Correlation between Protein Sequence Similarity and Crystallization Reagents in the Biological Macromolecule Crystallization Database


Abstract
:1. Introduction
2. Results and Discussion
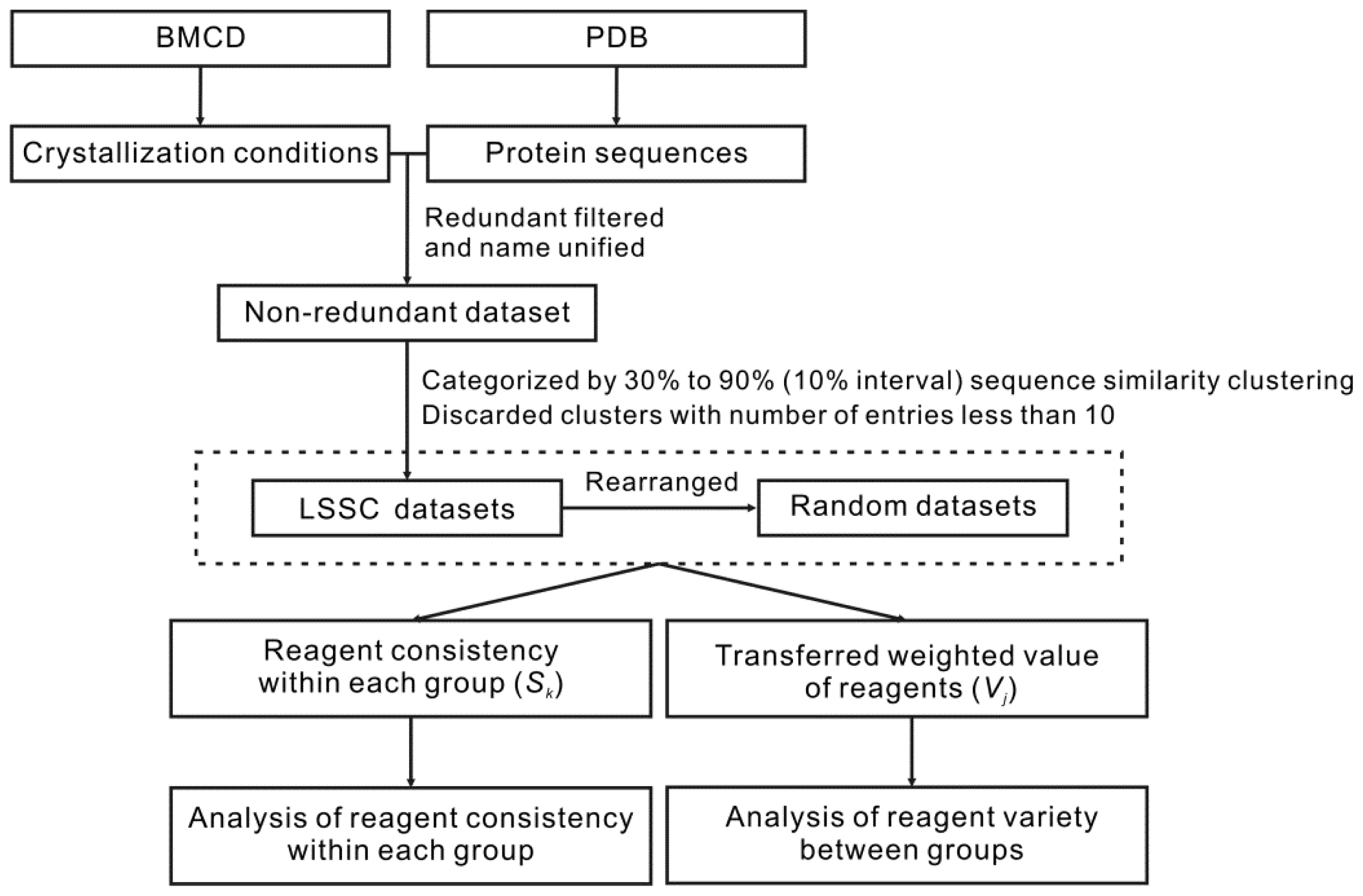
2.1. Datasets
2.2. Establishing the Large Sequence Similarity Clusters (LSSC) Dataset and Random Datasets
2.3. Statistical Analysis of Reagent Consistency within Each LSSC and Random Group
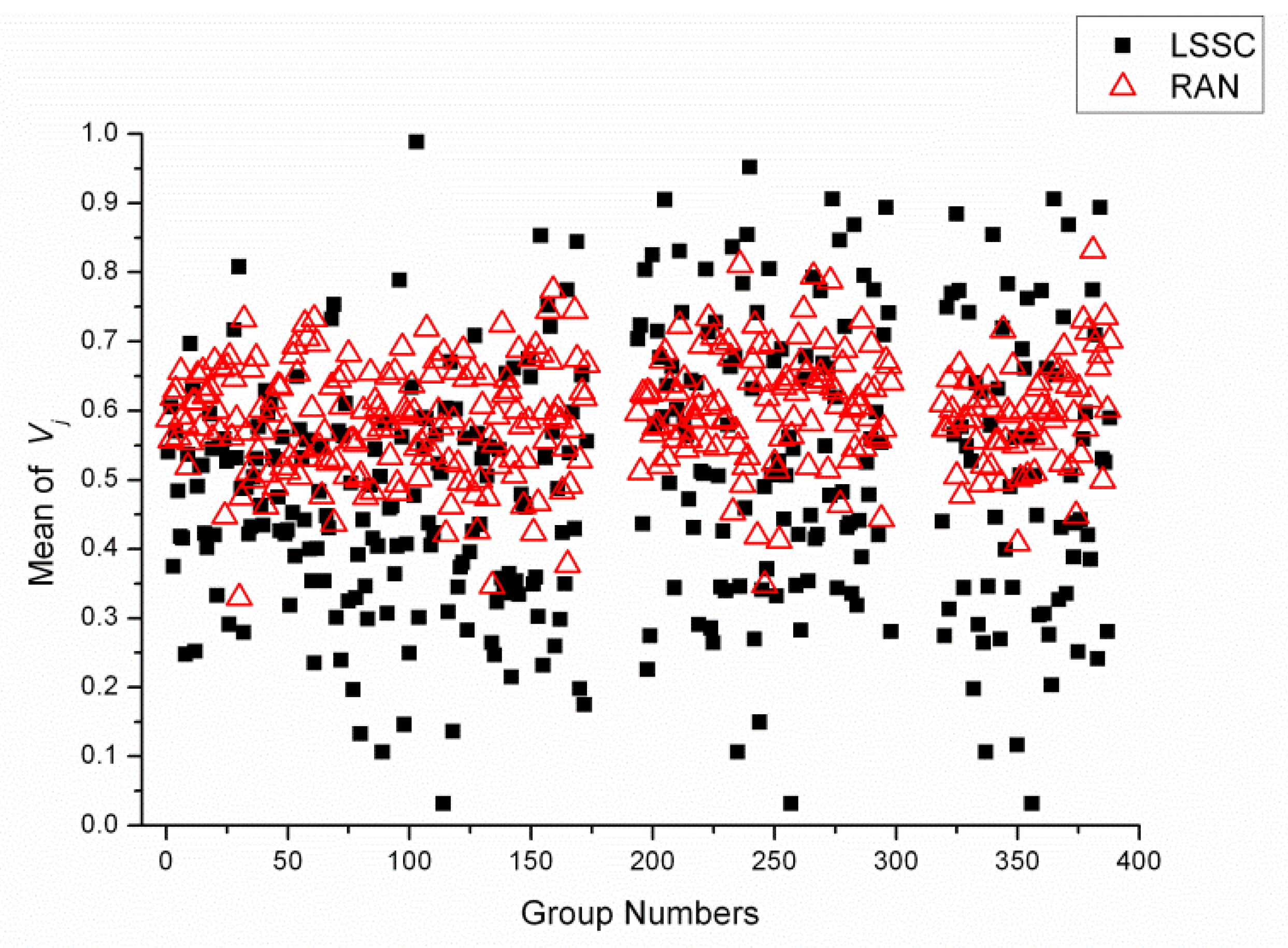
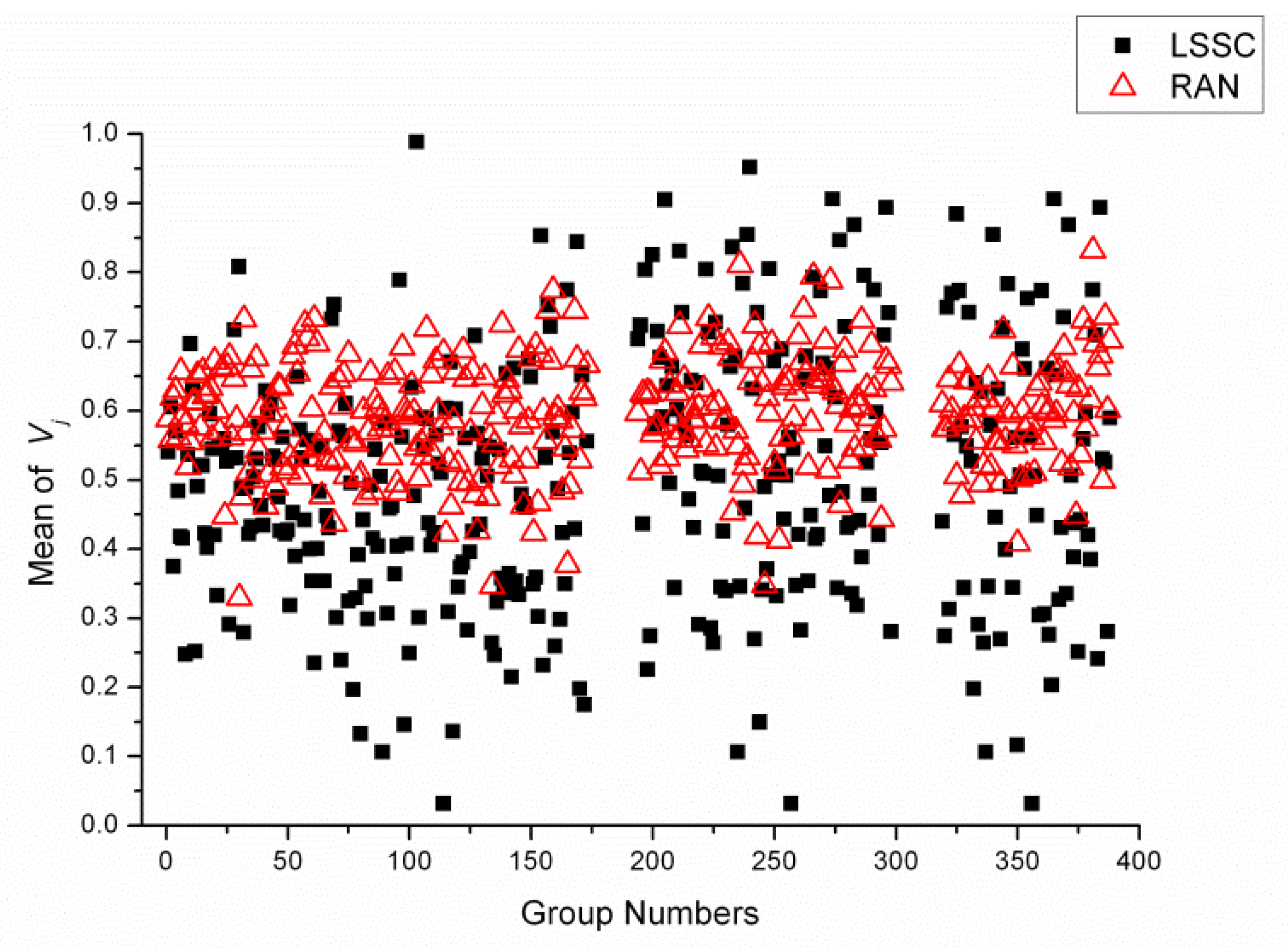
2.4. Statistical Analysis of Reagent Variety between Each LSSC and Random Group
3. Methods and Experimental Section
3.1. Datasets
3.2. Calculating the Frequency of each Kind of Crystallization Reagent
3.3. Clustering by Sequence Similarity to Establish the LSSC Datasets
3.4. Establishing the Random Datasets from the LSSC Datasets for Robust Analysis
3.5. Statistical Analysis of Reagent Consistency within Each Group
3.6. Statistical Analysis of Reagent Variety between Groups
4. Conclusions
Acknowledgments
References and Notes
- Berman, H.; Henrick, K.; Nakamura, H.; Markley, J.L. The worldwide Protein Data Bank (wwPDB): Ensuring a single, uniform archive of PDB data. Nucleic. Acids. Res 2007, 35, D301–D303. [Google Scholar]
- Wu, C.H.; Apweiler, R.; Bairoch, A.; Natale, D.A.; Barker, W.C.; Boeckmann, B.; Ferro, S.; Gasteiger, E.; Huang, H.; Lopez, R.; Magrane, M.; Martin, M.J.; Mazumder, R.; O’Donovan, C.; Redaschi, N.; Suzek, B. The Universal Protein Resource (UniProt): An expanding universe of protein information. Nucleic. Acids. Res 2006, 34, D187–D191. [Google Scholar]
- Chayen, N.E. Tackling the bottleneck of protein crystallization in the post-genomic era. Trends Biotechnol 2002, 20, 98. [Google Scholar]
- Malkin, A.J.; Thorne, R.E. Growth and disorder of macromolecular crystals: Insights from atomic force microscopy and X-ray diffraction studies. Methods 2004, 34, 273–299. [Google Scholar]
- Pusey, M.L.; Liu, Z.J.; Tempel, W.; Praissman, J.; Lin, D.; Wang, B.C.; Gavira, J.A.; Ng, J.D. Life in the fast lane for protein crystallization and X-ray crystallography. Prog. Biophys. Mol. Biol 2005, 88, 359–386. [Google Scholar]
- Caffrey, M. Membrane protein crystallization. J. Struct. Biol 2003, 142, 108–132. [Google Scholar]
- Zhang, C.Y.; Yin, D.C.; Lu, Q.Q.; Guo, Y.Z.; Guo, W.H.; Wang, X.K.; Li, H.S.; Lu, H.M.; Ye, Y.J. Cycling temperature strategy: A method to improve the efficiency of crystallization condition screening of proteins. Cryst. Growth Des 2008, 8, 4227–4232. [Google Scholar]
- Lu, H.M.; Yin, D.C.; Li, H.S.; Geng, L.Q.; Zhang, C.Y.; Lu, Q.Q.; Guo, Y.Z.; Guo, W.H.; Shang, P.; Wakayama, N.I. A containerless levitation setup for liquid processing in a superconducting magnet. Rev. Sci. Instrum 2008, 79, 093903. [Google Scholar]
- McPherson, A. Introduction to protein crystallization. Methods 2004, 34, 254–265. [Google Scholar]
- Wang, X.K.; Yin, D.C.; Zhang, C.Y.; Lu, Q.Q.; Guo, Y.Z.; Guo, W.H. Effect of temperature programmes on protein crystallisation. Cryst. Res. Technol 2010, 45, 479–489. [Google Scholar]
- Lu, Q.Q.; Yin, D.C.; Chen, R.Q.; Xie, S.X.; Liu, Y.M.; Zhang, X.F.; Zhu, L.; Liu, Z.T.; Shang, P. Replacing a reservoir solution with desiccant in vapor diffusion protein crystallization screening. J. Appl. Crystallogr 2010, 43, 1021–1026. [Google Scholar]
- Lu, Q.Q.; Yin, D.C.; Liu, Y.M.; Wang, X.K.; Yang, P.F.; Liu, Z.T.; Shang, P. Effect of mechanical vibration on protein crystallization. J. Appl. Crystallogr 2010, 43, 473–482. [Google Scholar]
- Guo, Y.Z.; Yin, D.C.; Lu, Q.Q.; Wang, X.K.; Liu, J. Enhancement of nucleation during hanging drop protein crystallization using HF Treatment of cover glasses. Cryst. Res. Technol 2010, 45, 158–166. [Google Scholar]
- Tung, M.; Gallagher, D.T. The Biomolecular Crystallization Database Version 4: Expanded content and new features. Acta Crystallogr. D Biol. Crystallogr 2009, 65, 18–23. [Google Scholar]
- Charles, M.; Veesler, S.; Bonnete, F. MPCD: A new interactive on-line crystallization data bank for screening strategies. Acta Crystallogr. D 2006, 62, 1311–1318. [Google Scholar]
- Meining, W. XtalBase—A comprehensive data management system for macromolecular crystallography. J. Appl. Crystallogr 2006, 39, 759–766. [Google Scholar]
- the Biological Macromolecule Crystallization Database (BMCD). http://xpdb.nist.gov:8060/BMCD4.
- the Protein Data Bank (PDB). http://www.rcsb.org/.
- Jaroszewski, L.; Slabinski, L.; Wooley, J.; Deacon, A.M.; Lesley, S.A.; Wilson, I.A.; Godzik, A. Genome pool strategy for structural coverage of protein families. Structure 2008, 16, 1659–1667. [Google Scholar]
- Anfinsen, C.B. Principles that govern the folding of protein chains. Science 1973, 181, 223–230. [Google Scholar]
- Rost, B. Protein structure prediction in 1D, 2D, and 3D. In Encyclopedia of Computational Chemistry; Rague-Schleyer, P.V., Allinger, N.L., Clark, T., Gasteiger, J., Kollman, P.A., Schaefer, H.F., Eds.; John Wiley: Sussex, UK, 1998; pp. 2242–2255. [Google Scholar]
- Latek, D.; Ekonomiuk, D.; Kolinski, A. Protein structure prediction: Combining de novo modeling with sparse experimental data. J. Comput. Chem 2007, 28, 1668–1676. [Google Scholar]
- Kundrotas, P.J.; Alexov, E. Predicting 3D structures of transient protein-protein complexes by homology. Biochim. Biophys. Acta 2006, 1764, 1498–1511. [Google Scholar]
- Kinjo, A.R. Protein secondary structure prediction. Tanpakushitsu Kakusan Koso 2007, 52, 50–56. [Google Scholar]
- Smialowski, P.; Martin-Galiano, A.J.; Mikolajka, A.; Girschick, T.; Holak, T.A.; Frishman, D. Protein solubility: Sequence based prediction and experimental verification. Bioinformatics 2007, 23, 2536–2542. [Google Scholar]
- Schneider, M.; Tognolli, M.; Bairoch, A. The Swiss-Prot protein knowledgebase and ExPASy: Providing the plant community with high quality proteomic data and tools. Plant Physiol. Bioch 2004, 42, 1013–1021. [Google Scholar]
- Lu, H.M.; Yin, D.C.; Ye, Y.J.; Luo, H.M.; Geng, L.Q.; Li, H.S.; Guo, W.H.; Shang, P. Correlation between protein sequence similarity and X-ray diffraction quality in the protein data bank. Protein Peptide Lett 2009, 16, 50–55. [Google Scholar]
- Mount, D.W. Using the Basic and Local Alignment Search Tool (BLAST). CSH Protoc 2007, 2007. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol 1990, 215, 403–410. [Google Scholar]
- Baker, D.; Sali, A. Protein structure prediction and structural genomics. Science 2001, 294, 93–96. [Google Scholar]
- Sanchez, R.; Sali, A. Large-scale protein structure modeling of the Saccharomyces cerevisiae genome. Proc. Natl. Acad. Sci. USA 1998, 95, 13597–13602. [Google Scholar]
- Koehl, P.; Levitt, M. A brighter future for protein structure prediction. Nat. Struct. Biol 1999, 6, 108–111. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| LSSC datasets | Number of clusters | Amount of entries | Average size of each cluster |
|---|---|---|---|
| LSSC30 | 173 | 3,921 | 22.7 |
| LSSC40 | 144 | 3,006 | 20.9 |
| LSSC50 | 122 | 2,433 | 19.9 |
| LSSC60 | 105 | 2,068 | 19.7 |
| LSSC70 | 87 | 1,757 | 20.2 |
| LSSC80 | 81 | 1,586 | 19.6 |
| LSSC90 | 70 | 1,340 | 19.1 |
| Datasets | VARtotal | Group number under VARtotal | Group size | Proportion of lower VARj than VARtotal |
|---|---|---|---|---|
| LSSC30 | 0.109 | 132 | 173 | 76.3% |
| LSSC60 | 0.111 | 81 | 105 | 77.1% |
| LSSC90 | 0.097 | 53 | 70 | 75.7% |
| RAN30 | 0.099 | 93 | 173 | 53.8% |
| RAN60 | 0.101 | 58 | 105 | 55.2% |
| RAN90 | 0.098 | 38 | 70 | 54.3% |
© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Lu, H.-M.; Yin, D.-C.; Liu, Y.-M.; Guo, W.-H.; Zhou, R.-B. Correlation between Protein Sequence Similarity and Crystallization Reagents in the Biological Macromolecule Crystallization Database. Int. J. Mol. Sci. 2012, 13, 9514-9526. https://doi.org/10.3390/ijms13089514
Lu H-M, Yin D-C, Liu Y-M, Guo W-H, Zhou R-B. Correlation between Protein Sequence Similarity and Crystallization Reagents in the Biological Macromolecule Crystallization Database. International Journal of Molecular Sciences. 2012; 13(8):9514-9526. https://doi.org/10.3390/ijms13089514
Chicago/Turabian StyleLu, Hui-Meng, Da-Chuan Yin, Yong-Ming Liu, Wei-Hong Guo, and Ren-Bin Zhou. 2012. "Correlation between Protein Sequence Similarity and Crystallization Reagents in the Biological Macromolecule Crystallization Database" International Journal of Molecular Sciences 13, no. 8: 9514-9526. https://doi.org/10.3390/ijms13089514