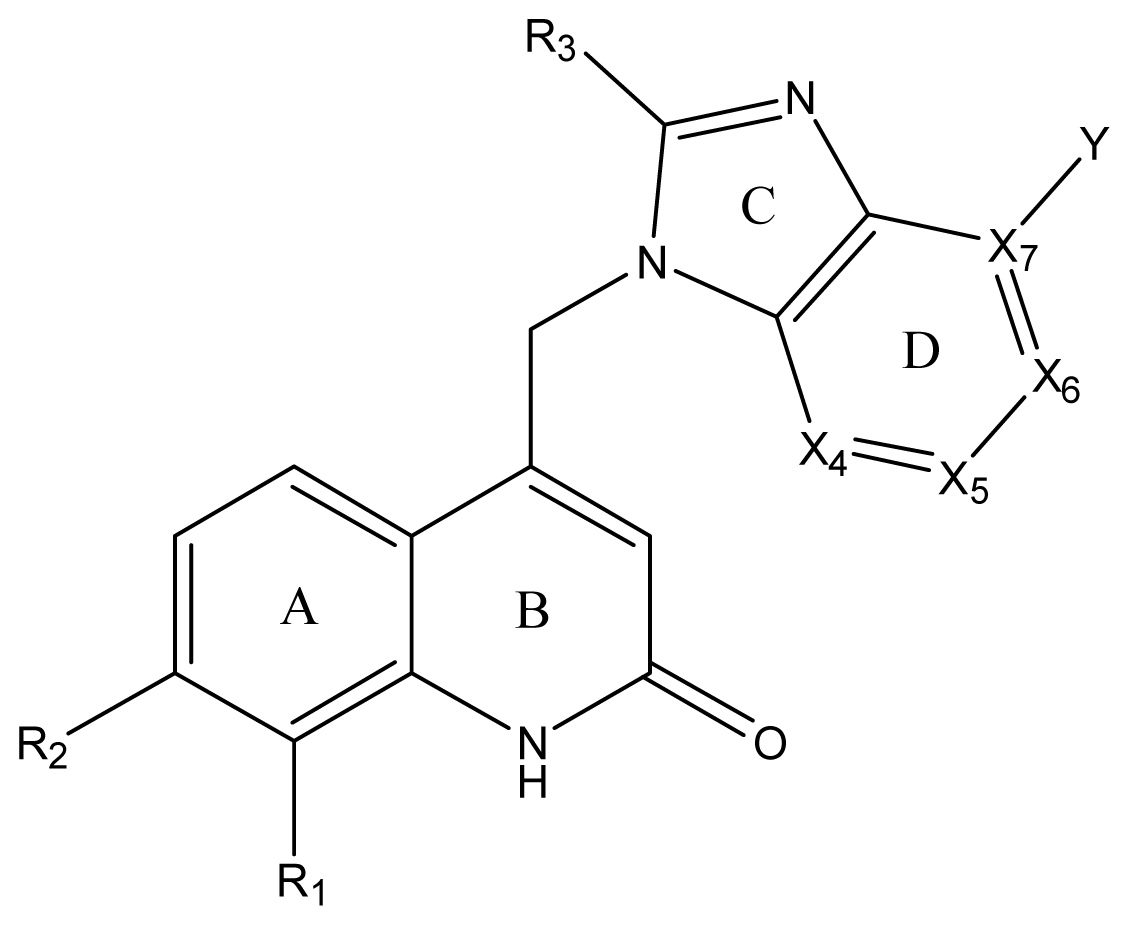 | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Compound NO. | X4 | X5 | X6 | X7 | R1 | R2 | Y | R3 | EC50 (mmol/L) | pEC50 a (expt.) | pEC50 (pred.) | Pharm set |
| 1 | C | C | C | C | F | H | H | 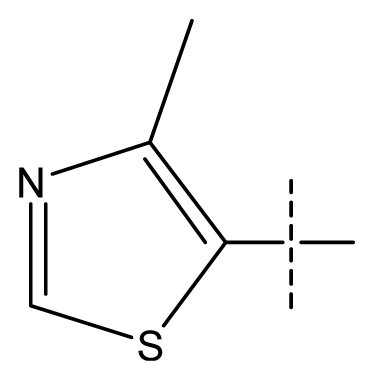 | 0.0037 | 2.43 | 2.29 | inactive |
| 2 * | C | C | C | C | F | F | H |  | 0.0018 | 2.75 | 2.61 | |
| 3 | C | C | C | C | F | F | H |  | 0.0086 | 2.07 | 2.04 | inactive |
| 4 | C | C | C | C | F | F | H | 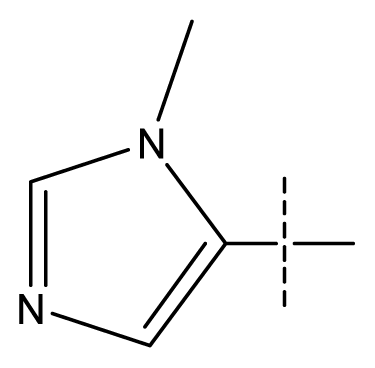 | 0.0059 | 2.23 | 2.09 | inactive |
| 5 * | C | C | C | C | F | F | H | 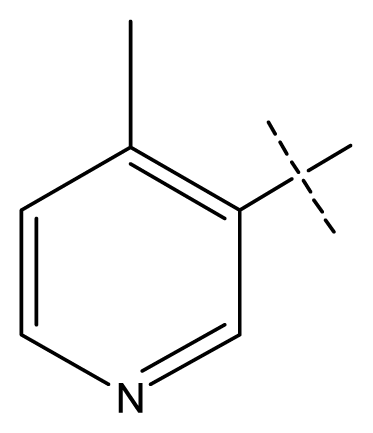 | 0.012 | 1.92 | 2.22 | inactive |
| 6 | C | C | C | C | F | F | H | 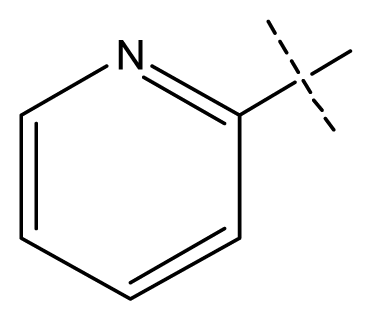 | 0.018 | 1.75 | 1.72 | inactive |
| 7 | C | C | C | C | F | F | H | 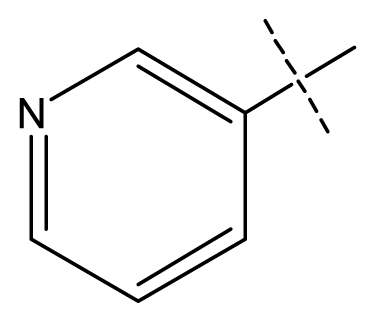 | 0.0093 | 2.03 | 2.11 | inactive |
| 8 | C | C | C | C | F | F | H |  | 0.0022 | 2.66 | 2.55 | |
| 9 | C | C | C | C | F | F | H |  | 0.00034 | 3.47 | 3.40 | active |
| 10 | C | C | C | C | F | F | H | 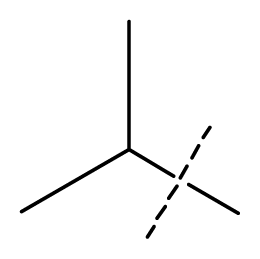 | 0.00080 | 3.10 | 2.79 | active |
| 11 * | C | C | C | C | F | F | H | 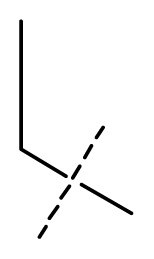 | 0.0043 | 2.37 | 2.49 | inactive |
| 12 | C | C | C | C | F | F | H |  | 0.048 | 1.32 | 1.37 | inactive |
| 13 | C | C | C | C | F | F | H | 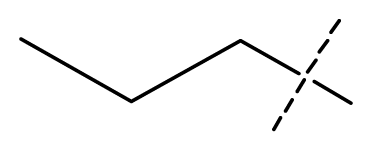 | 0.00073 | 3.14 | 3.35 | active |
| 14 * | C | C | C | C | F | F | H |  | 0.0016 | 2.80 | 2.83 | |
| 15 | C | C | C | C | F | F | H | 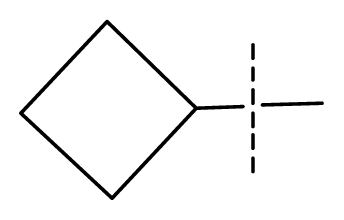 | 0.00026 | 3.59 | 3.21 | active |
| 16 | C | C | C | C | F | F | H |  | 0.00075 | 3.13 | 3.12 | active |
| 17 | C | C | C | C | F | F | H | 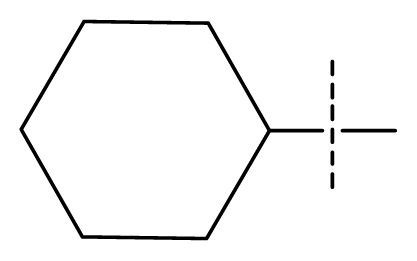 | 0.0089 | 2.05 | 2.33 | inactive |
| 18 * | C | C | C | C | F | F | H | 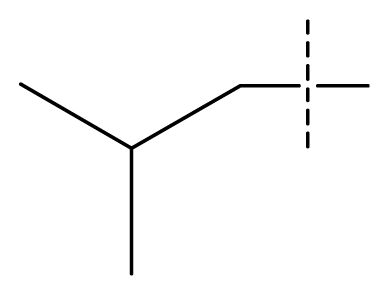 | 0.00038 | 3.42 | 3.05 | active |
| 19 | C | C | C | C | F | F | H |  | 0.00136 | 2.87 | 2.84 | |
| 20 | C | C | C | C | F | F | H | 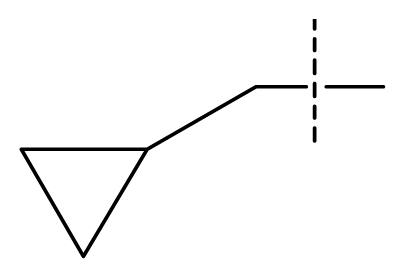 | 0.00060 | 3.22 | 3.50 | active |
| 21 | C | C | C | C | F | F | H | 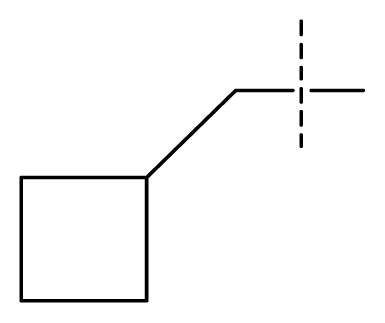 | 0.00054 | 3.27 | 3.24 | active |
| 22 | C | C | C | C | F | F | H | 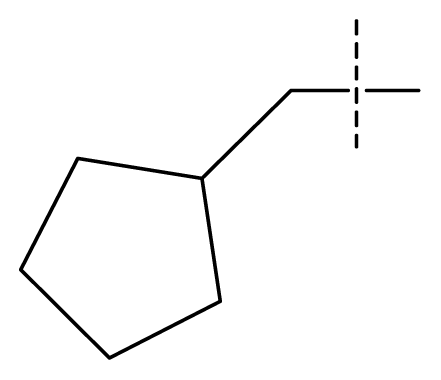 | 0.00177 | 2.75 | 2.76 | |
| 23 * | C | C | C | N | F | F | 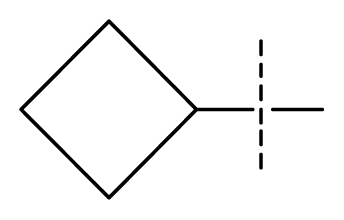 | 0.00059 | 3.23 | 3.01 | active | |
| 24 | C | C | N | C | F | F | H | 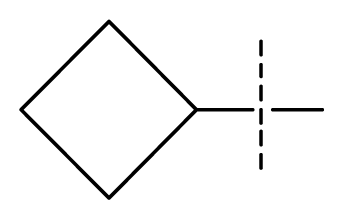 | 0.018 | 1.75 | 2.06 | inactive |
| 25 * | C | N | C | C | F | F | H | 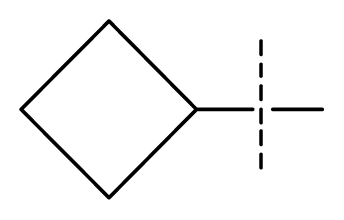 | 0.00046 | 3.34 | 2.90 | active |
| 26 | N | C | C | C | F | F | H |  | 0.00018 | 3.75 | 3.69 | active |
| 27 | N | C | C | C | F | F | Me |  | 0.00027 | 3.57 | 3.56 | active |
| 28 | N | C | C | C | F | F | Cl | 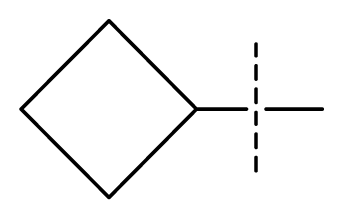 | 0.00027 | 3.57 | 3.61 | active |
| 29 * | N | C | C | C | F | F | OMe |  | 0.00021 | 3.68 | 3.78 | active |
| 30 | N | C | C | C | F | F | NMe2 | 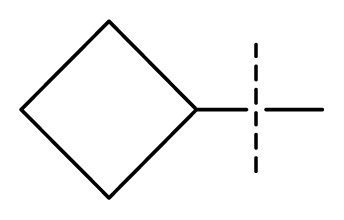 | 0.00017 | 3.77 | 3.83 | active |
| 31 * | N | C | C | C | F | F | CF3 | 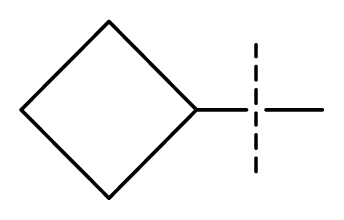 | 0.00015 | 3.82 | 3.69 | active |
| 32 * | N | C | C | N | F | F | 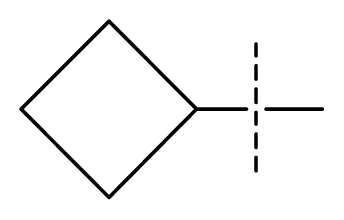 | 0.000091 | 4.04 | 3.70 | active | |
| 33 | N | C | C | N | F | F |  | 0.00028 | 3.55 | 3.67 | active | |
| 34 | N | C | C | N | F | F |  | 0.000088 | 4.06 | 3.70 | active | |
| 35 | N | C | C | N | F | F | 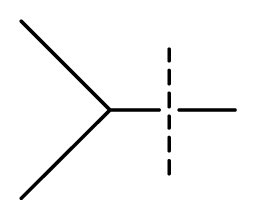 | 0.00085 | 3.07 | 3.12 | active | |
| 36 | N | C | C | N | F | F | 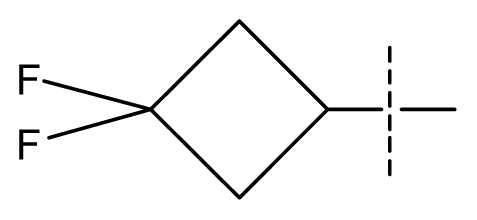 | 0.00012 | 3.92 | 3.58 | active | |
| 37 | N | C | C | N | F | H |  | 0.00019 | 3.72 | 3.88 | active | |
| 38 | N | C | C | N | F | H |  | 0.00029 | 3.54 | 3.68 | active | |
| 39 | N | C | C | N | F | H | 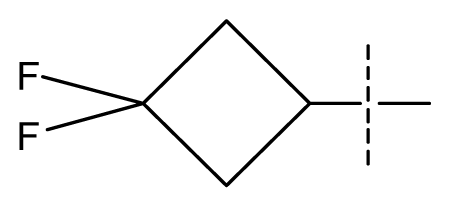 | 0.00022 | 3.66 | 3.58 | active | |
© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Zhang, H.; Zan, J.; Yu, G.; Jiang, M.; Liu, P. A Combination of 3D-QSAR, Molecular Docking and Molecular Dynamics Simulation Studies of Benzimidazole-Quinolinone Derivatives as iNOS Inhibitors. Int. J. Mol. Sci. 2012, 13, 11210-11227. https://doi.org/10.3390/ijms130911210
Zhang H, Zan J, Yu G, Jiang M, Liu P. A Combination of 3D-QSAR, Molecular Docking and Molecular Dynamics Simulation Studies of Benzimidazole-Quinolinone Derivatives as iNOS Inhibitors. International Journal of Molecular Sciences. 2012; 13(9):11210-11227. https://doi.org/10.3390/ijms130911210
Chicago/Turabian StyleZhang, Hao, Jinhang Zan, Guangyun Yu, Ming Jiang, and Peixun Liu. 2012. "A Combination of 3D-QSAR, Molecular Docking and Molecular Dynamics Simulation Studies of Benzimidazole-Quinolinone Derivatives as iNOS Inhibitors" International Journal of Molecular Sciences 13, no. 9: 11210-11227. https://doi.org/10.3390/ijms130911210