Evaluation of Sex-Specific Gene Expression in Archived Dried Blood Spots (DBS)

Abstract
:
1. Introduction
2. Results and Discussion
2.1. Differentially-Expressed Genes can Be Determined in DBS
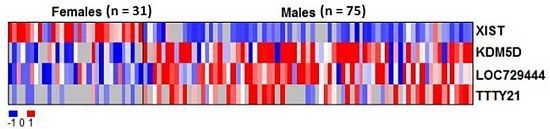
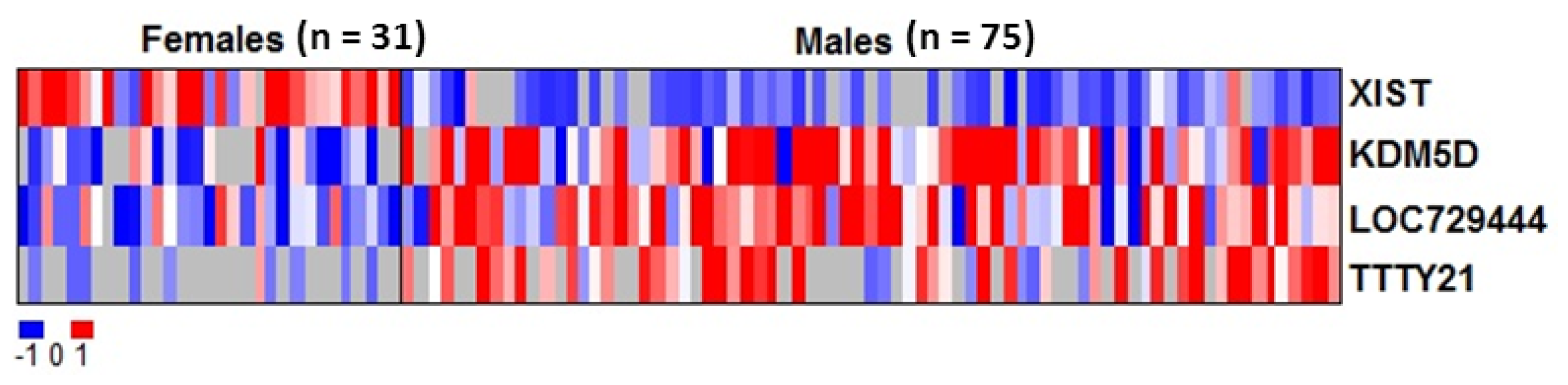
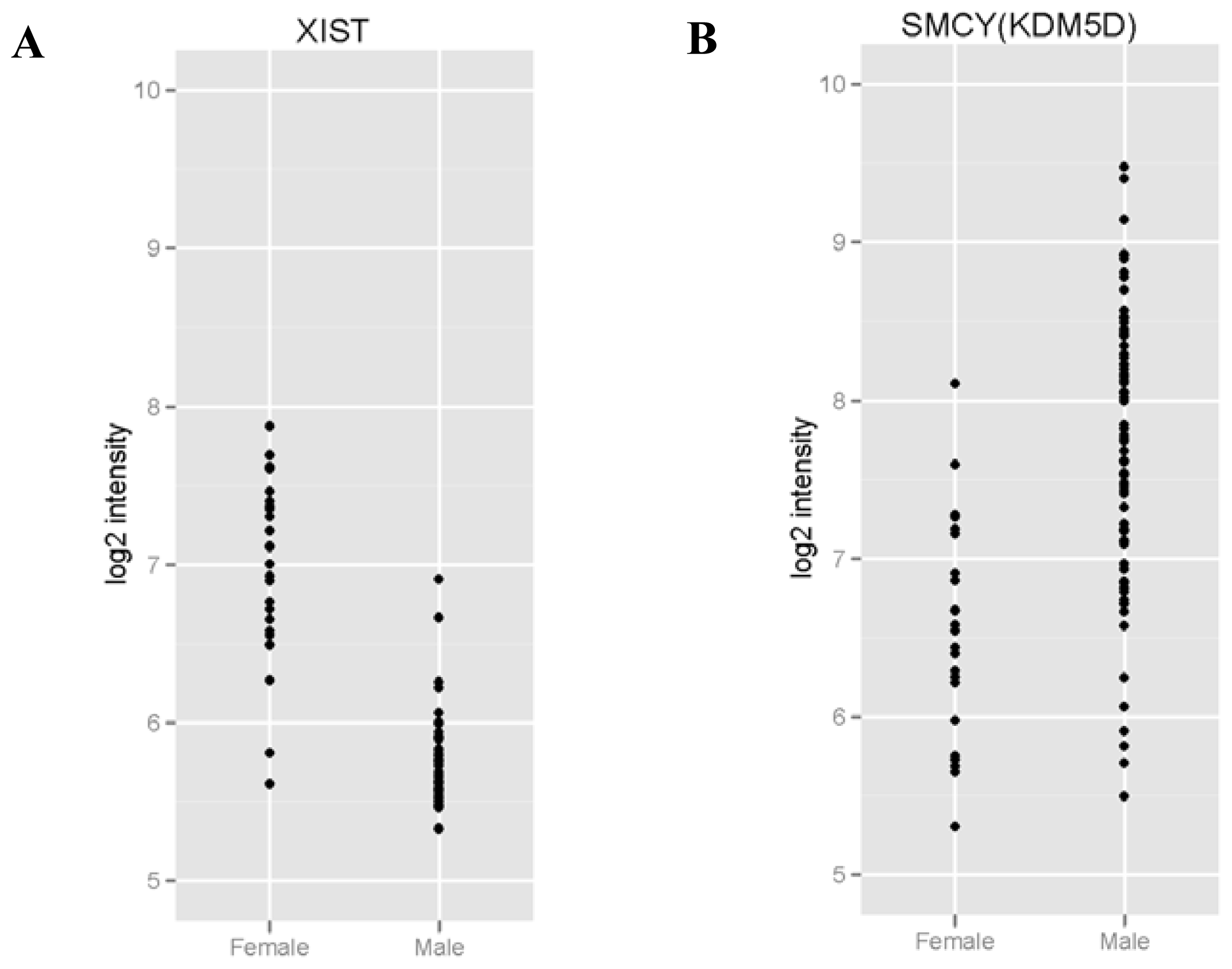
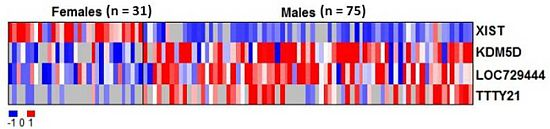
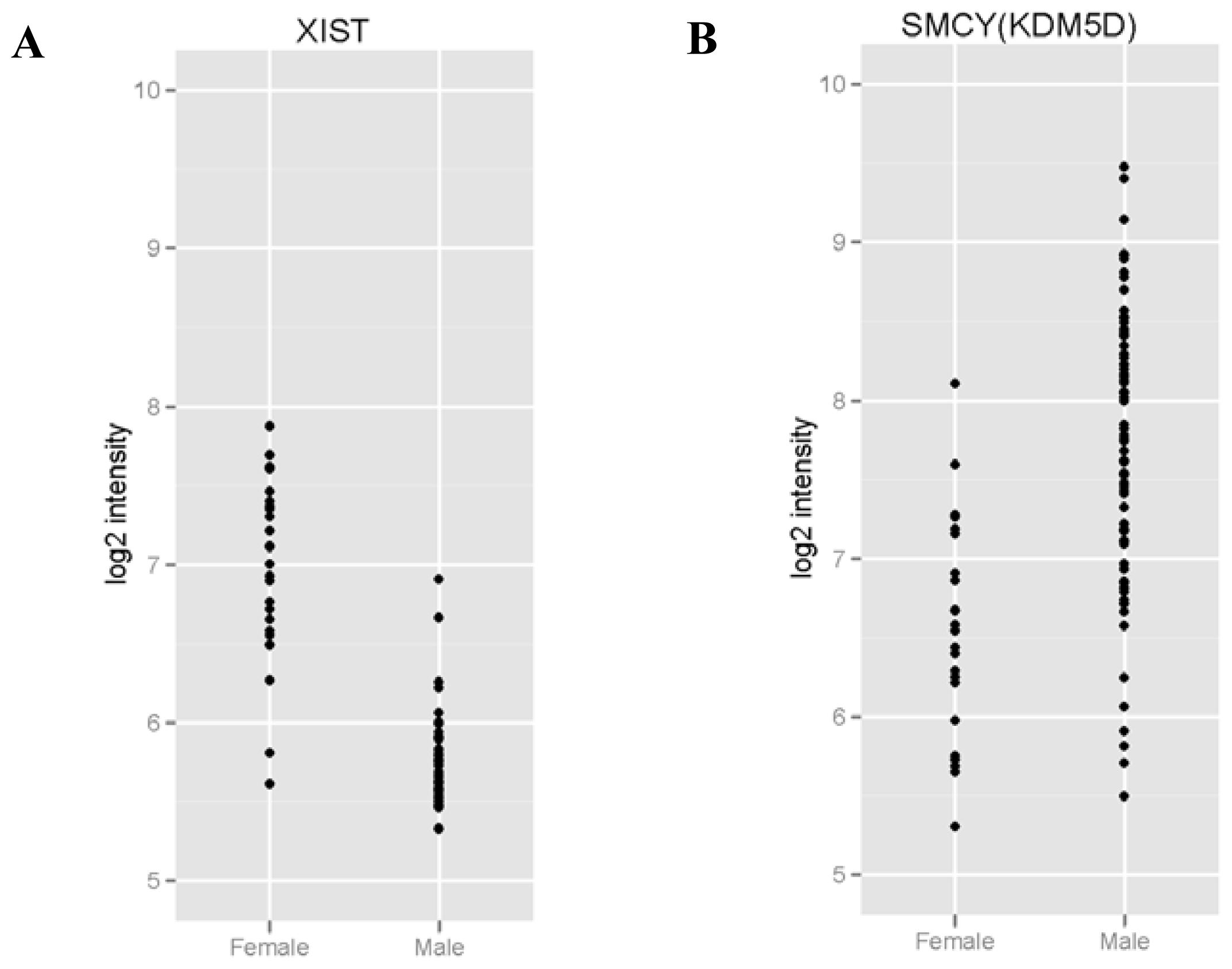
2.2. RNAs Related to Sex Are Preserved in DBS
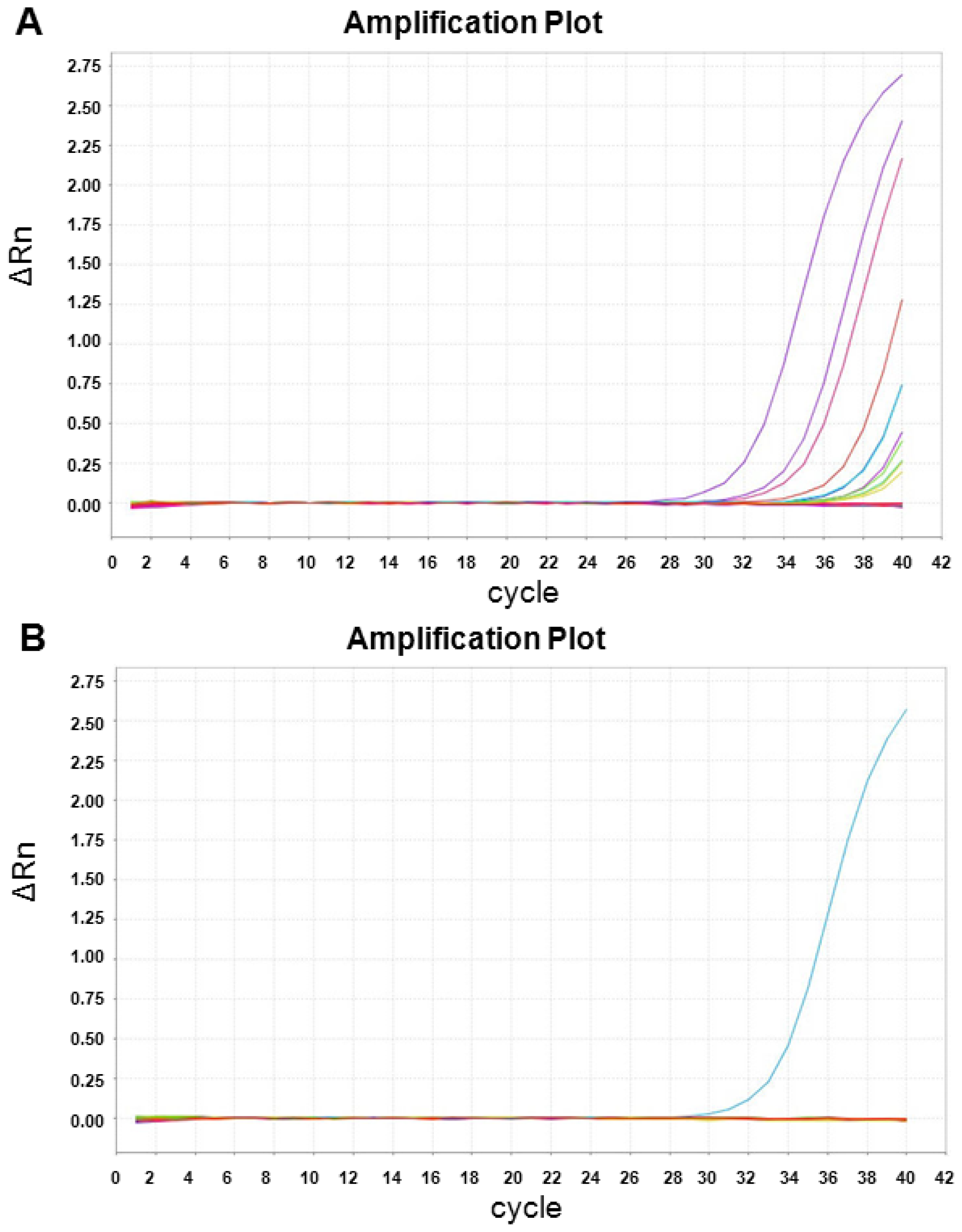
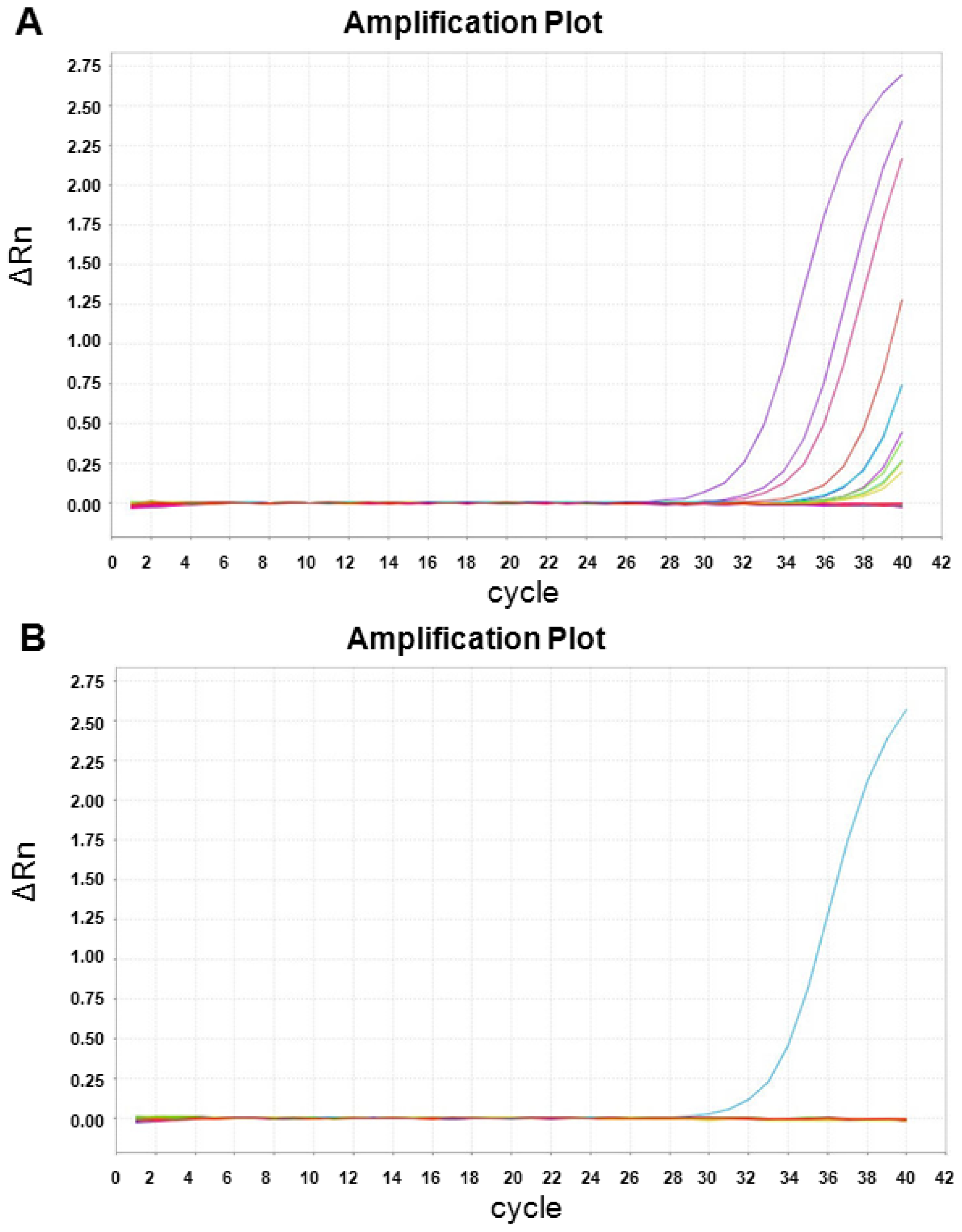
2.3. XIST Expression can Be Validated Using qRT-PCR
2.4. Discussion
3. Experimental Section
3.1. Archived Dried Blood Spots (DBS)
3.2. RNA and cDNA Preparation
3.3. Labeling, Hybridization, and Scanning Procedures
3.4. Gene Expression Microarray and Statistical Analyses
3.5. Quantitative Real-Time PCR Analysis (qRT-PCR)
4. Conclusions
Acknowledgments
- Conflict of InterestThe authors declare no conflict of interest.
References
- Green, N.S.; Pass, K.A. Neonatal screening by DNA microarray: Spots and chips. Nat. Rev. Genet 2005, 6, 147–151. [Google Scholar]
- Wilcken, B.; Wiley, V. Newborn screening. Pathology 2008, 40, 104–115. [Google Scholar]
- Kammesheidt, A.; Kharrazi, M.; Graham, S.; Young, S.; Pearl, M.; Dunlop, C.; Keiles, S. Comprehensive genetic analysis of the cystic fibrosis transmembrane conductance regulator from dried blood specimens—Implications for newborn screening. Genet. Med 2006, 8, 557–562. [Google Scholar]
- Streetly, A.; Clarke, M.; Downing, M.; Farrar, L.; Foo, Y.; Hall, K.; Kemp, H.; Newbold, J.; Walsh, P.; Yates, J.; et al. Implementation of the newborn screening programme for sickle cell disease in England: Results for 2003–2005. J. Med. Screen 2008, 15, 9–13. [Google Scholar]
- Hollegaard, M.V.; Grove, J.; Thorsen, P.; Norgaard-Pedersen, B.; Hougaard, D.M. High-throughput genotyping on archived dried blood spot samples. Genet. Test. Mol. Biomark. 2009, 13, 173–179. [Google Scholar]
- Hollegaard, M.V.; Thorsen, P.; Norgaard-Pedersen, B.; Hougaard, D.M. Genotyping whole-genome-amplified DNA from 3- to 25-year-old neonatal dried blood spot samples with reference to fresh genomic DNA. Electrophoresis 2009, 30, 2532–2535. [Google Scholar]
- Hollegaard, M.V.; Grauholm, J.; Borglum, A.; Nyegaard, M.; Norgaard-Pedersen, B.; Orntoft, T.; Mortensen, P.B.; Wiuf, C.; Mors, O.; Didriksen, M.; et al. Genome-wide scans using archived neonatal dried blood spot samples. BMC Genomics 2009, 10, 297. [Google Scholar]
- Hardin, J.; Finnell, R.H.; Wong, D.; Hogan, M.E.; Horovitz, J.; Shu, J.; Shaw, G.M. Whole genome microarray analysis, from neonatal blood cards. BMC Genet 2009, 10, 38. [Google Scholar]
- Ji, H.; Li, Y.; Graham, M.; Liang, B.B.; Pilon, R.; Tyson, S.; Peters, G.; Tyler, S.; Merks, H.; Bertagnolio, S.; et al. Next-generation sequencing of dried blood spot specimens: A novel approach to HIV drug-resistance surveillance. Antivir. Ther 2011, 16, 871–878. [Google Scholar]
- Winkel, B.G.; Hollegaard, M.V.; Olesen, M.S.; Svendsen, J.H.; Haunso, S.; Hougaard, D.M.; Tfelt-Hansen, J. Whole-genome amplified DNA from stored dried blood spots is reliable in high resolution melting curve and sequencing analysis. BMC Med. Genet 2011, 12, 22. [Google Scholar]
- Matsubara, Y.; Ikeda, H.; Endo, H.; Narisawa, K. Dried blood spot on filter paper as a source of mRNA. Nucleic Acids Res 1992, 20, 1998. [Google Scholar]
- De Crignis, E.; Re, M.C.; Cimatti, L.; Zecchi, L.; Gibellini, D. HIV-1 and HCV detection in dried blood spots by SYBR Green multiplex real-time RT-PCR. J. Virol. Methods 2010, 165, 51–56. [Google Scholar]
- Lang, P.O.; Mitchell, W.A.; Govind, S.; Aspinall, R. Real time-PCR assay estimating the naive T-cell pool in whole blood and dried blood spot samples: pilot study in young adults. J. Immunol. Methods 2011, 369, 133–140. [Google Scholar]
- Haak, P.T.; Busik, J.V.; Kort, E.J.; Tikhonenko, M.; Paneth, N.; Resau, J.H. Archived unfrozen neonatal blood spots are amenable to quantitative gene expression analysis. Neonatology 2009, 95, 210–216. [Google Scholar]
- Khoo, S.K.; Dykema, K.; Vadlapatla, N.M.; LaHaie, D.; Valle, S.; Satterthwaite, D.; Ramirez, S.A.; Carruthers, J.A.; Haak, P.T.; Resau, J.H. Acquiring genome-wide gene expression profiles in Guthrie card blood spots using microarrays. Pathol. Int 2011, 61, 1–6. [Google Scholar]
- Brockdorff, N.; Ashworth, A.; Kay, G.F.; Cooper, P.; Smith, S.; McCabe, V.M.; Norris, D.P.; Penny, G.D.; Patel, D.; Rastan, S. Conservation of position and exclusive expression of mouse Xist from the inactive X chromosome. Nature 1991, 351, 329–331. [Google Scholar]
- Wutz, A.; Gribnau, J. X inactivation Xplained. Curr. Opin. Genet. Dev 2007, 17, 387–393. [Google Scholar]
- Agulnik, A.I.; Mitchell, M.J.; Lerner, J.L.; Woods, D.R.; Bishop, C.E. A mouse Y chromosome gene encoded by a region essential for spermatogenesis and expression of male-specific minor histocompatibility antigens. Hum. Mol. Genet 1994, 3, 873–878. [Google Scholar]
- Kent-First, M.G.; Maffitt, M.; Muallem, A.; Brisco, P.; Shultz, J.; Ekenberg, S.; Agulnik, A.I.; Agulnik, I.; Shramm, D.; Bavister, B.; et al. Gene sequence and evolutionary conservation of human SMCY. Nat. Genet 1996, 14, 128–129. [Google Scholar]
- Kim, S.J.; Dix, D.J.; Thompson, K.E.; Murrell, R.N.; Schmid, J.E.; Gallagher, J.E.; Rockett, J.C. Effects of storage, RNA extraction, genechip type, and donor sex on gene expression profiling of human whole blood. Clin. Chem 2007, 53, 1038–1045. [Google Scholar]
- Vawter, M.P.; Evans, S.; Choudary, P.; Tomita, H.; Meador-Woodruff, J.; Molnar, M.; Li, J.; Lopez, J.F.; Myers, R.; Cox, D.; et al. Gender-specific gene expression in post-mortem human brain: Localization to sex chromosomes. Neuropsychopharmacology 2004, 29, 373–384. [Google Scholar]
- Watson, J.D.; Wang, S.; von Stetina, S.E.; Spencer, W.C.; Levy, S.; Dexheimer, P.J.; Kurn, N.; Heath, J.D.; Miller, D.M., III. Complementary RNA amplification methods enhance microarray identification of transcripts expressed in the C. elegans nervous system. BMC Genomics 2008, 9, 84. [Google Scholar]
- Patterson, T.A.; Lobenhofer, E.K.; Fulmer-Smentek, S.B.; Collins, P.J.; Chu, T.M.; Bao, W.; Fang, H.; Kawasaki, E.S.; Hager, J.; Tikhonova, I.R.; et al. Performance comparison of one-color and two-color platforms within the MicroArray Quality Control (MAQC) project. Nat. Biotechnol 2006, 24, 1140–1150. [Google Scholar]
- Bolstad, B.M.; Irizarry, R.A.; Astrand, M.; Speed, T.P. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics 2003, 19, 185–193. [Google Scholar]
- Smyth, G.K. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat. Appl. Genet. Mol. Biol 2004, 3. [Google Scholar] [CrossRef]
- Smyth, G.K. Limma: Linear Models for Microarray Data. In Bioinformatics and Computational Biology Solutions using R and Bioconductor; Gentleman, R., Carey, V., Dudoit, S., Irizarry, R., Huber, W., Eds.; Springer: New York, NY, USA, 2005; pp. 397–420. [Google Scholar]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. B 1995, 57, 289–300. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene ID | Average intensity in males (mean log2) | Average intensity in females (mean log2) | Adjusted p value |
|---|---|---|---|
| XIST | 5.73 | 6.92 | 1.6 × 10−16*** |
| KDM5D | 7.64 | 6.54 | 0.0036 ** |
| LOC729444 | 7.99 | 6.86 | 0.042 * |
| TTTY21 | 6.66 | 5.65 | 0.042 * |
© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Resau, J.H.; Ho, N.T.; Dykema, K.; Faber, M.S.; Busik, J.V.; Nickolov, R.Z.; Furge, K.A.; Paneth, N.; Jewell, S.; Khoo, S.K. Evaluation of Sex-Specific Gene Expression in Archived Dried Blood Spots (DBS). Int. J. Mol. Sci. 2012, 13, 9599-9608. https://doi.org/10.3390/ijms13089599
Resau JH, Ho NT, Dykema K, Faber MS, Busik JV, Nickolov RZ, Furge KA, Paneth N, Jewell S, Khoo SK. Evaluation of Sex-Specific Gene Expression in Archived Dried Blood Spots (DBS). International Journal of Molecular Sciences. 2012; 13(8):9599-9608. https://doi.org/10.3390/ijms13089599
Chicago/Turabian StyleResau, James H., Nhan T. Ho, Karl Dykema, Matthew S. Faber, Julia V. Busik, Radoslav Z. Nickolov, Kyle A. Furge, Nigel Paneth, Scott Jewell, and Sok Kean Khoo. 2012. "Evaluation of Sex-Specific Gene Expression in Archived Dried Blood Spots (DBS)" International Journal of Molecular Sciences 13, no. 8: 9599-9608. https://doi.org/10.3390/ijms13089599