Selenium Compounds, Apoptosis and Other Types of Cell Death: An Overview for Cancer Therapy




Abstract
:1. Introduction
2. Apoptosis and Kinases Modulation
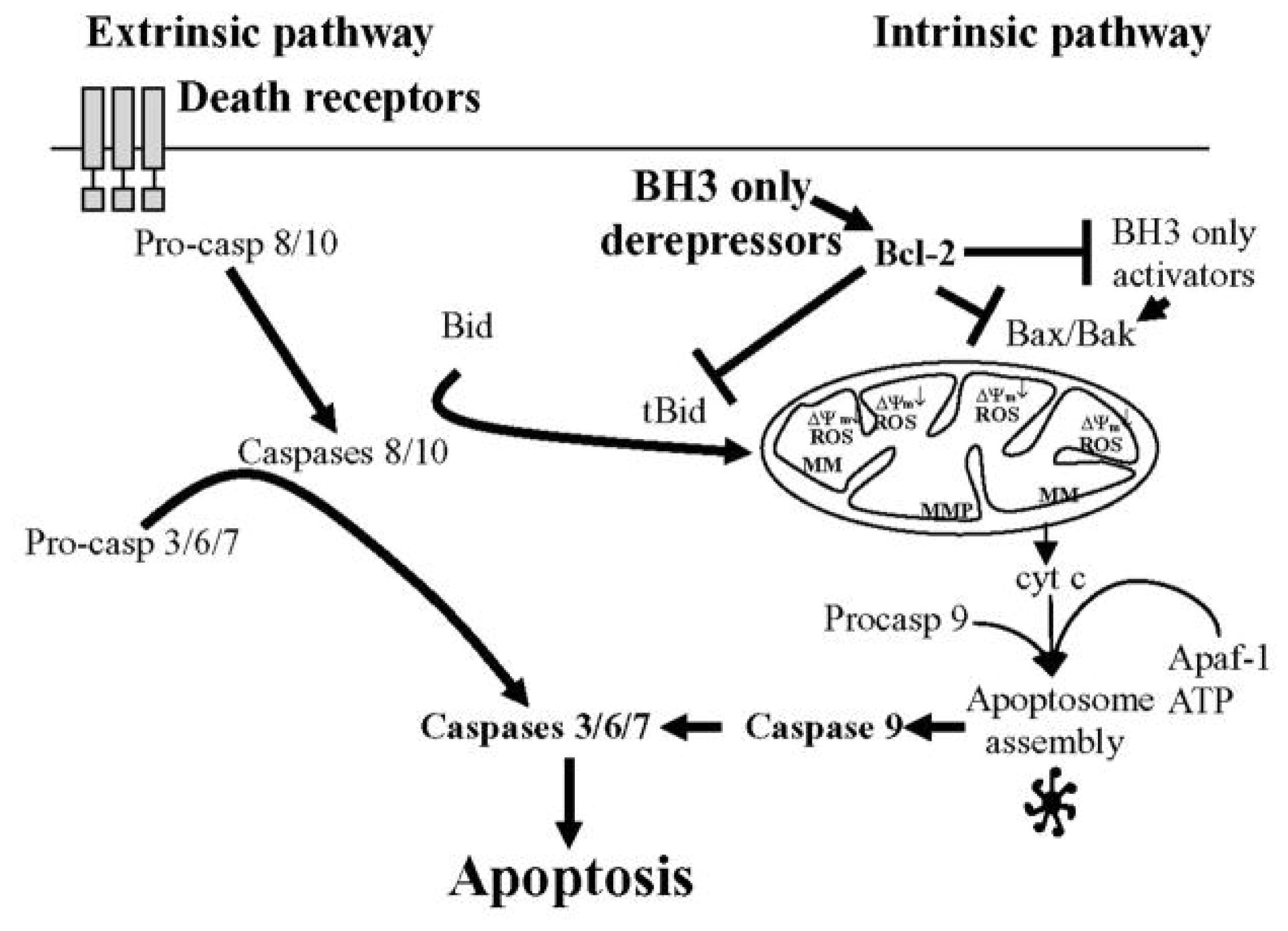
3. Apoptosis and Caspases
4. Apoptosis and Reactive Oxygen Species
4.1. Apoptosis and Glutathione System
4.2. Apoptosis and Thioredoxin System
5. Apoptosis and p53 Regulation
6. Apoptosis and Miscellaneous Mechanisms and Structures
7. Apoptosis versus Autophagy
8. Selenium, Cancer and Cell Death Types: Conclusions, Paradox and Perspectives
Acknowledgments
References
- Thomson, C.D. Assessment of requirements for selenium and adequacy of selenium status: A review. Eur. J. Clin. Nutr 2004, 58, 391–402. [Google Scholar]
- Goldhaber, S.B. Trace element risk assessment: Essentiality vs. toxicity. Regul. Toxicol. Pharmacol 2003, 38, 232–242. [Google Scholar]
- Navarro-Alarcon, M.; Cabrera-Vique, C. Selenium in food and the human body: A review. Sci. Total Environ 2008, 400, 115–141. [Google Scholar]
- Council, N.R. Dietary Reference Intakes for Vitamin C, Vitamin E, Selenium, and Carotenoids; The National Academies Press: Whasington, DC, USA; p. 2000.
- Lu, J.; Holmgren, A. Selenoproteins. J. Biol. Chem 2009, 284, 723–727. [Google Scholar]
- Papp, L.V.; Lu, J.; Holmgren, A.; Khanna, K.K. From selenium to selenoproteins: Synthesis, identity, and their role in human health. Antioxid. Redox Signal 2007, 9, 775–806. [Google Scholar]
- Brozmanova, J.; Manikova, D.; Vlckova, V.; Chovanec, M. Selenium: A double-edged sword for defense and offence in cancer. Arch. Toxicol 2010, 84, 919–938. [Google Scholar]
- Zeng, H.; Combs, G.F., Jr. Selenium as an anticancer nutrient: Roles in cell proliferation and tumor cell invasion. J. Nutr. Biochem. 2008, 19, 1–7. [Google Scholar]
- Cole-Ezea, P.; Swan, D.; Shanley, D.; Hesketh, J. Glutathione peroxidase 4 has a major role in protecting mitochondria from oxidative damage and maintaining oxidative phosphorylation complexes in gut epithelial cells. Free Radic. Biol. Med 2012, 53, 488–497. [Google Scholar]
- Sanmartin, C.; Plano, D.; Font, M.; Palop, J.A. Kinase regulation by sulfur and selenium containing compounds. Curr. Cancer Drug Targets 2011, 11, 496–523. [Google Scholar]
- Wu, M.; Kang, M.M.; Schoene, N.W.; Cheng, W.H. Selenium compounds activate early barriers of tumorigenesis. J. Biol. Chem 2010, 285, 12055–12062. [Google Scholar]
- Zeng, H. Selenium as an essential micronutrient: Roles in cell cycle and apoptosis. Molecules 2009, 14, 1263–1278. [Google Scholar]
- Valdiglesias, V.; Pasaro, E.; Mendez, J.; Laffon, B. In vitro evaluation of selenium genotoxic, cytotoxic, and protective effects: A review. Arch. Toxicol 2010, 84, 337–351. [Google Scholar]
- Rana, S.V. Metals and apoptosis: Recent developments. J. Trace Elem. Med. Biol 2008, 22, 262–284. [Google Scholar]
- Clark, L.C.; Combs, G.F., Jr; Turnbull, B.W.; Slate, E.H.; Chalker, D.K.; Chow, J.; Davis, L.S.; Glover, R.A.; Graham, G.F.; Gross, E.G.; et al. JAMA 1996, 276, 1957–1963.
- Duffield-Lillico, A.J.; Dalkin, B.L.; Reid, M.E.; Turnbull, B.W.; Slate, E.H.; Jacobs, E.T.; Marshall, J.R.; Clark, L.C. Selenium supplementation, baseline plasma selenium status and incidence of prostate cancer: An analysis of the complete treatment period of the nutritional prevention of cancer trial. BJU Int 2003, 91, 608–612. [Google Scholar]
- Lippman, S.M.; Klein, E.A.; Goodman, P.J.; Lucia, M.S.; Thompson, I.M.; Ford, L.G.; Parnes, H.L.; Minasian, L.M.; Gaziano, J.M.; Hartline, J.A.; et al. Effect of selenium and vitamin E on risk of prostate cancer and other cancers: The selenium and vitamin E cancer prevention trial (select). JAMA 2009, 301, 39–51. [Google Scholar]
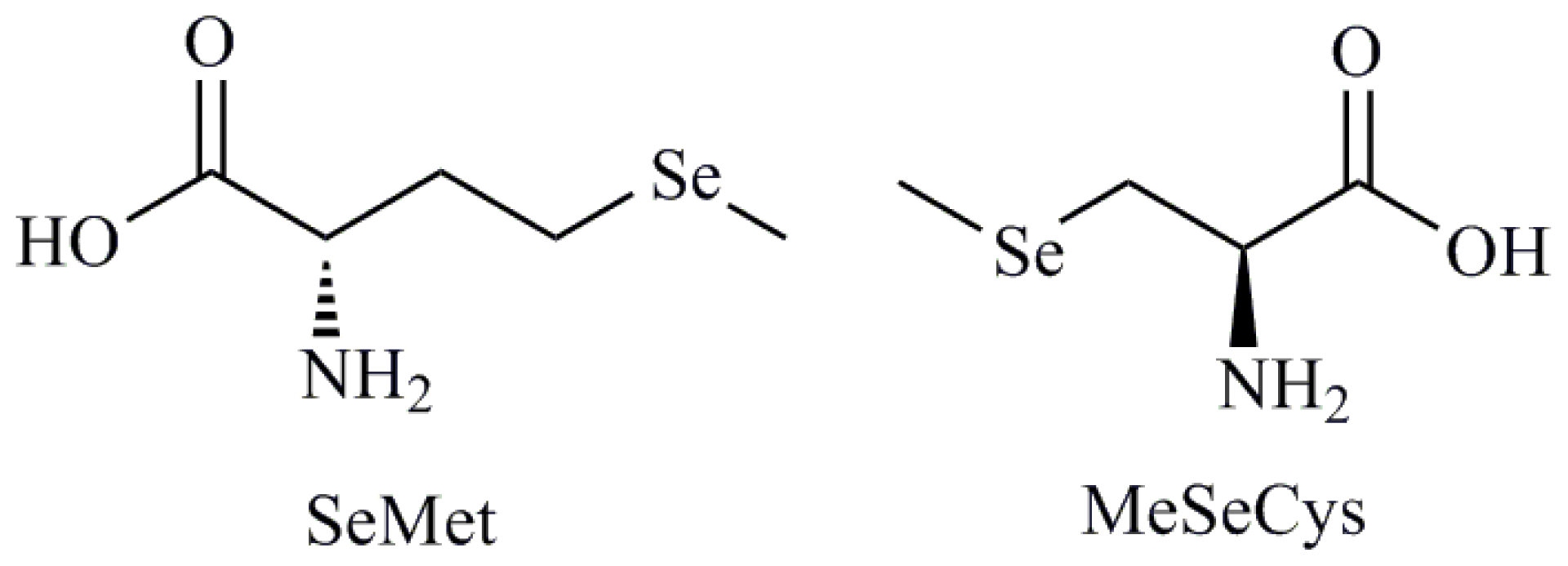
- Weekley, C.M.; Aitken, J.B.; Musgrave, I.F.; Harris, H.H. Methylselenocysteine treatment leads to diselenide formation in human cancer cells: Evidence from X-ray absorption spectroscopy studies. Biochemistry 2012, 51, 736–738. [Google Scholar]
- Tapiero, H.; Townsend, D.M.; Tew, K.D. The antioxidant role of selenium and seleno-compounds. Biomed. Pharmacother 2003, 57, 134–144. [Google Scholar]
- Zeng, H.; Briske-Anderson, M.; Wu, M.; Moyer, M.P. Methylselenol, a selenium metabolite, plays common and different roles in cancerous colon HCT116 cell and noncancerous NCM460 colon cell proliferation. Nutr. Cancer 2012, 64, 128–135. [Google Scholar]
- Singletary, K.; Milner, J. Diet, autophagy, and cancer: A review. Cancer Epidemiol. Biomark. Prev 2008, 17, 1596–1610. [Google Scholar]
- Ren, Y.; Huang, F.; Liu, Y.; Yang, Y.; Jiang, Q.; Xu, C. Autophagy inhibition through PI3K/Akt increases apoptosis by sodium selenite in NB4 cells. BMB Rep 2009, 42, 599–604. [Google Scholar]
- Suzuki, M.; Endo, M.; Shinohara, F.; Echigo, S.; Rikiishi, H. Rapamycin suppresses ros-dependent apoptosis caused by selenomethionine in A549 lung carcinoma cells. Cancer Chemother. Pharmacol 2011, 67, 1129–1136. [Google Scholar]
- Zeng, H.; Wu, M.; Botnen, J.H. Methylselenol, a selenium metabolite, induces cell cycle arrest in G1 phase and apoptosis via the extracellular-regulated kinase 1/2 pathway and other cancer signaling genes. J. Nutr 2009, 139, 1613–1618. [Google Scholar]
- Fang, W.; Han, A.; Bi, X.; Xiong, B.; Yang, W. Tumor inhibition by sodium selenite is associated with activation of c-jun NH2-terminal kinase 1 and suppression of beta-catenin signaling. Int. J. Cancer 2010, 127, 32–42. [Google Scholar]
- Facompre, N.D.; El-Bayoumy, K.; Sun, Y.W.; Pinto, J.T.; Sinha, R. 1,4-phenylenebis (methylene)selenocyanate, but not selenomethionine, inhibits androgen receptor and Akt signaling in human prostate cancer cells. Cancer Prev. Res 2010, 3, 975–984. [Google Scholar]
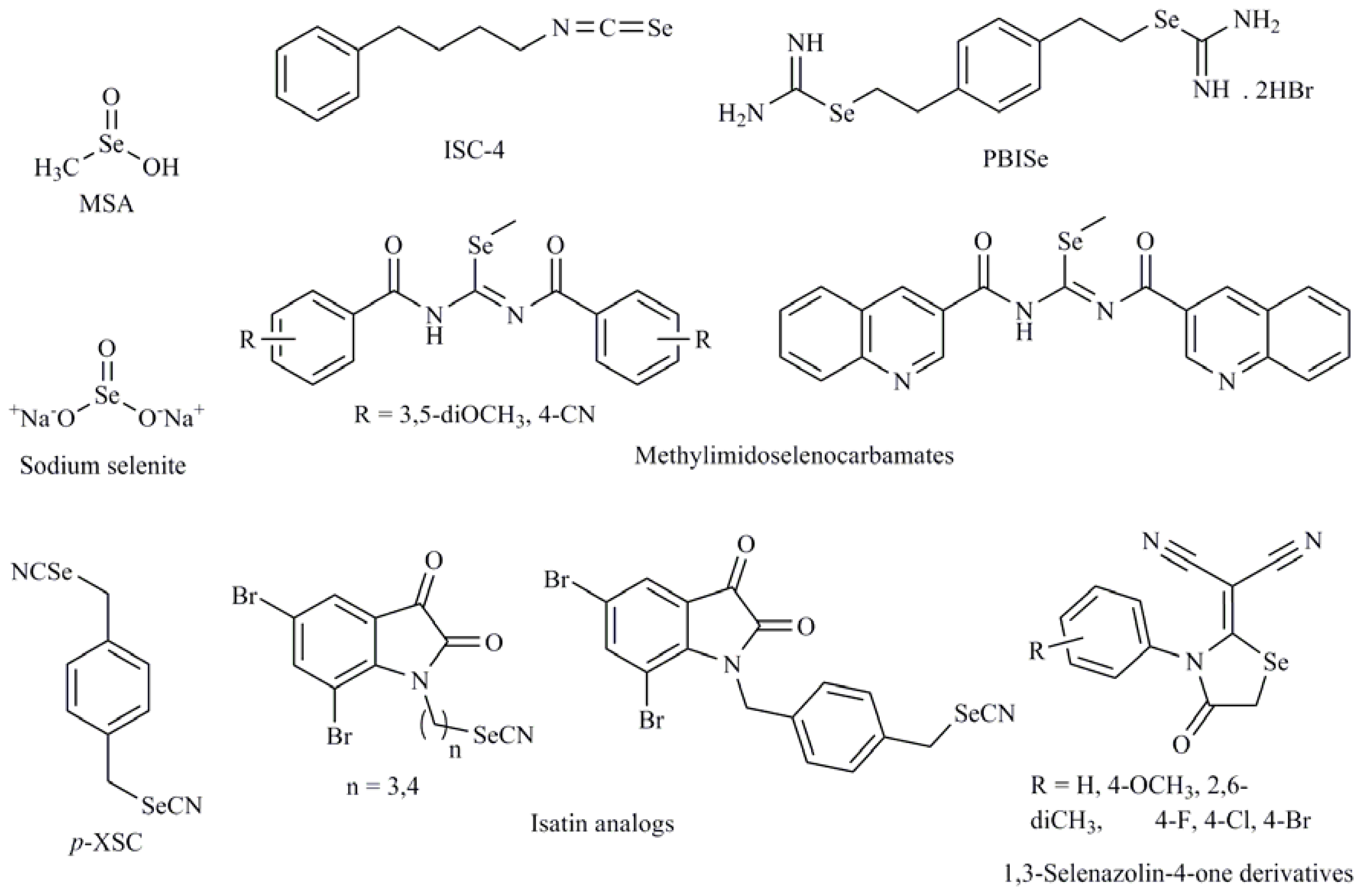
- Nguyen, N.; Sharma, A.; Sharma, A.K.; Desai, D.; Huh, S.J.; Amin, S.; Meyers, C.; Robertson, G.P. Melanoma chemoprevention in skin reconstructs and mouse xenografts using isoselenocyanate-4. Cancer Prev. Res 2011, 4, 248–258. [Google Scholar]
- Sharma, A.K.; Kline, C.L.; Berg, A.; Amin, S.; Irby, R.B. The Akt inhibitor ISC-4 activates prostate apoptosis response protein-4 and reduces colon tumor growth in a nude mouse model. Clin. Cancer Res 2011, 17, 4474–4483. [Google Scholar]
- Krishnegowda, G.; Prakasha Gowda, A.S.; Tagaram, H.R.; Carroll, K.F.; Irby, R.B.; Sharma, A.K.; Amin, S. Synthesis and biological evaluation of a novel class of isatin analogs as dual inhibitors of tubulin polymerization and Akt pathway. Bioorg. Med. Chem 2011, 19, 6006–6014. [Google Scholar]
- Luo, H.; Yang, Y.; Huang, F.; Li, F.; Jiang, Q.; Shi, K.; Xu, C. Selenite induces apoptosis in colorectal cancer cells via Akt-mediated inhibition of beta-catenin survival axis. Cancer Lett 2012, 315, 78–85. [Google Scholar]
- Plano, D.; Ibáñez, E.; Calvo, A.; Palop, J.A.; Sanmartín, C. Novel library of selenocompounds as kinase modulators. Molecules 2011, 16, 6349–6364. [Google Scholar]
- Ibanez, E.; Agliano, A.; Prior, C.; Nguewa, P.; Redrado, M.; Gonzalez-Zubeldia, I.; Plano, D.; Palop, J.A.; Sanmartin, C.; Calvo, A. The quinoline imidoselenocarbamate EI201 blocks the akt/mtor pathway and targets cancer stem cells leading to a strong antitumor activity. Curr. Med. Chem 2012, 19, 3031–3043. [Google Scholar]
- Kong, L.; Yuan, Q.; Zhu, H.; Li, Y.; Guo, Q.; Wang, Q.; Bi, X.; Gao, X. The suppression of prostate LNCaP cancer cells growth by selenium nanoparticles through Akt/MDM2/AR controlled apoptosis. Biomaterials 2011, 32, 6515–6522. [Google Scholar]
- Nishina, A.; Kimura, H.; Kozawa, K.; Sommen, G.; Nakamura, T.; Heimgartner, H.; Koketsu, M.; Furukawa, S. A superoxide anion-scavenger, 1,3-selenazolidin-4-one suppresses serum deprivation-induced apoptosis in PC12 cells by activating MAP kinase. Toxicol. Appl. Pharmacol 2011, 257, 388–395. [Google Scholar] [Green Version]
- Ranawat, P.; Bansal, M.P. Decreased glutathione levels potentiate the apoptotic efficacy of selenium: Possible involvement of p38 and JNK MAPKs—In vitro studies. Mol. Cell Biochem 2008, 309, 21–32. [Google Scholar]
- Ranawat, P.; Bansal, M.P. Apoptosis induced by modulation in selenium status involves p38 MAPK and ROS: Implications in spermatogenesis. Mol. Cell Biochem 2009, 330, 83–95. [Google Scholar]
- Chung, C.Y.; Madhunapantula, S.V.; Desai, D.; Amin, S.; Robertson, G.P. Melanoma prevention using topical PBISe. Cancer Prev. Res 2011, 4, 935–948. [Google Scholar]
- Wang, Z.; Lee, H.J.; Chai, Y.; Hu, H.; Wang, L.; Zhang, Y.; Jiang, C.; Lu, J. Persistent p21Cip1 induction mediates G1 cell cycle arrest by methylseleninic acid in DU145 prostate cancer cells. Curr. Cancer Drug Targets 2010, 10, 307–318. [Google Scholar]
- Li, Z.S.; Shi, K.J.; Guan, L.Y.; Jiang, Q.; Yang, Y.; Xu, C.M. Downregulation of protein kinase Calpha was involved in selenite-induced apoptosis of NB4 cells. Oncol. Res 2010, 19, 77–83. [Google Scholar]
- MacKenzie, S.H.; Schipper, J.L.; Clark, A.C. The potential for caspases in drug discovery. Curr. Opin. Drug Discov. Dev 2010, 13, 568–576. [Google Scholar]
- Wlodkowic, D.; Telford, W.; Skommer, J.; Darzynkiewicz, Z. Apoptosis and beyond: Cytometry in studies of programmed cell death. Methods Cell Biol 2011, 103, 55–98. [Google Scholar]
- Park, H.H. Structural features of caspase-activating complexes. Int. J. Mol. Sci 2012, 13, 4807–4818. [Google Scholar]
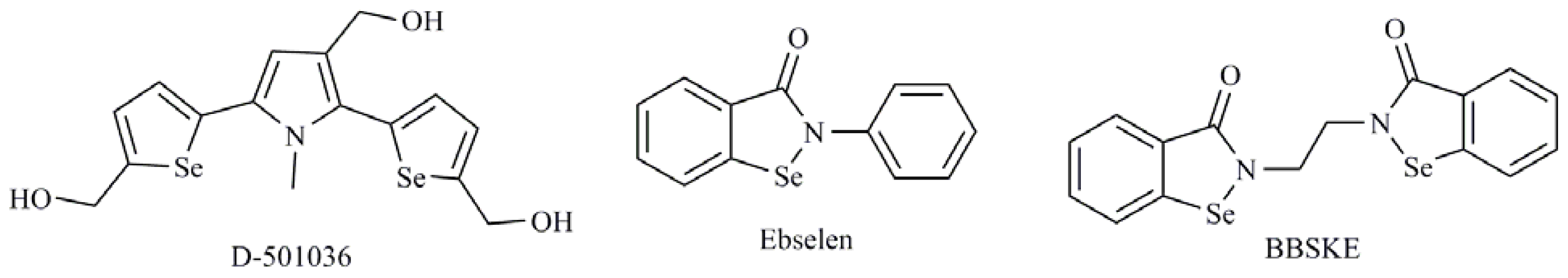
- Xing, F.; Li, S.; Ge, X.; Wang, C.; Zeng, H.; Li, D.; Dong, L. The inhibitory effect of a novel organoselenium compound BBSKE on the tongue cancer Tca8113 in vitro and in vivo. Oral Oncol 2008, 44, 963–969. [Google Scholar]
- Shiah, H.S.; Lee, W.S.; Juang, S.H.; Hong, P.C.; Lung, C.C.; Chang, C.J.; Chou, K.M.; Chang, J.Y. Mitochondria-mediated and p53-associated apoptosis induced in human cancer cells by a novel selenophene derivative, D-501036. Biochem. Pharmacol 2007, 73, 610–619. [Google Scholar]
- Li, Z.; Carrier, L.; Rowan, B.G. Methylseleninic acid synergizes with tamoxifen to induce caspase-mediated apoptosis in breast cancer cells. Mol. Cancer Ther 2008, 7, 3056–3063. [Google Scholar]
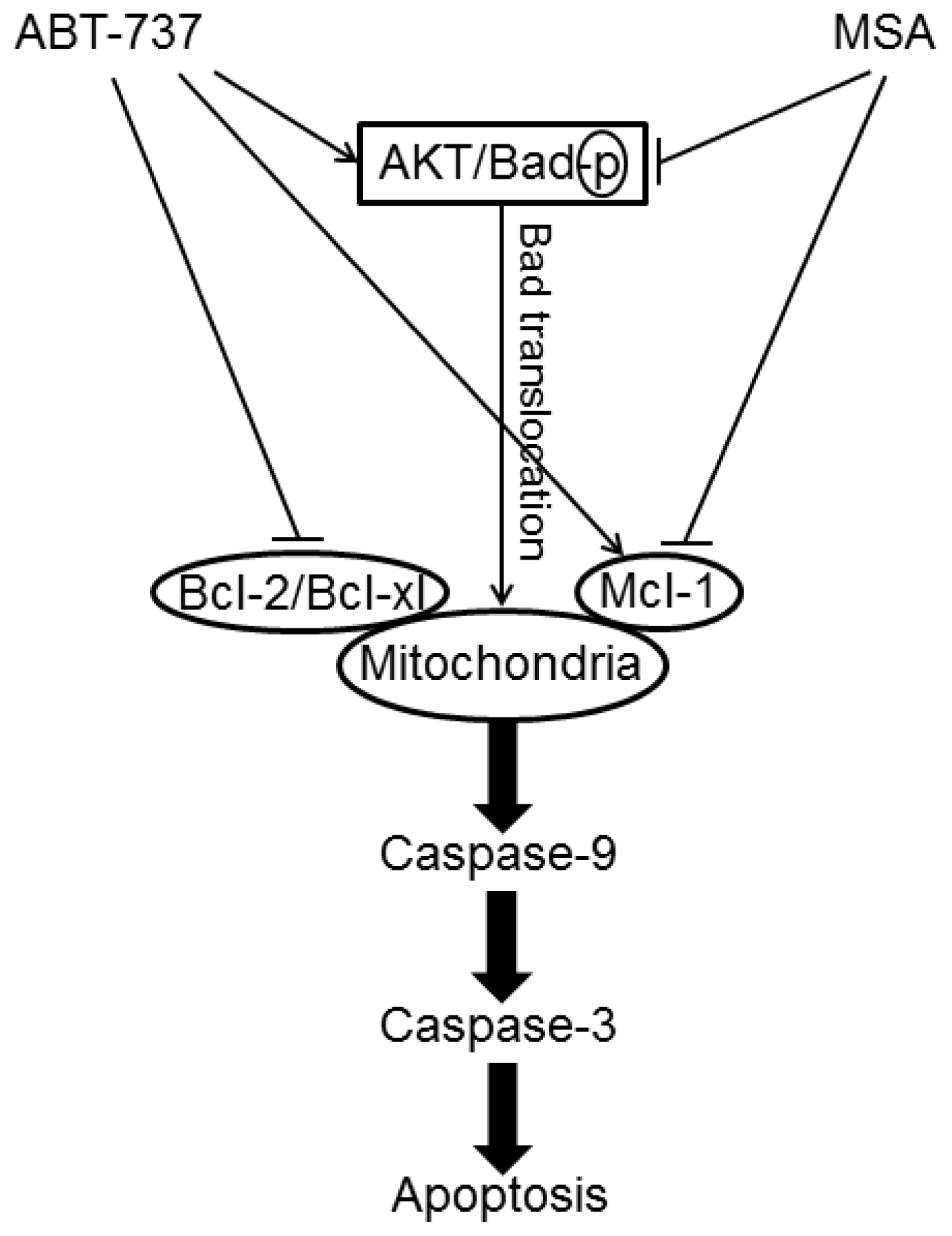
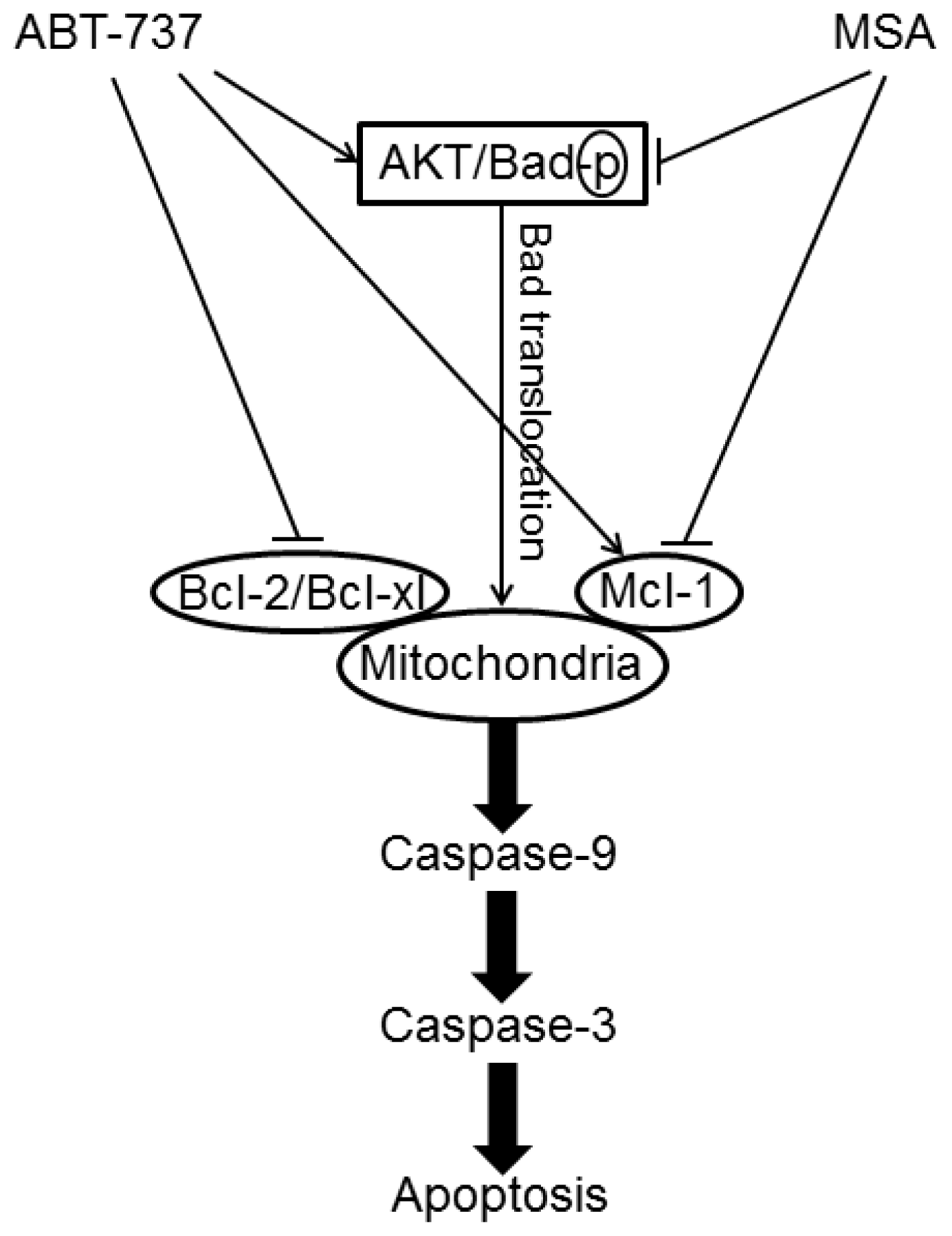
- Yin, S.; Dong, Y.; Li, J.; Fan, L.; Wang, L.; Lu, J.; Vang, O.; Hu, H. Methylseleninic acid potentiates multiple types of cancer cells to ABT-737-induced apoptosis by targeting Mcl-1 and Bad. Apoptosis 2012, 17, 388–399. [Google Scholar]
- Qi, Y.; Fu, X.; Xiong, Z.; Zhang, H.; Hill, S.M.; Rowan, B.G.; Dong, Y. Methylseleninic acid enhances paclitaxel efficacy for the treatment of triple-negative breast cancer. PLoS One 2012, 7, e31539. [Google Scholar]
- Rudolf, E.; Rudolf, K.; Cervinka, M. Selenium activates p53 and p38 pathways and induces caspase-independent cell death in cervical cancer cells. Cell Biol. Toxicol 2008, 24, 123–141. [Google Scholar]
- Freitas, M.; Alves, V.; Sarmento-Ribeiro, A.B.; Mota-Pinto, A. Combined effect of sodium selenite and docetaxel on Pc3 metastatic prostate cancer cell line. Biochem. Biophys. Res. Commun 2011, 408, 713–719. [Google Scholar]
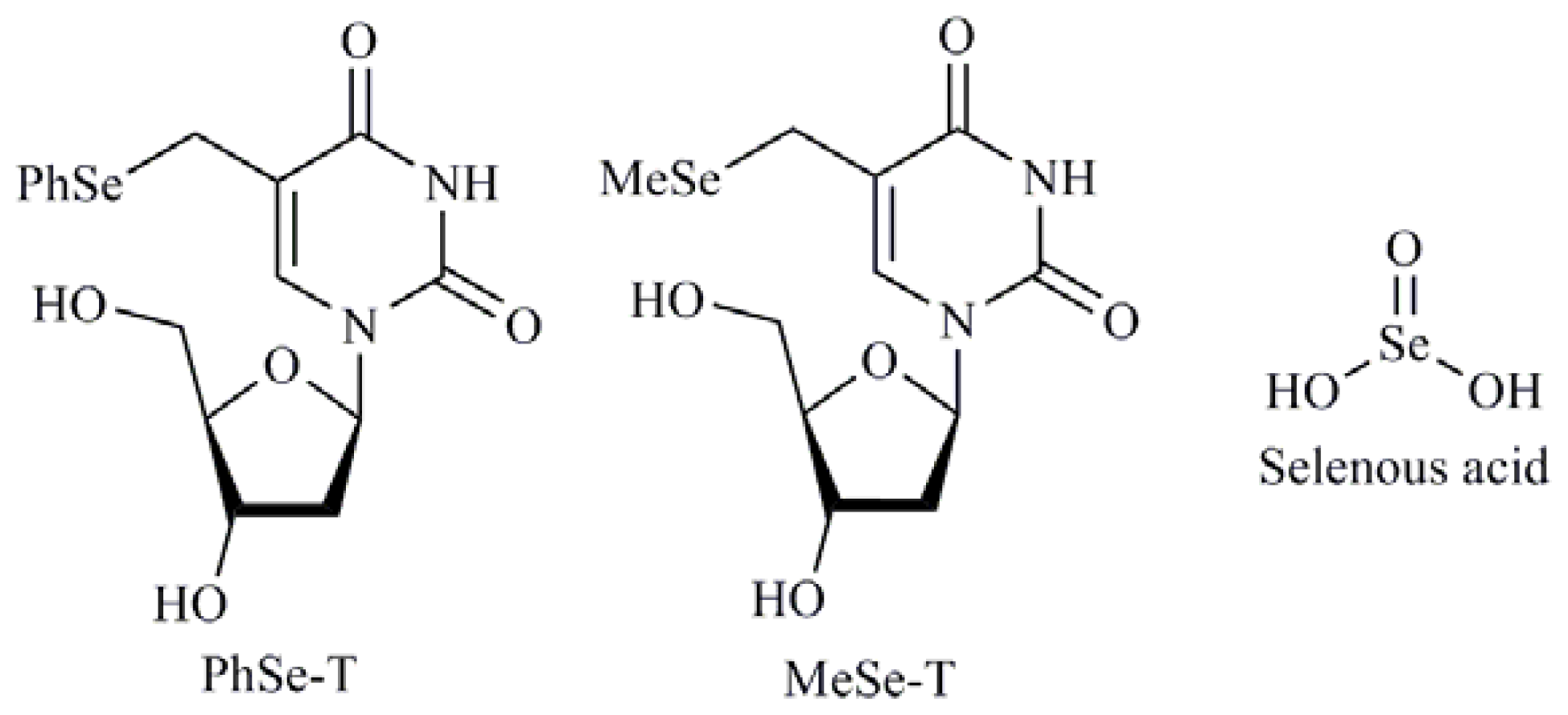
- Kim, B.; Rode, A.; Han, E.; Hong, I.; Hong, S. 5-phenylselenyl- and 5-methylselenyl-methyl-2′-deoxyuridine induce oxidative stress, DNA damage, and caspase-2-dependent apoptosis in cancer cells. Apoptosis 2012, 17, 200–216. [Google Scholar]
- Kim, B.M.; Lee, K.H.; Hong, I.S.; Hong, S.H. P38 mitogen-activated protein kinase is a key regulator of 5-phenylselenyl- and 5-methylselenyl-methyl-2′-deoxyuridine-induced apoptosis in human HL-60 cells. Biochem. Biophys. Res. Commun 2012, 417, 237–244. [Google Scholar]
- Thant, A.A.; Wu, Y.; Lee, J.; Mishra, D.K.; Garcia, H.; Koeffler, H.P.; Vadgama, J.V. Role of caspases in 5-FU and selenium-induced growth inhibition of colorectal cancer cells. Anticancer Res 2008, 28, 3579–3592. [Google Scholar]
- Montero, A.J.; Jassem, J. Cellular redox pathways as a therapeutic target in the treatment of cancer. Drugs 2011, 71, 1385–1396. [Google Scholar]
- Ray, P.D.; Huang, B.W.; Tsuji, Y. Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell Signal 2012, 24, 981–990. [Google Scholar]
- Takahashi, M.; Sato, T.; Shinohara, F.; Echigo, S.; Rikiishi, H. Possible role of glutathione in mitochondrial apoptosis of human oral squamous cell carcinoma caused by inorganic selenium compounds. Int. J. Oncol 2005, 27, 489–495. [Google Scholar]
- Misra, S.; Niyogi, S. Selenite causes cytotoxicity in rainbow trout (Oncorhynchus mykiss) hepatocytes by inducing oxidative stress. Toxicol. Vitr 2009, 23, 1249–1258. [Google Scholar]
- Misra, S.; Hamilton, C.; Niyogi, S. Induction of oxidative stress by selenomethionine in isolated hepatocytes of rainbow trout (Oncorhynchus mykiss). Toxicol. Vitr 2012, 26, 621–629. [Google Scholar]
- Chen, J.J.; Boylan, L.M.; Wu, C.K.; Spallholz, J.E. Oxidation of glutathione and superoxide generation by inorganic and organic selenium compounds. Biofactors 2007, 31, 55–66. [Google Scholar]
- Plano, D.; Baquedano, Y.; Ibáñez, E.; Jiménez, I.; Palop, J.A.; Spallholz, J.E.; Sanmartín, C. Antioxidant-prooxidant properties of a new organoselenium compound library. Molecules 2010, 15, 7292–7312. [Google Scholar]
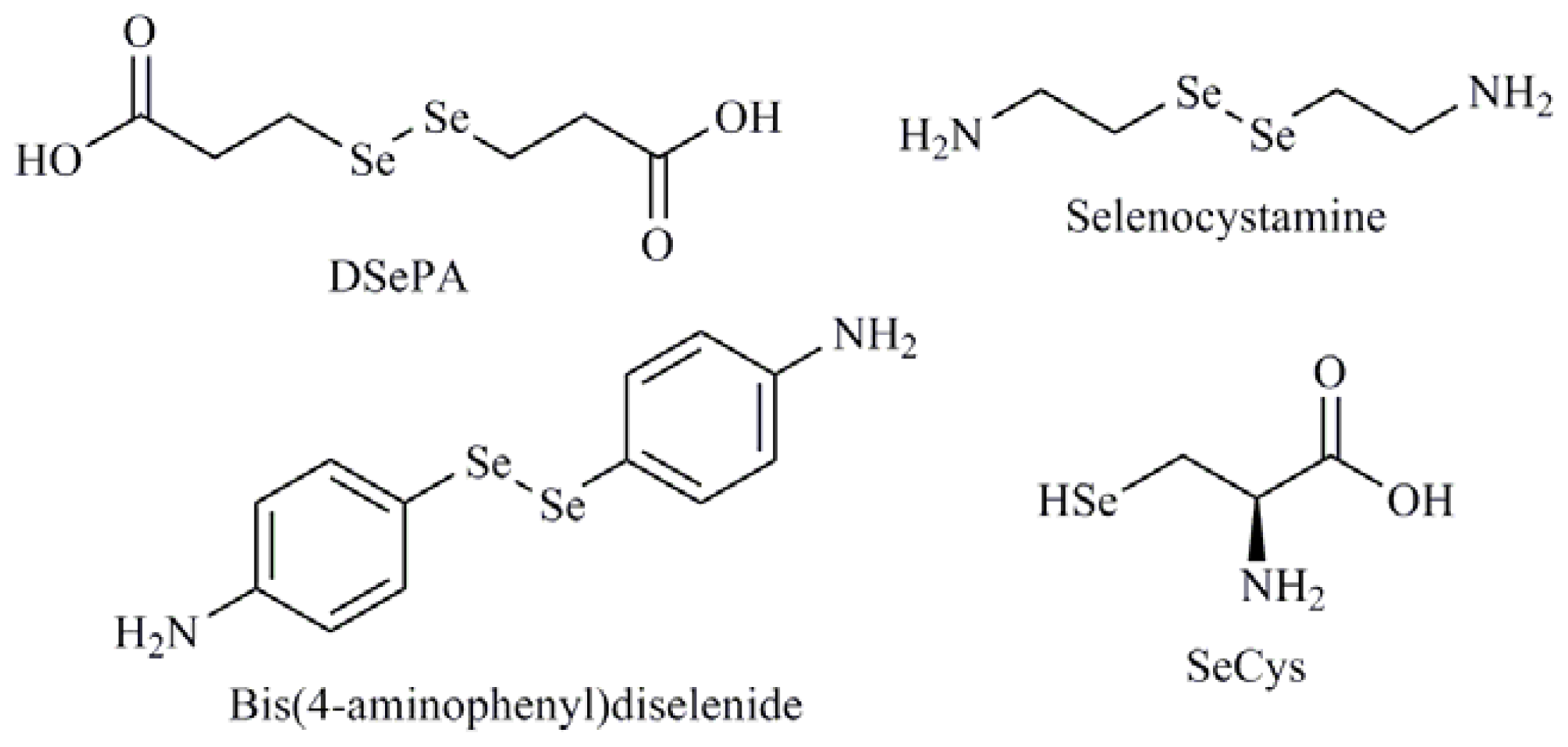
- Kunwar, A.; Bansal, P.; Kumar, S.J.; Bag, P.P.; Paul, P.; Reddy, N.D.; Kumbhare, L.B.; Jain, V.K.; Chaubey, R.C.; Unnikrishnan, M.K.; et al. In vivo radioprotection studies of 3,3′-diselenodipropionic acid, a selenocystine derivative. Free Radic. Biol. Med 2010, 48, 399–410. [Google Scholar]
- Liu, C.; Chen, J.; Wu, Z.; Li, Y.; Zhu, Y.; Ren, Y.; Zhou, Q. Selenium compounds induce ROS in human high-metastatic large cell lung cancer cell line L9981. Zhongguo Fei Ai Za Zhi 2008, 11, 354–358. [Google Scholar]
- Liu, C.; Liu, H.; Li, Y.; Wu, Z.; Zhu, Y.; Wang, T.; Gao, A.C.; Chen, J.; Zhou, Q. Intracellular glutathione content influences the sensitivity of lung cancer cell lines to methylseleninic acid. Mol. Carcinog 2011, 22. [Google Scholar] [CrossRef]
- Papp, L.V.; Lu, J.; Bolderson, E.; Boucher, D.; Singh, R.; Holmgren, A.; Khanna, K.K. SECIS-binding protein 2 promotes cell survival by protecting against oxidative stress. Antioxid. Redox Signal 2010, 12, 797–808. [Google Scholar]
- Poerschke, R.L.; Moos, P.J. Thioredoxin reductase 1 knockdown enhances selenazolidine cytotoxicity in human lung cancer cells via mitochondrial dysfunction. Biochem. Pharmacol 2011, 81, 211–221. [Google Scholar]
- Honeggar, M.; Beck, R.; Moos, P.J. Thioredoxin reductase 1 ablation sensitizes colon cancer cells to methylseleninate-mediated cytotoxicity. Toxicol. Appl. Pharmacol 2009, 241, 348–355. [Google Scholar]
- Wang, L.; Yang, Z.; Fu, J.; Yin, H.; Xiong, K.; Tan, Q.; Jin, H.; Li, J.; Wang, T.; Tang, W.; et al. Ethaselen: A potent mammalian thioredoxin reductase 1 inhibitor and novel organoselenium anticancer agent. Free Radic. Biol. Med 2012, 52, 898–908. [Google Scholar]
- Liu, X.; Pietsch, K.E.; Sturla, S.J. Susceptibility of the antioxidant selenoenyzmes thioredoxin reductase and glutathione peroxidase to alkylation-mediated inhibition by anticancer acylfulvenes. Chem. Res. Toxicol 2011, 24, 726–736. [Google Scholar]
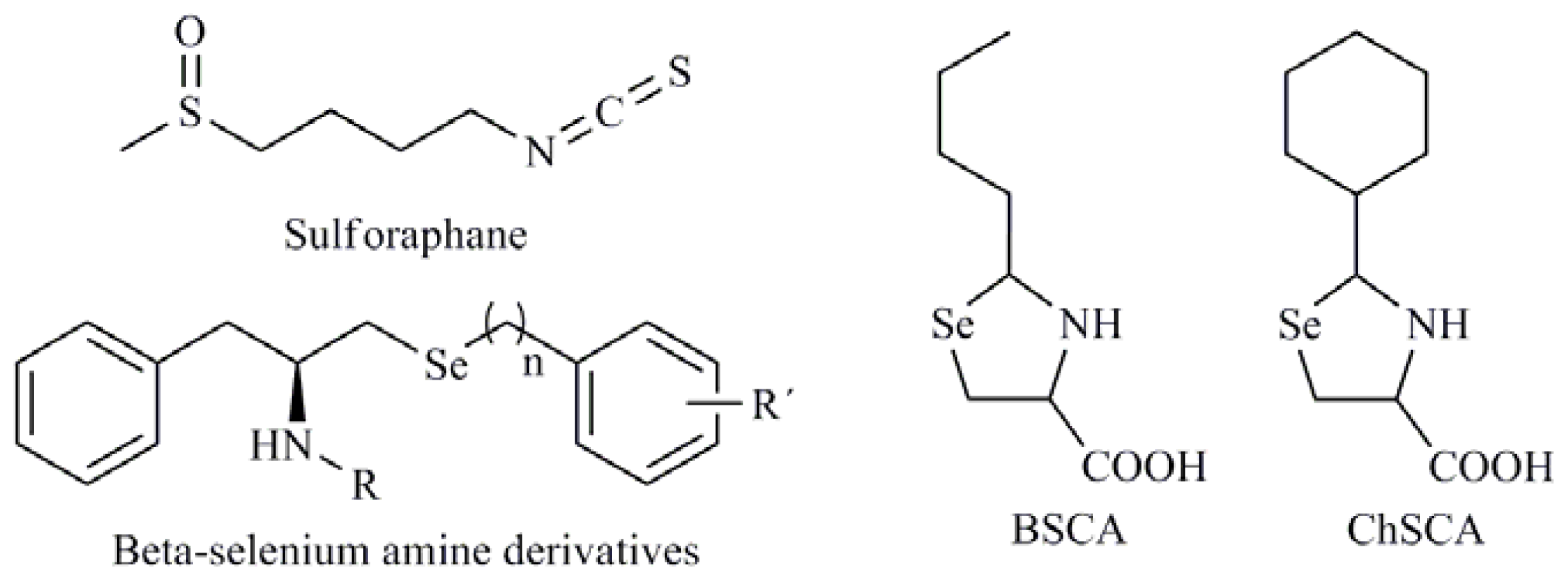
- De Souza Prestes, A.; Stefanello, S.T.; Salman, S.M.; Pazini, A.M.; Schwab, R.S.; Braga, A.L.; de Vargas Barbosa, N.B.; Rocha, J.B. Antioxidant activity of beta-selenoamines and their capacity to mimic different enzymes. Mol. Cell Biochem 2012, 365, 85–92. [Google Scholar]
- Gundimeda, U.; Schiffman, J.E.; Chhabra, D.; Wong, J.; Wu, A.; Gopalakrishna, R. Locally generated methylseleninic acid induces specific inactivation of protein kinase C isoenzymes: Relevance to selenium-induced apoptosis in prostate cancer cells. J. Biol. Chem 2008, 283, 34519–34531. [Google Scholar]
- Selenius, M.; Rundlof, A.K.; Olm, E.; Fernandes, A.P.; Bjornstedt, M. Selenium and the selenoprotein thioredoxin reductase in the prevention, treatment and diagnostics of cancer. Antioxid. Redox Signal 2010, 12, 867–880. [Google Scholar]
- Krehl, S.; Loewinger, M.; Florian, S.; Kipp, A.; Banning, A.; Wessjohann, L.; Brauer, M.; Iori, R.; Esworthy, R.S.; Chu, F.-F.; et al. Glutathione peroxidase-2 and selenium decreased inflammation and tumors in a mouse model of inflammation-associated carcinogenesis whereas sulforaphane effects differed with selenium supply. Carcinogenesis 2012, 33, 620–628. [Google Scholar]
- Hawkes, W.C.; Alkan, Z. Delayed cell cycle progression from SEPW1 depletion is p53- and p21-dependent in MCF-7 breast cancer cells. Biochem. Biophys. Res. Commun 2011, 413, 36–40. [Google Scholar]
- Bjelogrlic, S.; Todorovic, T.; Bacchi, A.; Zec, M.; Sladic, D.; Srdic-Rajic, T.; Radanovic, D.; Radulovic, S.; Pelizzi, G.; Andelkovic, K. Synthesis, structure and characterization of novel Cd(II) and Zn(II) complexes with the condensation product of 2-formylpyridine and selenosemicarbazide antiproliferative activity of the synthesized complexes and related selenosemicarbazone complexes. J. Inorg. Biochem 2010, 104, 673–682. [Google Scholar]
- Srdić-Rajić, T.; Zec, M.; Todorović, T.; Anđelković, K.; Radulović, S. Non-substituted n-heteroaromatic selenosemicarbazone metal complexes induce apoptosis in cancer cells via activation of mitochondrial pathway. Eur. J. Med. Chem 2011, 46, 3734–3747. [Google Scholar]
- Das, A.; Bortner, J.; Desai, D.; Amin, S.; El-Bayoumy, K. The selenium analog of the chemopreventive compound S,S′-(1,4-phenylenebis[1,2-ethanediyl])bisisothiourea is a remarkable inducer of apoptosis and inhibitor of cell growth in human non-small cell lung cancer. Chem. Biol. Interact 2009, 180, 158–164. [Google Scholar]
- Chen, T.; Wong, Y.S. Selenocystine induces caspase-independent apoptosis in MCF-7 human breast carcinoma cells with involvement of p53 phosphorylation and reactive oxygen species generation. Int. J. Biochem. Cell Biol 2009, 41, 666–676. [Google Scholar]
- Guan, L.; Huang, F.; Li, Z.; Han, B.; Jiang, Q.; Ren, Y.; Yang, Y.; Xu, C. P53 transcription-independent activity mediates selenite-induced acute promyelocytic leukemia NB4 cell apoptosis. BMB Rep 2008, 41, 745–750. [Google Scholar]
- Sarveswaran, S.; Liroff, J.; Zhou, Z.; Nikitin, A.Y.; Ghosh, J. Selenite triggers rapid transcriptional activation of p53, and p53-mediated apoptosis in prostate cancer cells: Implication for the treatment of early-stage prostate cancer. Int. J. Oncol 2010, 36, 1419–1428. [Google Scholar]
- Abdulah, R.; Kobayashi, K.; Yamazaki, C.; Koyama, H. Molecular targets of selenium in prostate cancer prevention (review). Int. J. Oncol 2011, 39, 301–309. [Google Scholar]
- Suzuki, M.; Endo, M.; Shinohara, F.; Echigo, S.; Rikiishi, H. Differential apoptotic response of human cancer cells to organoselenium compounds. Cancer Chemother. Pharmacol 2010, 66, 475–484. [Google Scholar]
- Jun-Ying, Y.; Cun-Shuan, X. Antitumor effects of a selenium heteropoly complex in K562 cells. Pharmacol. Rep 2009, 61, 288–295. [Google Scholar]
- Yang, J.; Yang, X.; Fan, J.; Zhao, Q.; Xu, C. The novel selenium heteropoly compound (NH4)4H4[Se2Mo2V4O24].7H2O induces apoptosis of K562 cells. Mol. Med. Rep 2011, 4, 1327–1332. [Google Scholar]
- Lee, J.T.; Lee, T.J.; Park, J.W.; Kwon, T.K. Se-methylselenocysteine sensitized TRAIL-mediated apoptosis via down-regulation of bcl-2 expression. Int. J. Oncol 2009, 34, 1455–1460. [Google Scholar]
- Lin, T.; Ding, Z.; Li, N.; Xu, J.; Luo, G.; Liu, J.; Shen, J. Seleno-cyclodextrin sensitises human breast cancer cells to TRAIL-induced apoptosis through DR5 induction and NF-kappaB suppression. Eur. J. Cancer 2011, 47, 1890–1907. [Google Scholar]
- Shang, D.; Zhang, J.; Wen, L.; Li, Y.; Cui, Q. Preparation, characterization, and antiproliferative activities of the se-containing polysaccharide SeGLP-2B-1 from Se-enriched Ganoderma lucidum. J. Agric. Food Chem 2009, 57, 7737–7742. [Google Scholar]
- Shang, D.; Li, Y.; Wang, C.; Wang, X.; Yu, Z.; Fu, X. A novel polysaccharide from Se-enriched Ganoderma lucidum induces apoptosis of human breast cancer cells. Oncol. Rep 2011, 25, 267–272. [Google Scholar]
- Abdulah, R.; Faried, A.; Kobayashi, K.; Yamazaki, C.; Suradji, E.; Ito, K.; Suzuki, K.; Murakami, M.; Kuwano, H.; Koyama, H. Selenium enrichment of broccoli sprout extract increases chemosensitivity and apoptosis of LNCaP prostate cancer cells. BMC Cancer 2009, 9. [Google Scholar] [CrossRef]
- Tsubura, A.; Lai, Y.C.; Kuwata, M.; Uehara, N.; Yoshizawa, K. Anticancer effects of garlic and garlic-derived compounds for breast cancer control. Anticancer Agents Med. Chem 2011, 11, 249–253. [Google Scholar]
- Kumi-Diaka, J.; Merchant, K.; Haces, A.; Hormann, V.; Johnson, M. Genistein-selenium combination induces growth arrest in prostate cancer cells. J. Med. Food 2010, 13, 842–850. [Google Scholar]
- Azrak, R.G.; Cao, S.; Durrani, F.A.; Toth, K.; Bhattacharya, A.; Rustum, Y.M. Augmented therapeutic efficacy of irinotecan is associated with enhanced drug accumulation. Cancer Lett 2011, 311, 219–229. [Google Scholar]
- Chakraborty, P.; Sk, U.H.; Bhattacharya, S. Chemoprotection and enhancement of cancer chemotherapeutic efficacy of cyclophosphamide in mice bearing ehrlich ascites carcinoma by diphenylmethyl selenocyanate. Cancer Chemother. Pharmacol 2009, 64, 971–980. [Google Scholar]
- Cheng, Y.; Sk, U.H.; Zhang, Y.; Ren, X.; Zhang, L.; Huber-Keener, K.J.; Sun, Y.-W.; Liao, J.; Amin, S.; Sharma, A.K.; et al. Rational incorporation of selenium into temozolomide elicits superior antitumor activity associated with both apoptotic and autophagic cell death. PLoS One 2012, 7, e35104. [Google Scholar]
- Kim, J.H.; Hue, J.J.; Kang, B.S.; Park, H.; Nam, S.Y.; Yun, Y.W.; Kim, J.S.; Lee, B.J. Effects of selenium on colon carcinogenesis induced by azoxymethane and dextran sodium sulfate in mouse model with high-iron diet. Lab. Anim. Res 2011, 27, 9–18. [Google Scholar]
- Rikiishi, H. Autophagic and apoptotic effects of HDAC inhibitors on cancer cells. J. Biomed. Biotechnol 2011, 2011. [Google Scholar] [CrossRef]
- Rikiishi, H. Novel insights into the interplay between apoptosis and autophagy. Int. J. Cell Biol 2012, 2012. [Google Scholar] [CrossRef]
- Long, J.; Zhao, J.; Yan, Z.; Liu, Z.; Wang, N. Antitumor effects of a novel sulfur-containing hydroxamate histone deacetylase inhibitor H40. Int. J. Cancer 2009, 124, 1235–1244. [Google Scholar]
- Gammoh, N.; Lam, D.; Puente, C.; Ganley, I.; Marks, P.A.; Jiang, X. Role of autophagy in histone deacetylase inhibitor-induced apoptotic and nonapoptotic cell death. Proc. Natl. Acad. Sci. USA 2012. [Google Scholar] [CrossRef]
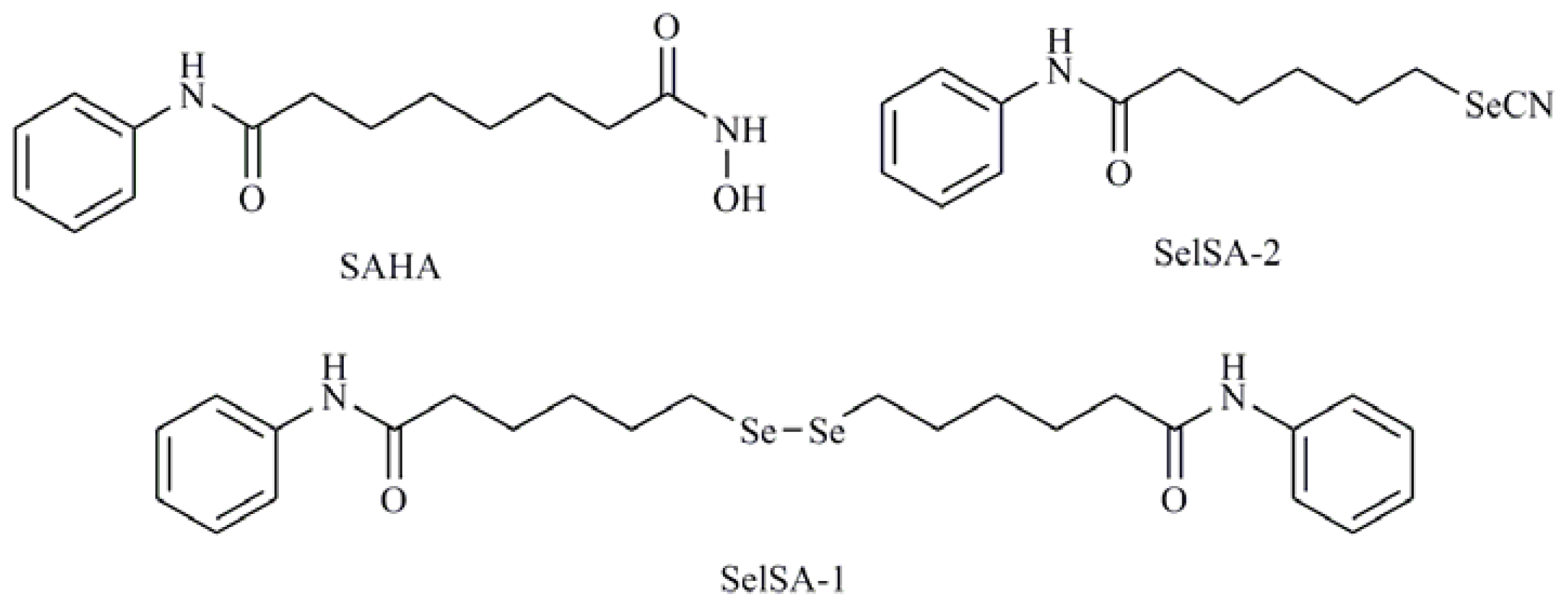
- Desai, D.; Salli, U.; Vrana, K.E.; Amin, S. SelSA, selenium analogs of SAHA as potent histone deacetylase inhibitors. Bioorg. Med. Chem. Lett 2010, 20, 2044–2047. [Google Scholar]
- Karelia, N.; Desai, D.; Hengst, J.A.; Amin, S.; Rudrabhatla, S.V.; Yun, J. Selenium-containing analogs of SAHA induce cytotoxicity in lung cancer cells. Bioorg. Med. Chem. Lett 2010, 20, 6816–6819. [Google Scholar]
- Sanmartin, C.; Plano, D.; Palop, J.A. Selenium compounds and apoptotic modulation: A new perspective in cancer therapy. Mini-Rev. Med. Chem 2008, 8, 1020–1031. [Google Scholar]
- Kim, Y.W.; Bae, S.M.; Liu, H.B.; Kim, I.W.; Chun, H.J.; Ahn, W.S. Selenium enhances the efficacy of radachlorin mediated-photodynamic therapy in TC-1 tumor development. Oncol. Rep 2012, 28, 576–584. [Google Scholar]
- Filomeni, G.; Piccirillo, S.; Rotilio, G.; Ciriolo, M.R. P38MAPK and ERK1/2 dictate cell death/survival response to different pro-oxidant stimuli via p53 and Nrf2 in neuroblastoma cells SH-SY5Y. Biochem. Pharmacol 2012, 83, 1349–1357. [Google Scholar]
- Huang, C.; Ding, G.; Gu, C.; Zhou, J.; Kuang, M.; Ji, Y.; He, Y.; Kondo, T.; Fan, J. Decreased selenium-binding protein 1 enhances glutathione peroxidase 1 activity and downregulates HIF-1alpha to promote hepatocellular carcinoma invasiveness. Clin. Cancer Res 2012, 18, 3042–3053. [Google Scholar]
- Wang, D.; Taylor, E.; Wang, Y.; Wan, X.; Zhang, J. Encapsulated nanoepigallocatechin-3-gallate and elemental selenium nanoparticles as paradigms for nanochemoprevention. Int. J. Nanomed 2012, 7, 1711–1721. [Google Scholar]
- Suradji, E.W.; Hatabu, T.; Kobayashi, K.; Yamazaki, C.; Abdulah, R.; Nakazawa, M.; Nakajima-Shimada, J.; Koyama, H. Selenium-induced apoptosis-like cell death in Plasmodium falciparum. Parasitology 2011, 19, 1–11. [Google Scholar]
- Afoulous, S.; Ferhout, H.; Raoelison, E.G.; Valentin, A.; Moukarzel, B.; Couderc, F.; Bouajila, J. Helichrysum gymnocephalum essential oil: Chemical composition and cytotoxic, antimalarial and antioxidant activities, attribution of the activity origin by correlations. Molecules 2011, 16, 8273–8291. [Google Scholar]
- Moreno, D.; Plano, D.; Baquedano, Y.; Jimenez-Ruiz, A.; Palop, J.A.; Sanmartin, C. Antileishmanial activity of imidothiocarbamates and imidoselenocarbamates. Parasitol. Res 2011, 108, 233–239. [Google Scholar]
- Singh, K.; Kaur, H.; Chibale, K.; Balzarini, J.; Little, S.; Bharatam, P.V. 2-aminopyrimidine based 4-aminoquinoline anti-plasmodial agents. Synthesis, biological activity, structure–activity relationship and mode of action studies. Eur. J. Med. Chem 2012, 52, 82–97. [Google Scholar]
- Jamier, V.; Ba, L.A.; Jacob, C. Selenium- and tellurium-containing multifunctional redox agents as biochemical redox modulators with selective cytotoxicity. Chemistry 2010, 16, 10920–10928. [Google Scholar]
- Ramoutar, R.R.; Brumaghim, J.L. Antioxidant and anticancer properties and mechanisms of inorganic selenium, oxo-sulfur, and oxo-selenium compounds. Cell Biochem. Biophys 2010, 58, 1–23. [Google Scholar]
- Lee, K.H.; Jeong, D. Bimodal actions of selenium essential for antioxidant and toxic pro-oxidant activities: The selenium paradox (review). Mol. Med. Rep 2012, 5, 299–304. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
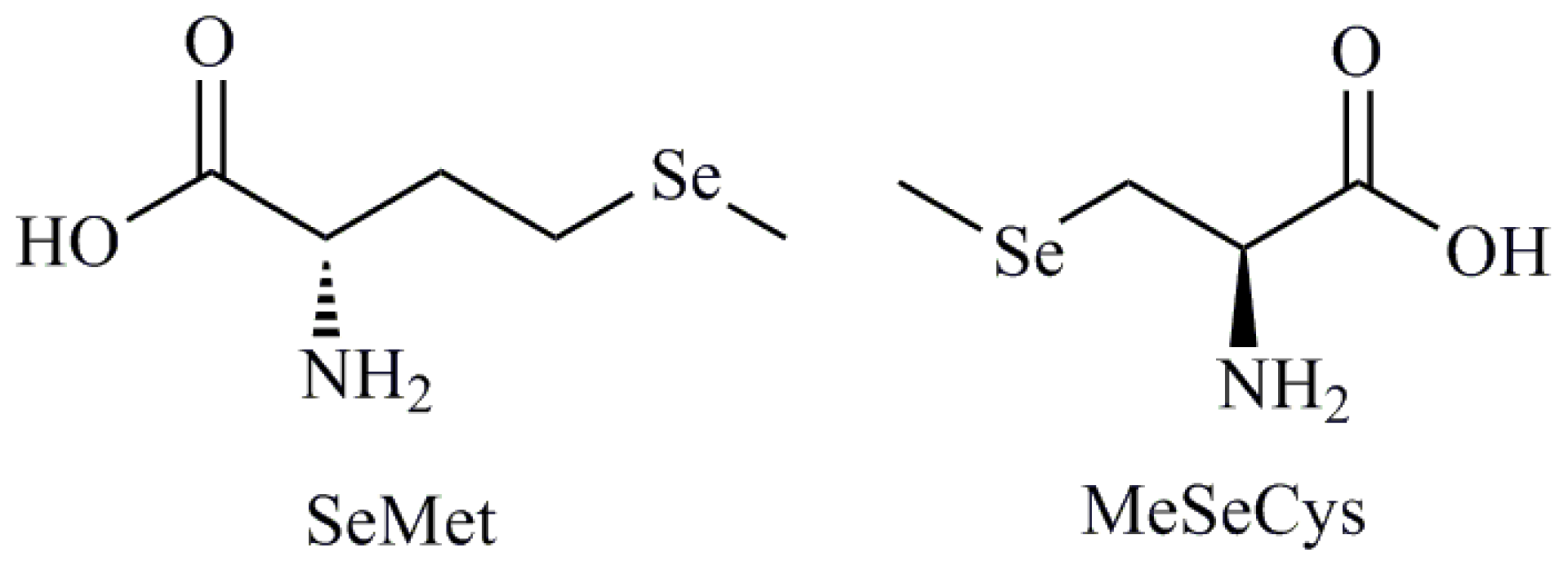
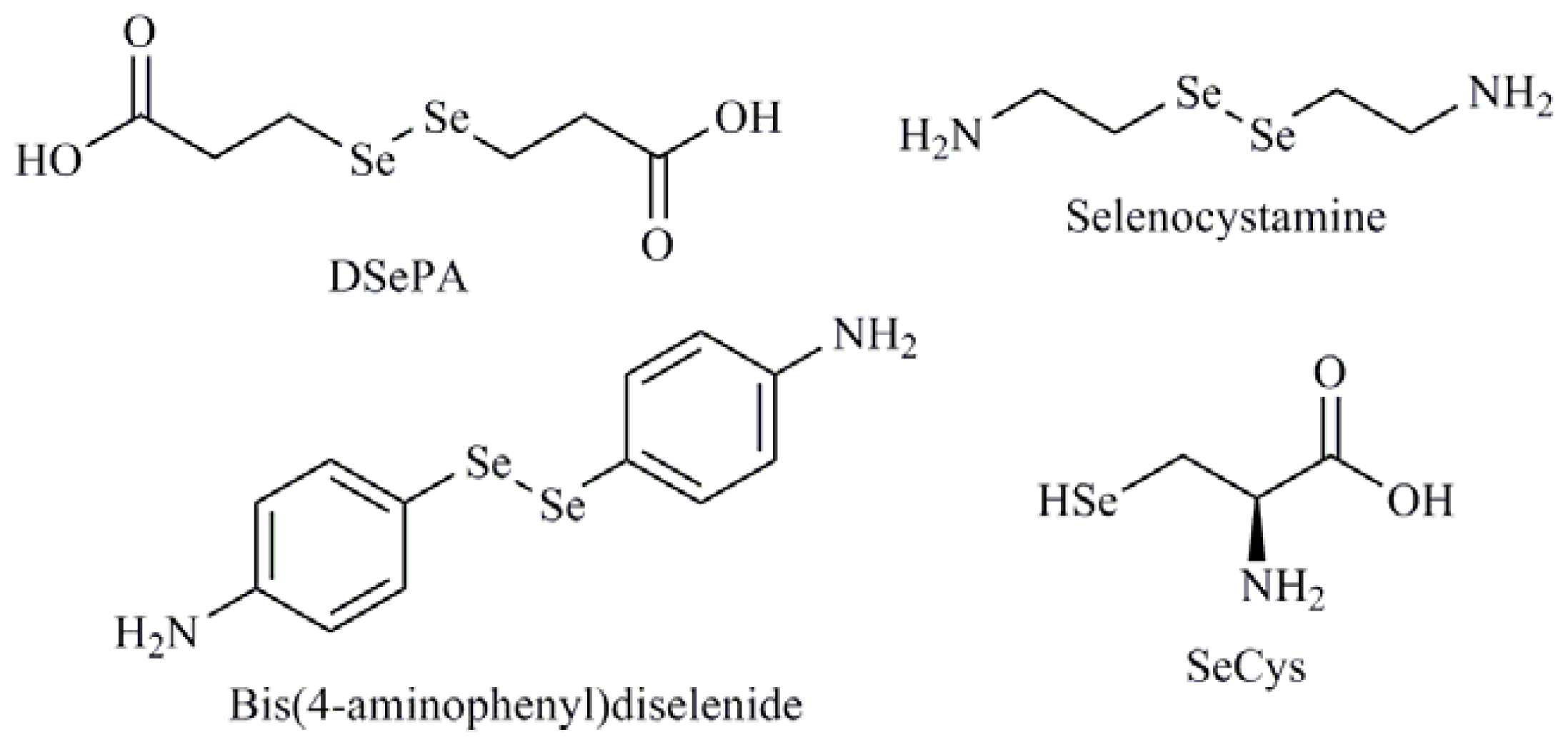
| Selenoaminoacids |
| Selenomethionine (SeMet); Methylselenocysteine (MeSeCys); Selenocystamine; Selenocysteine (SeCys) |
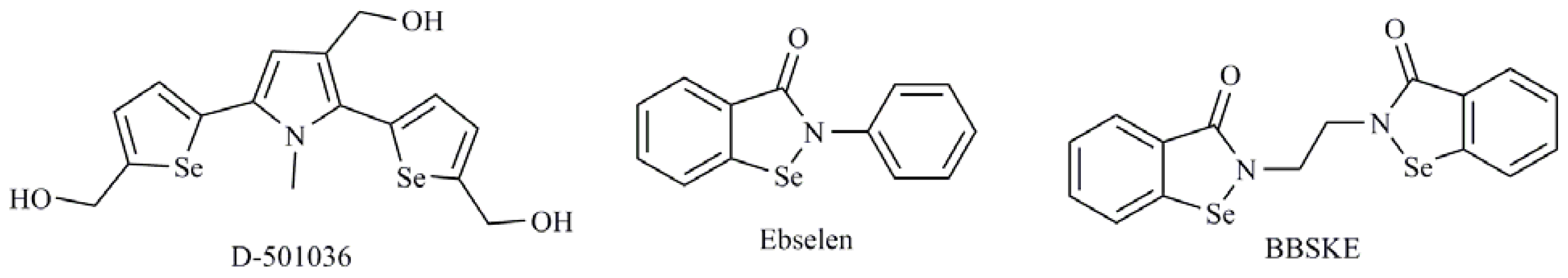
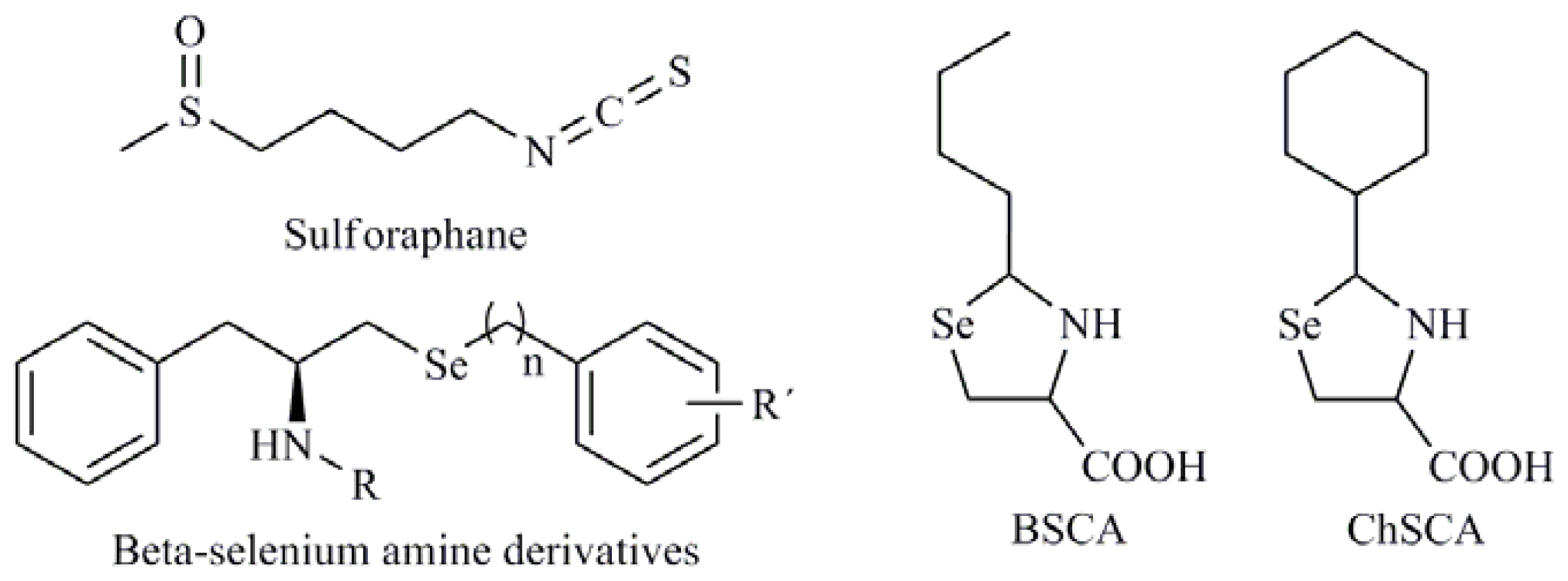
| Se-heterocyclic compounds |
| 1,3-Selenazolin-4-one derivatives; 2,5-Bis(5-hydroxymethyl-2-selenienyl)-3-hydroxymethyl-N-methylpyrrole (D-501036); 2-Phenyl-1,2-benzisoselenazol-3(2H)-one (ebselen); 1,2-[Bis(1,2-benzisoselenazolone-3-(2H)-ketone)]ethane (BBSKE); 2-Buthylselenazolidine-4-(R)-carboxylic acid (BSCA); 2-Cyclohexylselenazolidine-4-(R)-carboxylic acid (ChSCA) |
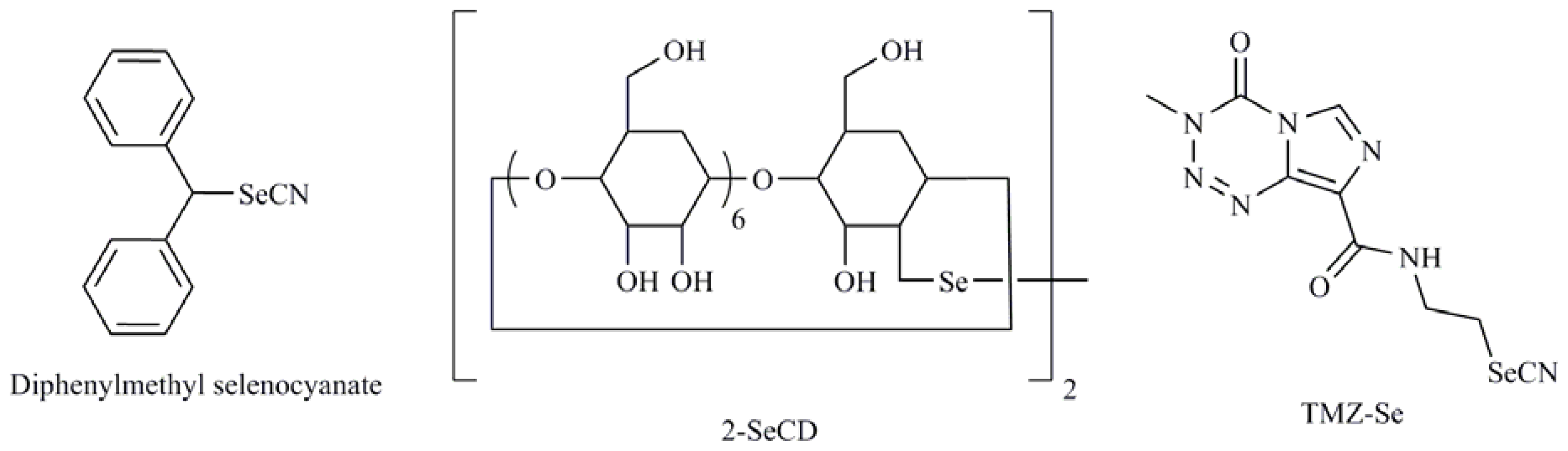
| Selenocyanates |
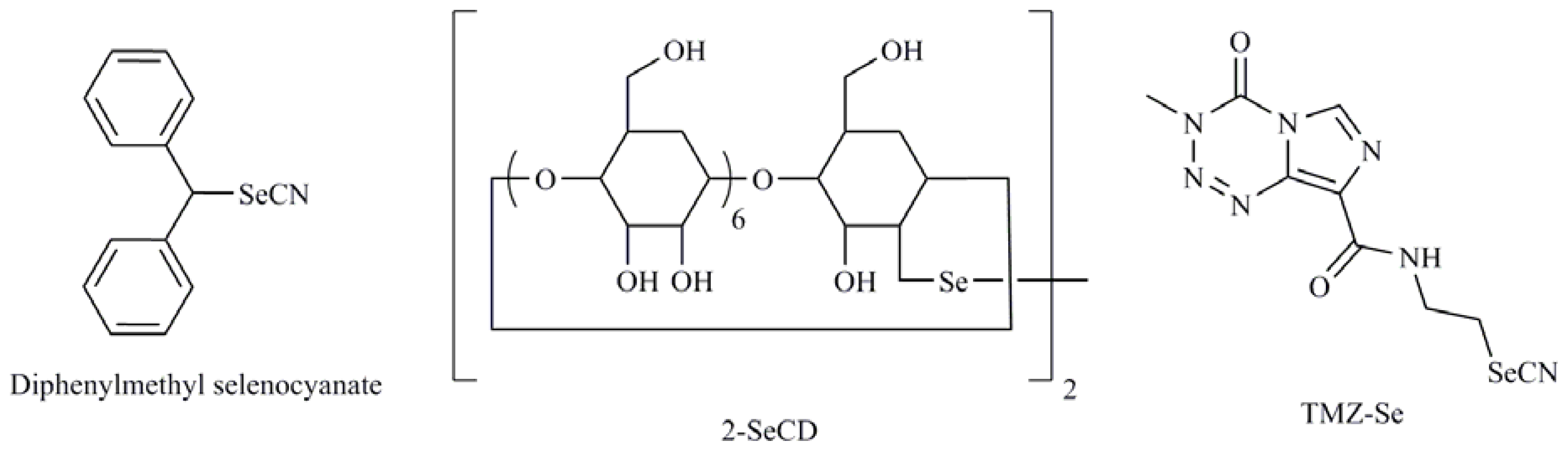
| 1-4-phenylenebis(methylene)selenocyanate (p-XSC); Isatin analogs; Diphenylmethylselenocyanate; TMZ-Se; 5-phenylcarbamoylpentyl selenocyanide (SelSA-2) |
| Isoselenocyanates |
| 4-Phenylbutylisoselenocyanate (ISC-4) |
| Diselenides |
| Diselenodipropionic acid (DSePA); Bis(4-aminophenyl)diselenide; 2-Selenium-bridged β-cyclodextrin (2-SeCD); Bis(5-phenylcarbamoylpentyl) diselenide (SelSA-1) |
| Selenides |
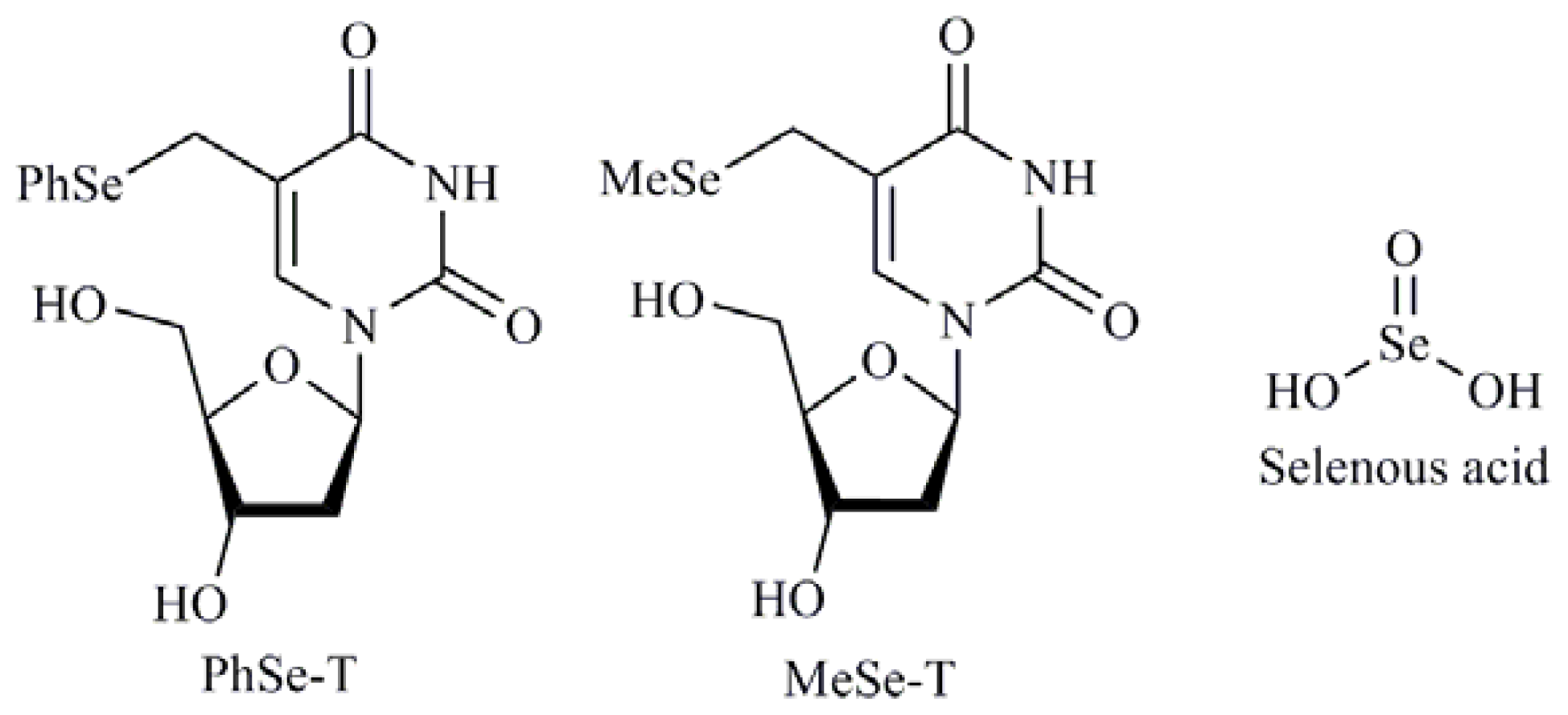
| Se,Se′-1,4-phenylenebis(1,2-ethanediyl)bisisoselenourea (PBISe); Methylimidoselenocarbamates; 5-Phenylselenyl-methyl-2′-deoxyuridine (PhSe-T); 5-Methylselenyl-methyl-2′-deoxyuridine (MeSe-T); β-selenium amine derivatives |
| Se(IV) compounds |
| Sodium selenite (Na2SeO3); Methylseleninic acid (MSA); Selenous acid; Selenium dioxide (SeO2) |
© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Sanmartín, C.; Plano, D.; Sharma, A.K.; Palop, J.A. Selenium Compounds, Apoptosis and Other Types of Cell Death: An Overview for Cancer Therapy. Int. J. Mol. Sci. 2012, 13, 9649-9672. https://doi.org/10.3390/ijms13089649
Sanmartín C, Plano D, Sharma AK, Palop JA. Selenium Compounds, Apoptosis and Other Types of Cell Death: An Overview for Cancer Therapy. International Journal of Molecular Sciences. 2012; 13(8):9649-9672. https://doi.org/10.3390/ijms13089649
Chicago/Turabian StyleSanmartín, Carmen, Daniel Plano, Arun K. Sharma, and Juan Antonio Palop. 2012. "Selenium Compounds, Apoptosis and Other Types of Cell Death: An Overview for Cancer Therapy" International Journal of Molecular Sciences 13, no. 8: 9649-9672. https://doi.org/10.3390/ijms13089649
APA StyleSanmartín, C., Plano, D., Sharma, A. K., & Palop, J. A. (2012). Selenium Compounds, Apoptosis and Other Types of Cell Death: An Overview for Cancer Therapy. International Journal of Molecular Sciences, 13(8), 9649-9672. https://doi.org/10.3390/ijms13089649