Molecular Mechanisms of Epigenetic Variation in Plants

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
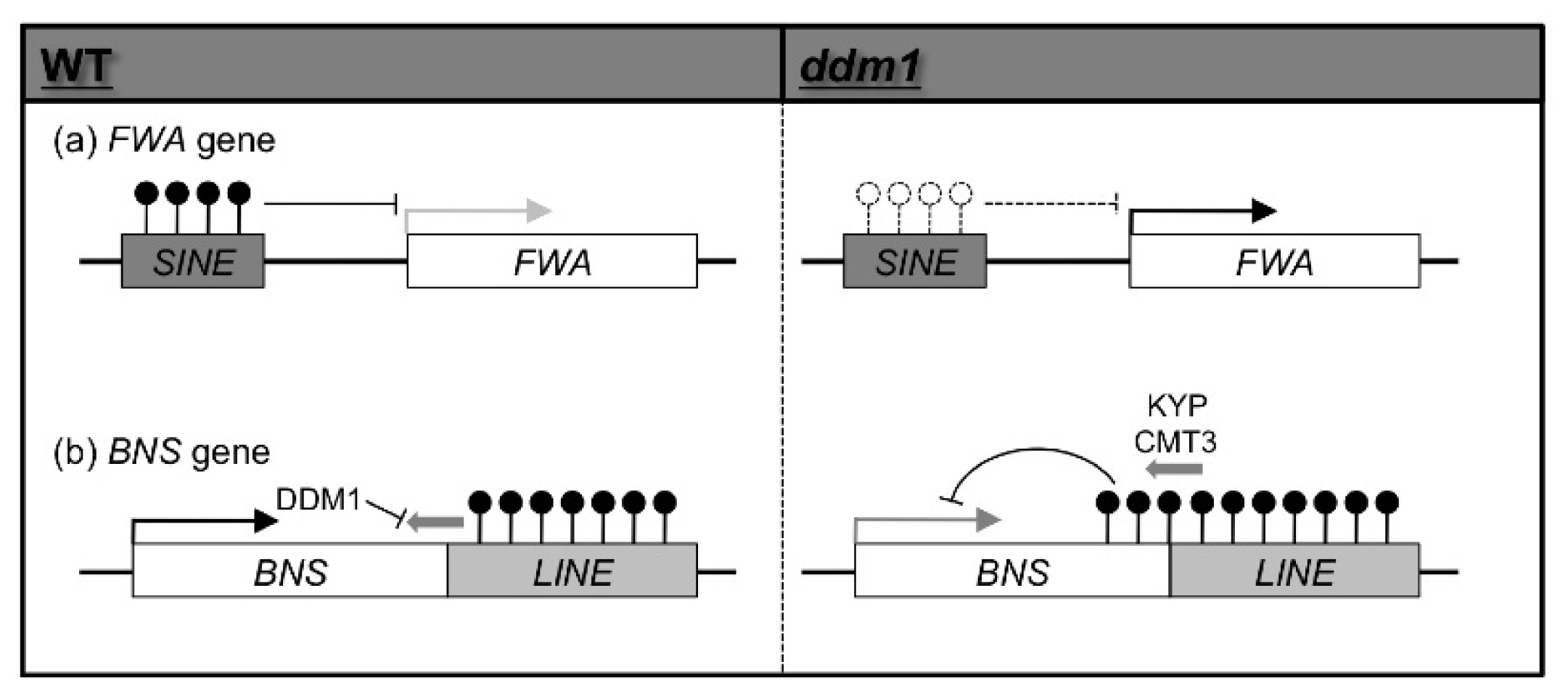
2. Epigenetic Variation Induced by Mutations of Genes Involved in Epigenetic Modification in A. thaliana
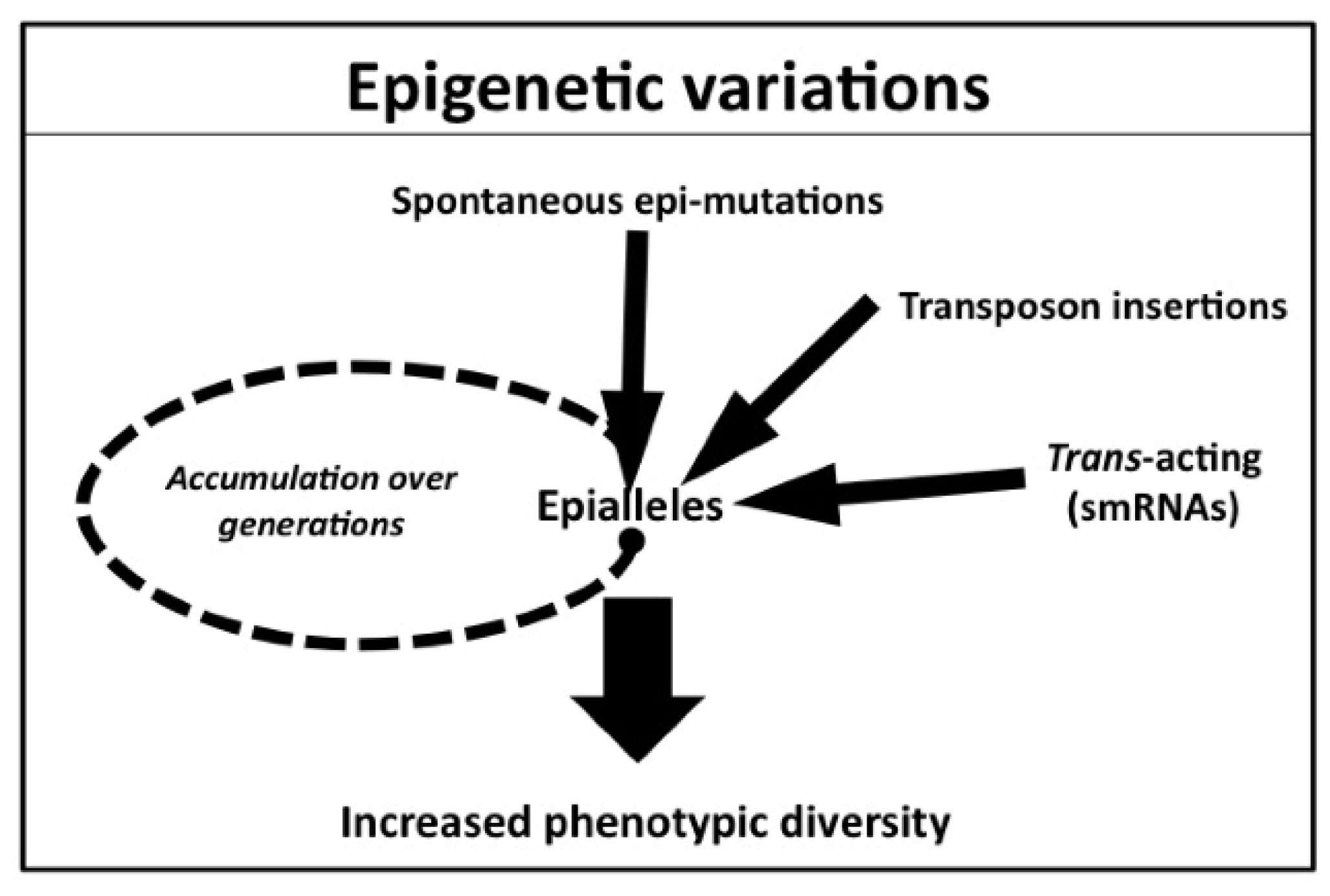
3. Natural Variation of Epigenetic Status
3.1. Spontaneous Epigenetic Mutants Occurring at Single Loci
3.2. Transposon Insertion Can Generate Epigenetic Alleles
3.3. Trans-Acting Epigenetic Modifications
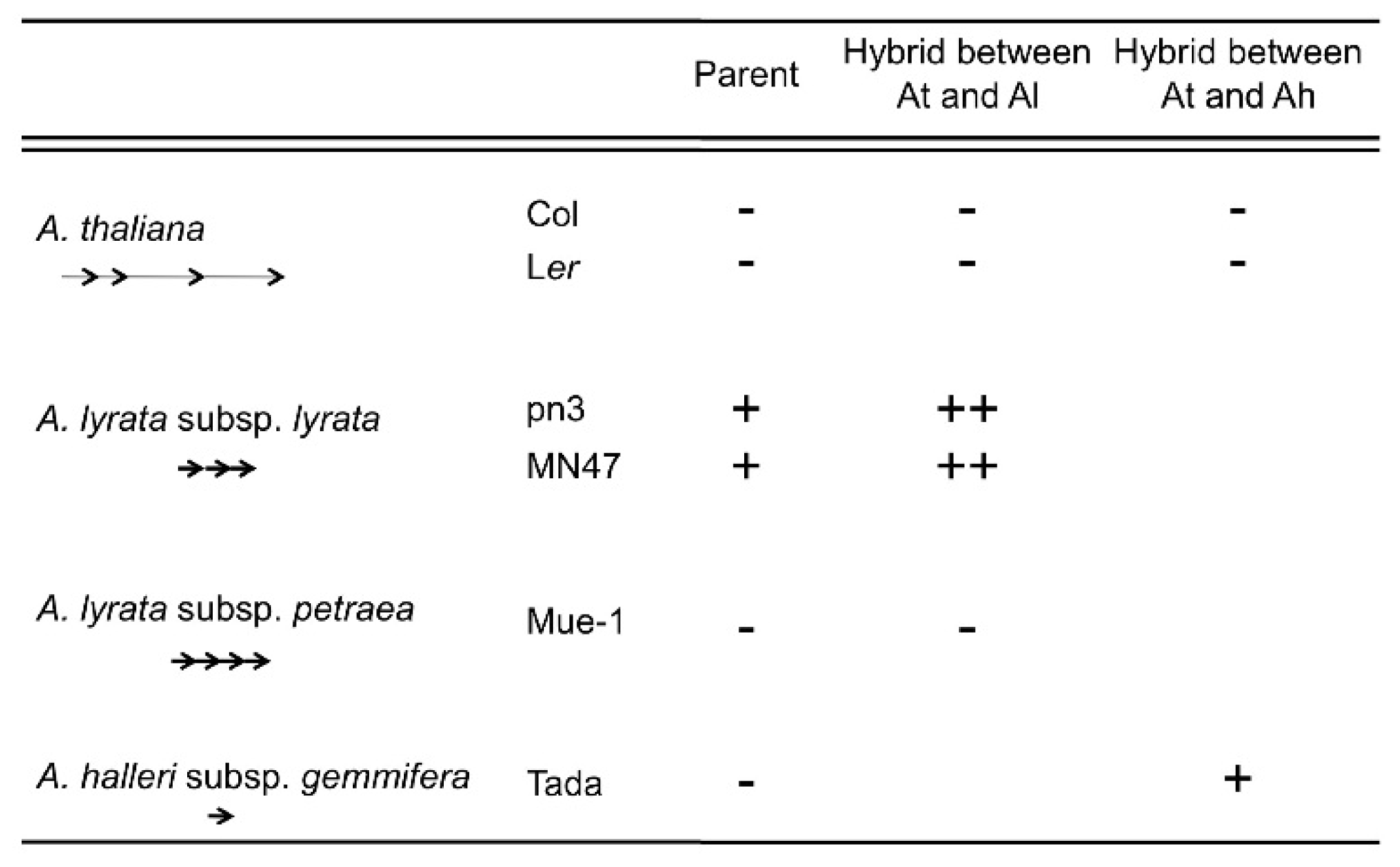
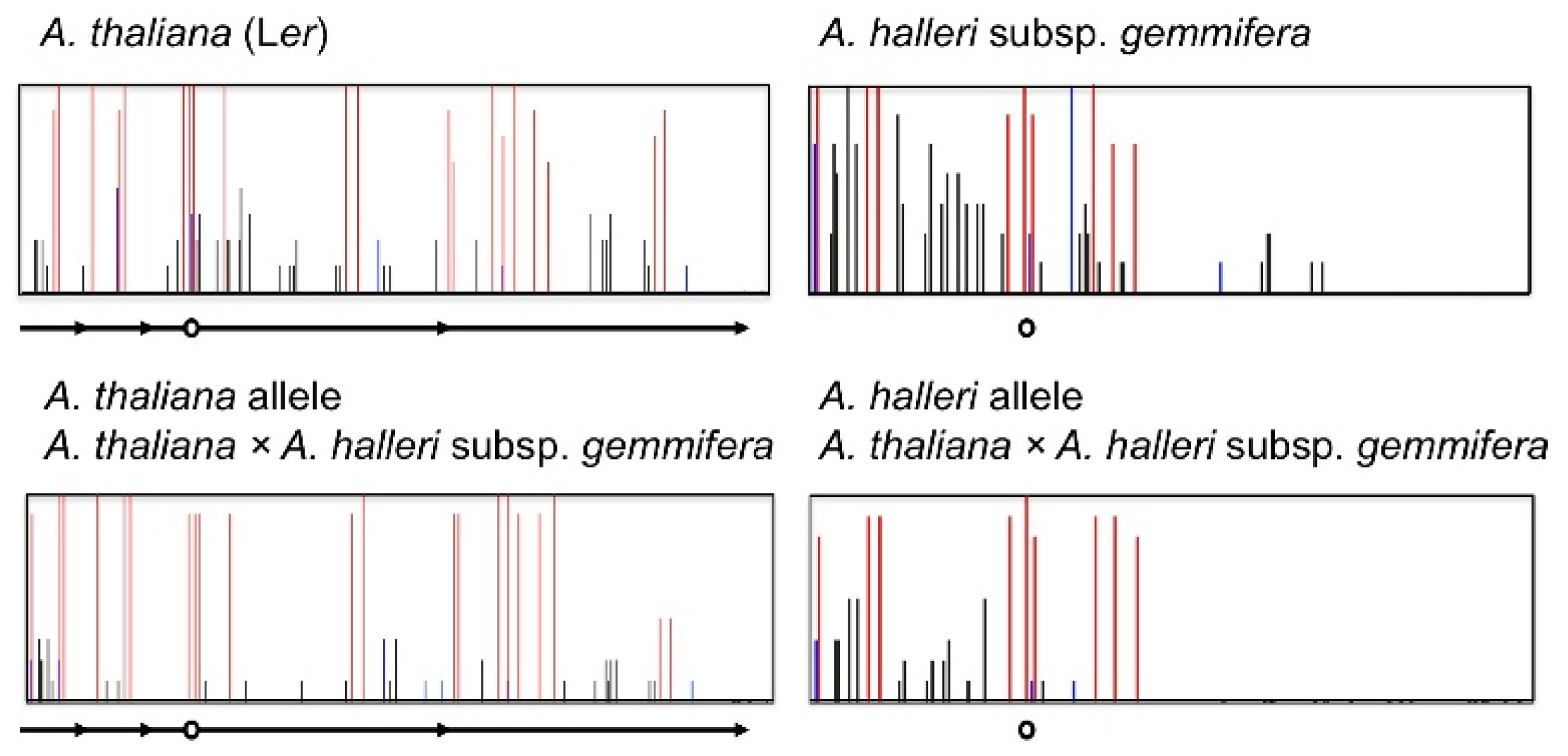
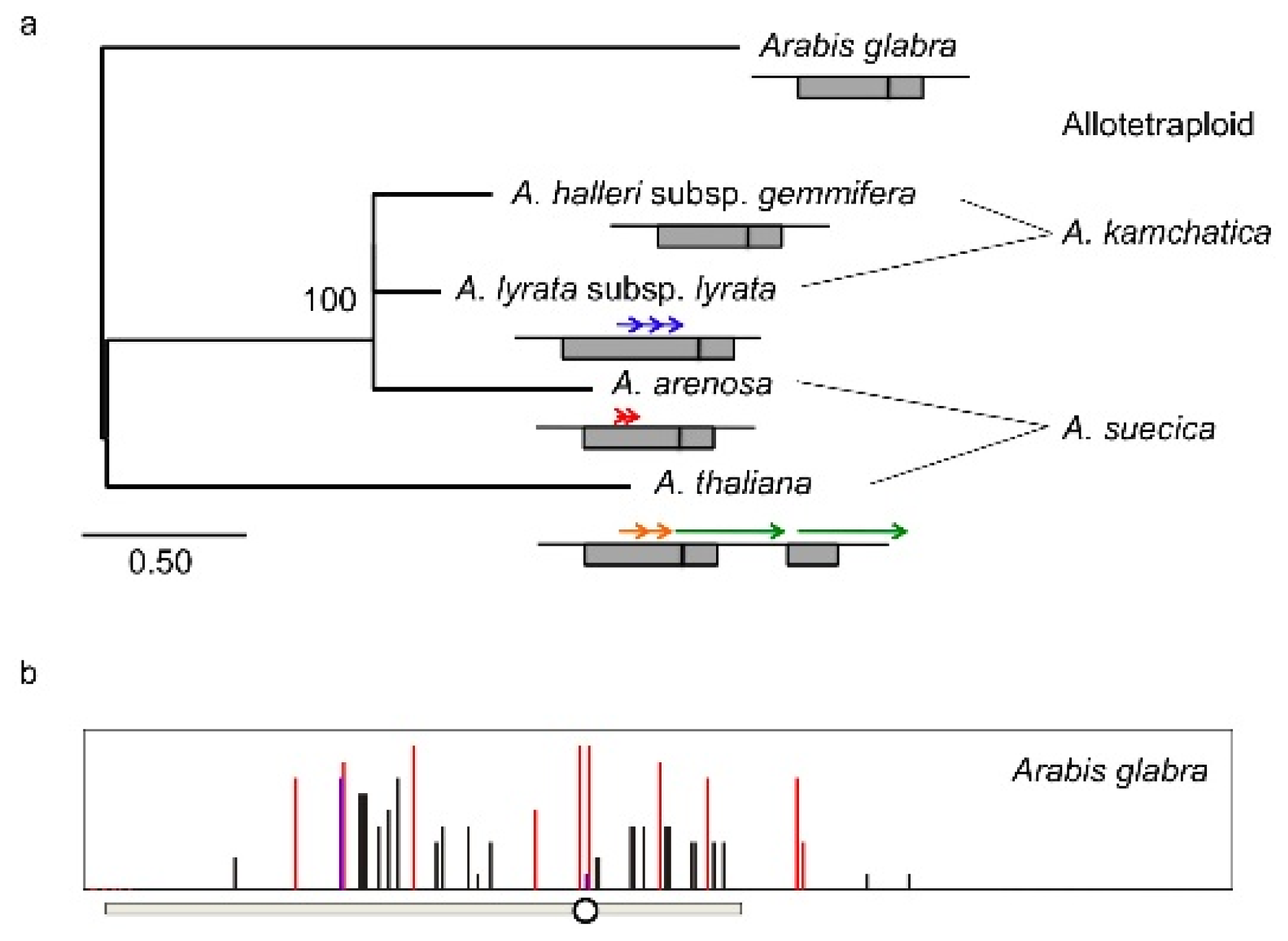
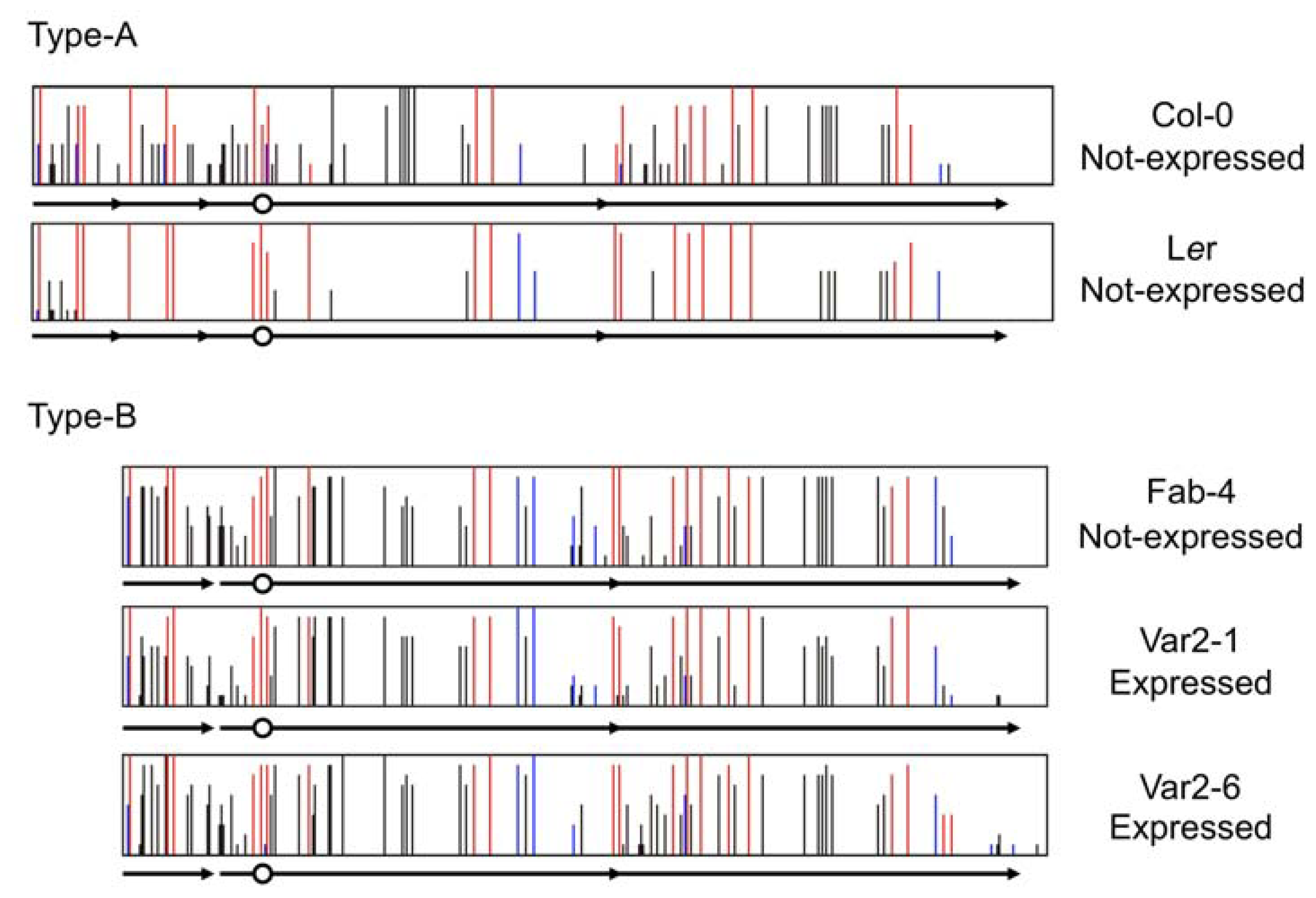
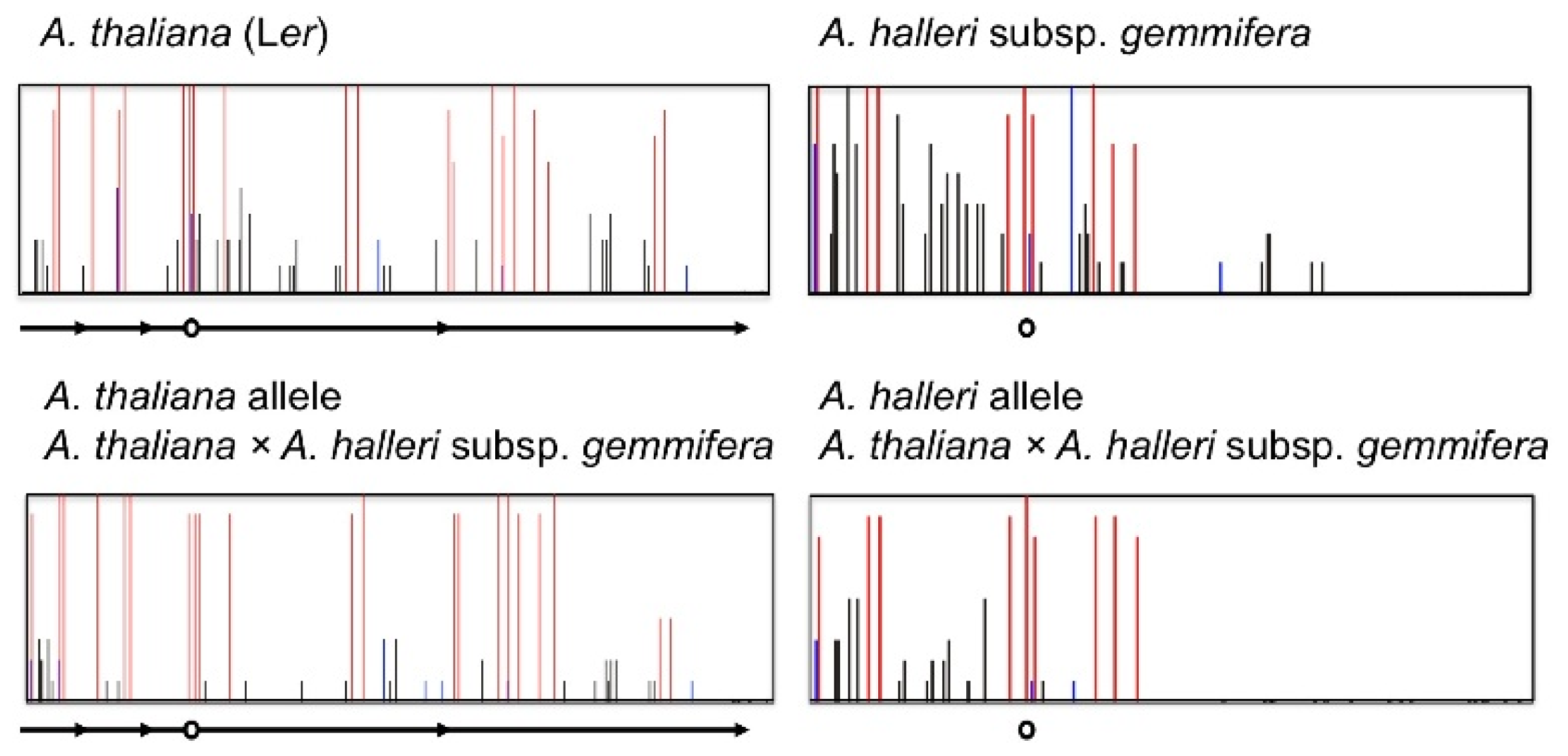
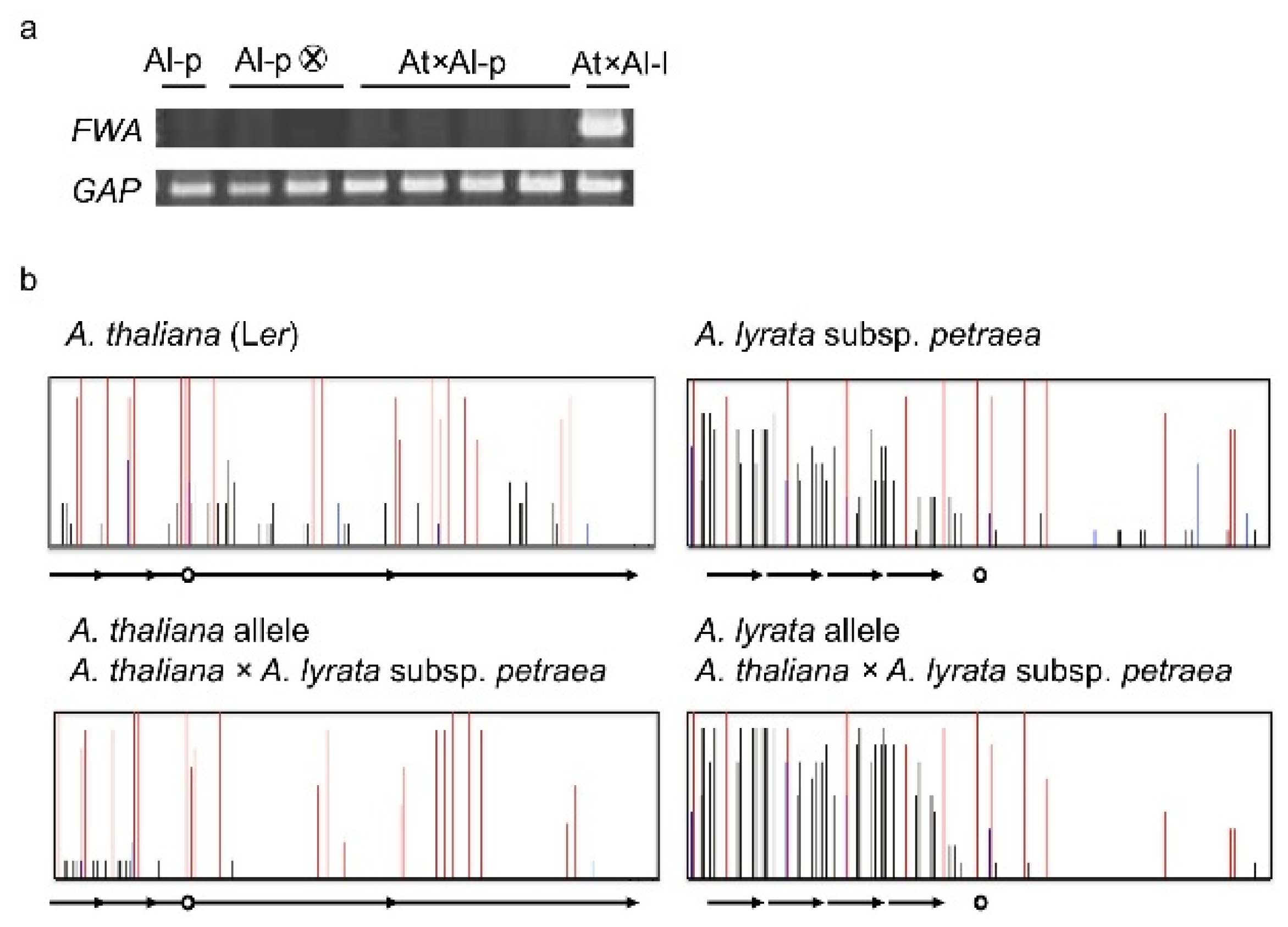
4. Natural Variation of Imprinted FWA Genes in the Genus Arabidopsis
5. Conclusions
Acknowledgments
References
- Cokus, S.J.; Feng, S.; Zhang, X.; Chen, Z.; Merriman, B.; Haudenschild, C.D.; Pradhan, S.; Nelson, S.F.; Pellegrini, M.; Jacobsen, S.E. Shotgun bisulphite sequencing of the Arabidopsis genome reveals DNA methylation patterning. Nature 2008, 452, 215–219. [Google Scholar]
- Lister, R.; O’Malley, R.C.; Tonti-Filippini, J.; Gregory, B.D.; Berry, C.C.; Millar, A.H.; Ecker, J.R. Highly integrated single-base resolution maps of the epigenome in Arabidopsis. Cell 2008, 133, 523–536. [Google Scholar]
- Law, J.A.; Jacobsen, S.E. Establishing, maintaining and modifying DNA methylation patterns in plants and animals. Nat. Rev. Genet 2010, 11, 204–220. [Google Scholar]
- Zhang, X.; Yazaki, J.; Sundaresan, A.; Cokus, S.; Chan, S.W.; Chen, H.; Henderson, I.R.; Shinn, P.; Pellegrini, M.; Jacobsen, S.E.; et al. Genome-Wide high-resolution mapping and functional analysis of DNA methylation in Arabidopsis. Cell 2006, 126, 1189–1201. [Google Scholar]
- Zilberman, D.; Gehring, M.; Tran, R.K.; Ballinger, T.; Henikoff, S. Genome-Wide analysis of Arabidopsis thaliana DNA methylation uncovers an interdependence between methylation and transcription. Nat. Genet 2007, 39, 61–69. [Google Scholar]
- Li, X.; Wang, X.; He, K.; Ma, Y.; Su, N.; He, H.; Stolc, V.; Tongprasit, W.; Jin, W.; Jiang, J.; et al. High-resolution mapping of epigenetic modifications of the rice genome uncovers interplay between DNA methylation, histone methylation, and gene expression. Plant Cell 2008, 20, 259–276. [Google Scholar]
- Wang, X.; Elling, A.A.; Li, X.; Li, N.; Peng, Z.; He, G.; Sun, H.; Qi, Y.; Liu, X.S.; Deng, X.W. Genome-Wide and organ-specific landscapes of epigenetic modifications and their relationships to mRNA and small RNA transcriptomes in maize. Plan Cell 2009, 21, 1053–1069. [Google Scholar]
- Miura, A.; Yonebayashi, S.; Watanabe, K.; Toyama, T.; Shimada, H.; Kakutani, T. Mobilization of transposons by a mutation abolishing full DNA methylation in Arabidopsis. Nature 2001, 411, 212–214. [Google Scholar]
- Singer, T.; Yordan, C.; Martienssen, R.A. Robertson’s Mutator transposons in A. thaliana are regulated by the chromatin-remodeling gene Decrease in DNA methylation (DDM1). Genes Dev 2001, 15, 591–602. [Google Scholar]
- Fujimoto, R.; Sasaki, T.; Inoue, H.; Nishio, T. Hypomethylation and transcriptional reactivation of retrotransposon-like sequences in ddm1 transgenic plants of Brassica rapa. Plant Mol. Biol 2008, 66, 463–473. [Google Scholar]
- Tsukahara, S.; Kobayashi, A.; Kawabe, A.; Mathieu, O.; Miura, A.; Kakutani, T. Bursts of retrotransposition reproduced in Arabidopsis. Nature 2009, 461, 423–426. [Google Scholar]
- Sasaki, T.; Fujimoto, R.; Kishitani, S.; Nishio, T. Analysis of target sequences of DDM1s in Brassica rapa by MSAP. Plant Cell Rep 2011, 30, 81–88. [Google Scholar]
- Feng, S.; Cokus, S.J.; Zhang, X.; Chen, P.Y.; Bostick, M.; Goll, M.G.; Hetzel, J.; Jaine, J.; Strauss, S.H.; Halpern, M.E.; et al. Conservation and divergence of methylation patterning in plants and animals. Proc. Natl. Acad. Sci. USA 2010, 107, 8689–8694. [Google Scholar]
- Zemach, A.; McDaniel, I.E.; Silva, P.; Zilberman, D. Genome-Wide evolutionary analysis of eularyotic DNA methylation. Science 2010, 328, 916–919. [Google Scholar]
- He, G.; Elling, A.A.; Deng, X.W. The epigenome and plant development. Annu. Rev. Plant Biol 2011, 62, 411–435. [Google Scholar]
- Lauria, M.; Rossi, V. Epigenetic control of gene regulation in plants. Biochim. Biophys. Acta 2011, 1809, 369–378. [Google Scholar]
- Fuchs, J.; Demidov, D.; Houben, A.; Schubert, I. Chromosomal histone modification patterns–from conservation to diversity. Trends Plant Sci 2006, 11, 199–208. [Google Scholar]
- Preuss, S.; Pikaard, C.S. rRNA gene silencing and nucleolar dominance: Insights into a chromosome-scale epigenetic on/off switch. Biochim. Biophys. Acta 2007, 1769, 383–392. [Google Scholar]
- Erhard, K.F., Jr; Hollick, J.B. Paramutation: A process for acquiring trans-generational regulatory states. Curr. Opin. Plant Biol. 2011, 14, 210–216. [Google Scholar]
- Groszmann, M.; Greaves, I.K.; Albert, N.; Fujimoto, R.; Helliwell, C.A.; Dennis, E.S.; Peacock, W.J. Epigenetics in plants-vernalisation and hybrid vigour. Biochem. Biophys. Acta 2011, 1809, 427–437. [Google Scholar]
- Wollmann, H.; Berger, F. Epigenetic reprogramming during pant reproduction and seed development. Curr. Opin. Plant Biol 2012, 15, 63–69. [Google Scholar]
- Vaughn, M.W.; Tanurdžić, M.; Lippman, Z.; Jiang, H.; Carrasquillo, R.; Rabinowicz, P.D.; Dedhia, N.; McCombie, W.R.; Agier, N.; Bulski, A.; et al. Epigenetic natural variation in Arabidopsis thaliana. PLoS Biol 2007, 5, e174. [Google Scholar]
- He, G.; Zhu, X.; Elling, A.A.; Chen, L.; Wang, X.; Guo, L.; Liang, M.; He, H.; Zhang, H.; Chen, F.; et al. Global epigenetic and transcriptional trends among two rice subspecies and their reciprocal hybrids. Plant Cell 2010, 22, 17–33. [Google Scholar]
- Eichten, S.R.; Swanson-Wagner, R.A.; Schnable, J.C.; Waters, A.J.; Hermanson, P.J.; Liu, S.; Yeh, C.T.; Jia, Y.; Gendler, K.; Freeling, M.; et al. Heritable epigenetic variation among maize inbreds. PLoS Genet 2011, 7, e1002372. [Google Scholar]
- Greaves, I.K.; Groszmann, M.; Ying, H.; Taylor, J.M.; Peacock, W.J.; Dennis, E.S. Trans chromosomal methylation in Arabidopsis hybrids. Proc. Natl. Acad. Sci. USA 2012, 109, 3570–3575. [Google Scholar]
- Shen, H.; He, H.; Li, J.; Chen, W.; Wang, X.; Guo, L.; Peng, Z.; He, G.; Zhong, S.; Qi, Y.; et al. Genome-Wide analysis of DNA methylation and gene expression changes in two Arabidopsis ecotypes and their reciprocal hybrids. Plant Cell 2012, 24, 875–892. [Google Scholar]
- Richards, E.J. Inherited epigenetic variation—Revisiting soft inheritance. Nat. Rev. Genet 2006, 7, 395–401. [Google Scholar]
- Paszkowski, J.; Grossniklaus, U. Selected aspects of transgenerational epigenetic inheritance and resetting in plants. Curr. Opin. Plant Biol 2011, 14, 195–203. [Google Scholar]
- Johannes, F.; Porcher, E.; Teixeira, F.K.; Saliba-Colombani, V.; Simon, M.; Agier, N.; Bulski, A.; Albuisson, J.; Heredia, F.; Audigier, P.; et al. Assessing the impact of transgenerational epigenetic variation on complex traits. PLoS Genet 2009, 5, e1000530. [Google Scholar]
- Cubas, P.; Vincent, C.; Coen, E. An epigenetic mutation responsible for natural variation in floral symmetry. Nature 1999, 401, 157–161. [Google Scholar]
- Manning, K.; Tör, M.; Poole, M.; Hong, Y.; Thompson, A.J.; King, G.J.; Giovannoni, J.J.; Seymour, G.B. A naturally occurring epigenetic mutation in a gene encoding an SBP-box transcription factor inhibits tomato fruit ripening. Nat. Genet 2006, 38, 948–952. [Google Scholar]
- Miura, K.; Agetsuma, M.; Kitano, H.; Yoshimura, A.; Matsuoka, M.; Jacobsen, S.E.; Ashikari, M. A metastable DWARF1 epigenetic mutant affecting plant stature in rice. Proc. Natl. Acad. Sci. USA 2009, 106, 11218–11223. [Google Scholar]
- Kakutani, T. Epi-Alleles in plants: Inheritance of epigenetic information over generations. Plant Cell Physiol 2002, 43, 1106–1111. [Google Scholar]
- Richards, E.J. Natural epigenetic variation in plant species: A view from the field. Curr. Opin. Plant Biol 2011, 14, 204–209. [Google Scholar]
- Saze, H. Epigenetic memory transmission through mitosis and meiosis in plants. Semin. Cell Dev. Biol 2008, 19, 527–536. [Google Scholar]
- Matzke, M.; Kanno, T.; Daxinger, L.; Huettel, B.; Matzke, A.J.M. RNA-Mediated chromatin-based silencing in plants. Curr. Opin. Cell Biol 2009, 21, 367–376. [Google Scholar]
- Haag, J.R.; Pikaard, C.S. Multisubunit RNA polymerases IV and V: Purveyors of non-coding RNA for plant gene silencing. Nat. Rev. Mol. Cell Biol 2011, 12, 483–492. [Google Scholar]
- Kakutani, T.; Jeddeloh, J.A.; Flowers, S.K.; Munakata, K.; Richards, E.J. Developmental abnormalities and epimutations associated with DNA hypomethylation mutations. Proc. Natl. Acad. Sci. USA 1996, 93, 12406–12411. [Google Scholar]
- Kakutani, T. Genetic characterization of late-flowering traits induced by DNA hypomethylation mutation in Arabidopsis thaliana. Plant J 1997, 12, 1447–1451. [Google Scholar]
- Kakutani, T.; Munakata, K.; Richards, E.J.; Hirochika, H. Meiotically and mitotically stable inheritance of DNA hypomethylation induced by ddm1 mutation of Arabidopsis thaliana. Genetics 1999, 151, 831–838. [Google Scholar]
- Kato, M.; Miura, A.; Bender, J.; Jacobsen, S.E.; Kakutani, T. Role of CG and non-CG methylation in immobilization of transposons in Arabidopsis. Curr. Biol 2003, 13, 421–426. [Google Scholar]
- Mirouze, M.; Reinders, J.; Bucher, E.; Nishimura, T.; Schneeberger, K.; Ossowski, S.; Cao, J.; Weigel, D.; Paszkowski, J.; Mathieu, O. Selective epigenetic control of retrotransposition in Arabidopsis. Nature 2009, 461, 427–430. [Google Scholar]
- Soppe, W.J.; Jacobsen, S.E.; Alonso-Blanco, C.; Jackson, J.P.; Kakutani, T.; Koornneef, M.; Peeters, A.J.M. The late flowering phenotype of fwa mutants is caused by gain-of-function epigenetic alleles of a homeodomain gene. Mol. Cell 2000, 6, 791–802. [Google Scholar]
- Kankel, M.W.; Ramsey, D.E.; Stokes, T.L.; Flowers, S.K.; Haag, J.R.; Jeddeloh, J.A.; Riddle, N.C.; Verbsky, M.L.; Richards, E.J. Arabidopsis MET1 cytosine methyltransferase mutants. Genetics 2003, 163, 1109–1122. [Google Scholar]
- Saze, H.; Scheid, O.M.; Paszkowski, J. Maintenance of CpG methylation is essential for epigenetic inheritance during plant gametogenesis. Nat. Genet 2003, 34, 65–69. [Google Scholar]
- Cao, X.; Jacobsen, S.E. Locus-specific control of asymmetric and CpNpG methylation by the DRM and CMT3 methyltransferase genes. Proc. Natl. Acad. Sci. USA 2002, 99, 16491–16498. [Google Scholar]
- Saze, H.; Kakutani, T. Heritable epigenetic mutation of a transposon-flanked Arabidopsis gene due to lack of the chromatin-remodeling factor DDM1. EMBO J 2007, 26, 3641–3652. [Google Scholar]
- Sasaki, T.; Kobayashi, A.; Saze, H.; Kakutani, T. RNAi-independent de novo DNA methylation revealed in Arabidopsis mutants of chromatin remodeling gene DDM1. Plant J 2012, 70, 750–758. [Google Scholar]
- Finnegan, E.J.; Peacock, W.J.; Dennis, E.S. Reduced DNA methylation in Arabidopsis thaliana results in abnormal plant development. Proc. Natl. Acad. Sci. USA 1996, 93, 8449–8454. [Google Scholar]
- Ronemus, M.J.; Galbiati, M.; Ticknor, C.; Chen, J.; Dellaporta, S.L. Demethylation-induced developmental pleiotropy in Arabidopsis. Science 1996, 273, 654–657. [Google Scholar]
- Reinders, J.; Wulff, B.B.H.; Mirouze, M.; Marí-Ordóñez, A.; Dapp, M.; Rozhon, W.; Bucher, E.; Theiler, G.; Paszkowski, J. Compromised stability of DNA methylation and transposon immobilization in mosaic Arabiopsis epigenomes. Genes Dev 2009, 23, 939–950. [Google Scholar]
- Jacobsen, S.E.; Meyerowitz, E.M. Hypermethylated SUPERMAN epigenetic alleles in Arabidopsis. Science 1997, 277, 1100–1103. [Google Scholar]
- Jacobsen, S.E.; Sakai, H.; Finnegan, E.J.; Cao, X.; Meyerowitz, E.M. Ectopic hypermethylation of flower-specific genes in Arabidopsis. Curr. Biol 2000, 10, 179–186. [Google Scholar]
- Mathieu, O.; Reinders, J.; Caikovski, M.; Smathajitt, C.; Paszkowski, J. Transgenerational stability of the Arabidopsis epigenome is coordinated by CG methylation. Cell 2007, 130, 851–862. [Google Scholar]
- Chan, S.W.; Henderson, I.R.; Zhang, X.; Shah, G.; Chien, J.S.; Jacobsen, S.E. RNAi, DRD1, and histone methylation actively target developmentally important non-CG DNA methylation in Arabidopsis. PLoS Genet 2006, 2, e83. [Google Scholar]
- Henderson, I.R.; Jacobsen, S.E. Tandem repeats upstream of the Arabidopsis endogene SDC recruit non-CG DNA methylation and initiate siRNA spreading. Genes Dev 2008, 22, 1597–1606. [Google Scholar]
- Zhang, X.; Shiu, S.H.; Cal, A.; Borevitz, J.O. Global analysis of genetic, epigenetic and transcriptional polymorphisms in Arabidopsis thaliana using whole genome tiling arrays. PLoS Genet 2008, 4, e1000032. [Google Scholar]
- Zhai, J.; Liu, J.; Liu, B.; Li, P.; Meyers, B.C.; Chen, X.; Cao, X. Small RNA-directed epigenetic natural variation in Arabidopsis thaliana. PLoS Genet 2008, 4, e1000056. [Google Scholar]
- Becker, C.; Hagmann, J.; Müller, J.; Koenig, D.; Stegle, O.; Borgwardt, K.; Weigel, D. Spontaneous epigenetic variation in the Arabidopsis thaliana methylome. Nature 2011, 480, 245–249. [Google Scholar]
- Schmitz, R.J.; Schultz, M.D.; Lewsey, M.G.; O’Malley, R.C.; Urich, M.A.; Libiger, O.; Schork, N.J.; Ecker, J.R. Transgenerational epigenetic instability is a source of novel methylation variants. Science 2011, 334, 369–373. [Google Scholar]
- Ossowski, S.; Schneeberger, K.; Lucas-Lledó, J.I.; Warthmann, N.; Clark, R.M.; Shaw, R.G.; Weigel, D.; Lynch, M. The rate and molecular spectrum of spontaneous mutations in Arabidopsis thaliana. Science 2010, 327, 92–94. [Google Scholar]
- Teixeira, F.K.; Heredia, F.; Sarazin, A.; Roudier, F.; Boccara, M.; Ciaudo, C.; Cruaud, C.; Poulain, J.; Berdasco, M.; Fraga, M.F.; et al. A role for RNAi in the selective correction of DNA methylation defects. Science 2009, 323, 1600–1604. [Google Scholar]
- Roux, F.; Colomé-Tatché, M.; Edelist, C.; Wardenaar, R.; Guerche, P.; Hospital, F.; Colot, V.; Jansen, R.C.; Johannes, F. Genome-Wide epigenetic perturbation jump-starts patterns of heritable variation found in nature. Genetics 2011, 88, 1015–1017. [Google Scholar]
- Kobayashi, S.; Goto-Yamamoto, N.; Hirochika, H. Retrotransposon-induced mutations in grape skin color. Science 2004, 304, 982. [Google Scholar]
- Fujimoto, R.; Sugimura, T.; Fukai, E.; Nishio, T. Suppression of gene expression of a recessive SP11/SCR allele by an untranscribed SP11/SCR allele in Brassica self-incompatibility. Plant Mol. Biol 2006, 61, 577–587. [Google Scholar]
- Naito, K.; Zhang, F.; Tsukiyama, T.; Saito, H.; Hancock, C.N.; Richardson, A.O.; Okumoto, Y.; Tanisaka, T.; Wessler, S.R. Unexpected consequences of a sudden and massive transposon amplification on rice gene expression. Nature 2009, 461, 1130–1134. [Google Scholar]
- Fernandez, L.; Torregrosa, L.; Segura, V.; Bouquet, A.; Martinez-Zapater, J.M. Transposon-induced gene activation as a mechanism generating cluster shape somatic variation in grapevine. Plant J 2010, 61, 545–557. [Google Scholar] [Green Version]
- Hollister, J.D.; Smith, L.M.; Guo, Y.L.; Ott, F.; Weigel, D.; Gaut, B.S. Transposable elements and small RNAs contribute to gene expression divergence between Arabidopsis thaliana and Arabidopsis lyrata. Proc. Natl. Acad. Sci. USA 2011, 108, 2322–2327. [Google Scholar]
- Butelli, E.; Licciardello, C.; Zhang, Y.; Liu, J.; Mackay, S.; Bailey, P.; Reforgiato-Recupero, G.; Martin, C. Retrotransposons control fruit-specific, cold-dependent accumulation of anthocyanins in blood oranges. Plant Cell 2012, 24, 1242–1255. [Google Scholar]
- Martin, A.; Troadec, C.; Boualem, A.; Rajab, M.; Fernandez, R.; Morin, H.; Pitrat, M.; Dogimont, C.; Bendahmane, A. A transposon-induced epigenetic change leads to sex determination in melon. Nature 2009, 461, 1135–1138. [Google Scholar]
- Michaels, S.D.; Ditta, G.; Gustafson-Brown, C.; Pelaz, S.; Yanofsky, M.; Amasino, R.M. AGL24 acts as a promoter of flowering in Arabidopsis and is positively regulated by vernalization. Plant J 2003, 33, 867–874. [Google Scholar]
- Gazzani, S.; Gendall, A.R.; Lister, C.; Dean, C. Analysis of the molecular basis of flowering time variation in Arabidopsis accessions. Plant Physiol 2003, 132, 1107–1114. [Google Scholar]
- Liu, J.; He, Y.; Amasino, R.; Chen, X. siRNAs targeting an intronic transposon in the regulation of natural flowering behavior in Arabidopsis. Genes Dev 2004, 18, 2873–2878. [Google Scholar]
- Rutter, M.T.; Cross, K.V.; van Woert, P.A. Birth, death and subfunctionalization in the Arabidopsis genome. Trends Plant Sci 2012, 17, 204–212. [Google Scholar]
- Hollister, J.D.; Smith, L.M.; Guo, Y.L.; Ott, F.; Weigel, D.; Gaut, B.S. Transposable elements and small RNAs contribute to gene expression divergence between Arabidopsis thaliana and Arabidopsis lyrata. Proc. Natl. Acad. Sci. USA 2011, 108, 2322–2327. [Google Scholar]
- Kawanabe, T.; Fujimoto, R.; Taku Sasaki, T.; Taylor, J.M.; Dennis, E.S. A comparison of transcriptome and epigenetic status between closely related species in the genus Arabidopsis. Gene 2012, 506, 301–309. [Google Scholar]
- Bender, J.; Fink, G.R. Epigenetic control of an endogenous gene family is revealed by a novel blue fluorescent mutant of Arabidopsis. Cell 1995, 83, 725–734. [Google Scholar]
- Melquist, S.; Luff, B.; Bender, J. Arabidopsis PAI gene arrangements, cytosine methylation and expression. Genetics 1999, 153, 401–413. [Google Scholar]
- Luff, B.; Pawlowski, L.; Bender, J. An inverted repeat triggers cytosine methylation of identical sequences in Arabidopsis. Mol. Cell 1999, 3, 505–511. [Google Scholar]
- Ebbs, M.L.; Bartee, L.; Bender, J. H3 Lysine 9 methylation is maintained on a transcribed inverted repeat by combined action of SUVH6 and SUVH4 methyltransferases. Mol. Cell Biol 2005, 25, 10507–10515. [Google Scholar]
- Ebbs, M.L.; Bender, J. Locus-Specific control of DNA methylation by the Arabidopsis SUVH5 histone methyltransferase. Plant Cell 2006, 18, 1166–1176. [Google Scholar]
- Melquist, S.; Bender, J. Transcription from an upstream promoter controls methylation signaling from an inverted repeat of endogenous genes in Arabidopsis. Genes Dev 2003, 17, 2036–2047. [Google Scholar]
- Enke, R.A.; Dong, Z.; Bender, J. Small RNAs prevent transcription-coupled loss of Histone H3 Lysine 9 methylation in Arabidopsis thaliana. PLoS Genet 2011, 7, e1002350. [Google Scholar]
- Durand, S.; Bouché, N.; Strand, E.P.; Loudet, O.; Camilleri, C. Rapid establishment of genetic incompatibility through natural epigenetic variation. Curr. Biol 2012, 22, 326–331. [Google Scholar]
- Slotkin, R.K.; Freeling, M.; Lisch, D. Mu killer causes the heritable inactivation of the Mutator family of transposable elements in Zea mays. Genetics 2003, 165, 781–797. [Google Scholar]
- Slotkin, R.K.; Freeling, M.; Lisch, D. Heritable transposon silencing initiated by a naturally occurring transposon inverted duplication. Nat. Genet 2005, 37, 641–644. [Google Scholar]
- Singh, J.; Freeling, M.; Lisch, D. A position effect on the heritability of epigenetic silencing. PLoS Genet 2008, 4, e1000216. [Google Scholar]
- Woodhouse, M.R.; Freeling, M.; Lisch, D. Initiation, establishment, and maintenance of heritable MuDR transposon silencing in maize are mediated by distinct factors. PLoS Biol 2006, 4, e339. [Google Scholar]
- Nobuta, K.; Lu, C.; Shrivastava, R.; Pillay, M.; de Paoli, E.; Accerbi, M.; Arteaga-Vazquez, M.; Sidorenko, L.; Jeong, D.H.; Yen, Y.; et al. Distinct size distribution of endogenous siRNAs in maize: Evidence from deep sequencing in the mop1-1 mutant. Proc. Natl. Acad. Sci. USA 2008, 105, 14958–14963. [Google Scholar]
- Tarutani, Y.; Shiba, H.; Ito, T.; Kakizaki, T.; Suzuki, G.; Watanabe, M.; Isogai, A.; Takayama, S. Trans-acting small RNA determines dominance relationships in Brassica self-incompatibility. Nature 2010, 466, 983–986. [Google Scholar]
- Takayama, S.; Isogai, A. Self-incompatibility in plants. Annu. Rev. Plant Biol 2005, 56, 231–251. [Google Scholar]
- Fujimoto, R.; Nishio, T. Self-incompatibility. Adv. Bot. Res 2007, 45, 139–154. [Google Scholar]
- Watanabe, M.; Takayama, S.; Isogai, A.; Hinata, K. Recent progresses on self-incompatibility research in Brassica species. Breed. Sci 2003, 53, 199–208. [Google Scholar]
- Shiba, H.; Kakizaki, T.; Iwano, M.; Tarutani, Y.; Watanabe, M.; Isogai, A.; Takayama, S. Dominance relationships between self-incompatibility alleles controlled by DNA methylation. Nat. Genet 2006, 38, 297–299. [Google Scholar]
- Tarutani, Y.; Takayama, S. Monoallelic gene expression and its mechanisms. Curr. Opin. Plant Biol 2011, 14, 608–613. [Google Scholar]
- Finnegan, E.J.; Liang, D.; Wang, M.B. Self-incompatibility: Smi silences through a novel sRNA pathway. Trends Plant Sci 2011, 16, 238–241. [Google Scholar]
- Koornneef, M.; Hanhart, C.J.; van der Veen, J.H. A genetic and physiological analysis of late flowering mutants in Arabidopsis thaliana. Mol. Gen. Genet 1991, 229, 57–66. [Google Scholar]
- Ikeda, Y.; Kobayashi, Y.; Yamaguchi, A.; Abe, M.; Araki, T. Molecular basis of late-flowering phenotype caused by dominant epi-alleles of the FWA locus in Arabidopsis. Plant Cell Physiol 2007, 48, 205–220. [Google Scholar]
- Kinoshita, T.; Miura, A.; Choi, Y.; Kinoshita, Y.; Cao, X.; Jacobsen, S.E.; Fischer, R.L.; Kakutani, T. One-way control of FWA imprinting in Arabidopsis endosperm by DNA methylation. Science 2004, 303, 521–523. [Google Scholar]
- Choi, Y.; Gehring, M.; Johnson, L.; Hannon, M.; Harada, J.J.; Goldberg, R.B.; Jacobsen, S.E.; Fischer, R.L. DEMETER, a DNA Glycosylase Domain Protein, is required for endosperm gene imprinting and seed viability in Arabidopsis. Cell 2002, 110, 33–42. [Google Scholar]
- Lippman, Z.; Gendrel, A.V.; Black, M.; Vaughn, M.W.; Dedhia, N.; McCombie, W.R.; Lavine, K.; Mittal, V.; May, B.; Kasschau, K.D.; et al. Role of transposable elements in heterochromatin and epigenetic control. Nature 2004, 430, 471–476. [Google Scholar]
- Chan, S.W.; Zhang, X.; Bernatavichute, Y.V.; Jacobsen, S.E. Two-Step recruitment of RNA-directed DNA methylation to tandem repeats. PLoS Biol 2006, 4, e363. [Google Scholar]
- Chan, S.W.; Zilberman, D.; Xie, Z.; Johansen, L.K.; Carrington, J.C.; Jacobsen, S.E. RNA silencing genes control de novo DNA methylation. Science 2004, 303. [Google Scholar] [CrossRef]
- Kinoshita, Y.; Saze, H.; Kinoshita, T.; Miura, A.; Soppe, W.J.; Koornneef, M.; Kakutani, T. Control of FWA gene silencing in Arabidopsis thaliana by SINE-related direct repeats. Plant J 2007, 49, 38–45. [Google Scholar]
- Fujimoto, R.; Kinoshita, Y.; Kawabe, A.; Kinoshita, T.; Takashima, K.; Nordborg, M.; Nasrallah, M.E.; Shimizu, K.K.; Kudoh, H.; Kakutani, T. Evolution and metastable epigenetic states of imprinted FWA genes in the genus Arabidopsis. PLoS Genet 2008, 4, e1000048. [Google Scholar]
- Gehring, M.; Bubb, K.L.; Henikoff, S. Extensive demethylation of repetitive elements during seed development underlies gene imprinting. Science 2009, 324, 1447–1451. [Google Scholar]
- Hsieh, T.F.; Ibarra, C.A.; Silva, P.; Zemach, A.; Eshed-Williams, L.; Fischer, R.L.; Zilberman, D. Genome-wide demethylation of Arabidopsis endosperm. Science 2009, 324, 1451–1454. [Google Scholar]
- Fujimoto, R.; Sasaki, T.; Kudoh, H.; Taylor, J.M.; Kakutani, T.; Dennis, E.S. Epigenetic variation in the FWA gene within the genus Arabidopsis. Plant J 2011, 66, 831–843. [Google Scholar]
- Fujimoto, R.; Taylor, J.M.; Sasaki, T.; Kawanabe, T.; Dennis, E.S. Genome wide gene expression in artificially synthesized amphidiploids of Arabidopsis. Plant Mol. Biol 2011, 77, 419–431. [Google Scholar]
- Kawanabe, T.; Fujimoto, R. Inflorescence abnormalities occur with overexpression of Arabidopsis lyrata FT in the fwa mutant of Arabidopsis thaliana. Plant Sci 2011, 181, 496–503. [Google Scholar]
- Angers, B.; Castonguay, E.; Massicotte, R. Environmentally induced phenotypes and DNA methylation: How to deal with unpredictable conditions until the next generation and after. Mol. Ecol 2010, 19, 1283–1295. [Google Scholar]
- Grativol, C.; Hemerly, A.S.; Ferreira, P.C. Genetic and epigenetic regulation of stress responses in natural plant populations. Biochim. Biophys. Acta 2012, 1819, 176–185. [Google Scholar]


© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Fujimoto, R.; Sasaki, T.; Ishikawa, R.; Osabe, K.; Kawanabe, T.; Dennis, E.S. Molecular Mechanisms of Epigenetic Variation in Plants. Int. J. Mol. Sci. 2012, 13, 9900-9922. https://doi.org/10.3390/ijms13089900
Fujimoto R, Sasaki T, Ishikawa R, Osabe K, Kawanabe T, Dennis ES. Molecular Mechanisms of Epigenetic Variation in Plants. International Journal of Molecular Sciences. 2012; 13(8):9900-9922. https://doi.org/10.3390/ijms13089900
Chicago/Turabian StyleFujimoto, Ryo, Taku Sasaki, Ryo Ishikawa, Kenji Osabe, Takahiro Kawanabe, and Elizabeth S. Dennis. 2012. "Molecular Mechanisms of Epigenetic Variation in Plants" International Journal of Molecular Sciences 13, no. 8: 9900-9922. https://doi.org/10.3390/ijms13089900