Recent Advances on the Neuroprotective Potential of Antioxidants in Experimental Models of Parkinson’s Disease



Abstract
:1. Introduction
2. PD and ROS
3. Role of Neuroprotective Antioxidant Compounds and Recent Discoveries in Experimental Models of PD
4. Antioxidant Compounds in Experimental Models of PD
5. Conclusions
Acknowledgments
References
- Przedborski, S.; Ischiropoulos, H. Reactive oxygen and nitrogen species: Weapons of neuronal destruction in models of Parkinson’s disease. Antioxid. Redox Signal 2005, 7, 685–693. [Google Scholar]
- Schapira, A.H. Pathogenesis of Parkinson’s disease. Baillieres Clin. Neurol 1997, 6, 15–36. [Google Scholar]
- Olanow, C.W.; Tatton, W.G. Etiology and pathogenesis of Parkinson’s disease. Annu. Rev. Neurosci 1999, 22, 123–144. [Google Scholar]
- Jankovic, J.; Tolosa, E. Parkinson’s Disease and Movement Disorders; Williams & Wilkins: Baltimore, MD, USA, 1998; pp. 67–103. [Google Scholar]
- Retz, W.; Gsell, W.; Münch, G.; Rosler, M.; Riederer, P. Free radicals in Alzheimer’s disease. J. Neural. Transm. Suppl 1998, 54, 221–236. [Google Scholar]
- Pappolla, M.A.; Chyan, Y.-J.; Poeggeler, B.; Bozner, P.; Ghiso, J.; LeDoux, S.P.; Wilson, G.L. Alzheimer β protein mediated oxidative damage of mitochondrial DNA: Prevention by melatonin. J. Pineal Res 1999, 27, 226–229. [Google Scholar]
- Sofic, E.; Lange, K.W.; Jellinger, K.; Riederer, P. Reduced and oxidized glutathione in the substantia nigra of patients with Parkinson’s disease. Neurosci. Lett 1992, 142, 128–130. [Google Scholar]
- Saggu, H.; Cooksey, J.; Dexter, D.; Wells, F.R.; Lees, A.; Jenner, P.; Marsden, C.D. A selective increase in particulate superoxide dismutase activity in parkinsonian substantia nigra. J. Neurochem 1989, 53, 692–697. [Google Scholar]
- Dexter, D.T.; Wells, F.R.; Lees, A.J.; Agid, F.; Agid, Y.; Jenner, P.; Marsden, C.D. Increased nigral iron content and alterations in other metal ions occurring in brain in Parkinson’s disease. J. Neurochem 1989, 52, 1830–1836. [Google Scholar]
- Beal, M.F. Mitochondrial dysfunction and oxidative damage in Alzheimer’s and Parkinson’s diseases and coenzyme Q10 as a potential treatment. J. Bioenerg. Biomembr 2004, 36, 381–386. [Google Scholar]
- Graham, D.G. Oxidative pathways for catecholamines in the genesis of neuromelanin and cytotoxic quinones. Mol. Pharmacol 1978, 14, 633–643. [Google Scholar]
- Weiner, W.J. Is levodopa toxic? Arch. Neurol 2000, 57, 408–410. [Google Scholar]
- Shulman, L.M. Levodopa toxicity in Parkinson disease: Reality or myth? Reality-practice patterns should change. Arch. Neurol 2000, 57, 406–408. [Google Scholar]
- Fahn, S. Levodopa-induced neurotoxicity: Does it represent a problem for the treatment of Parkinson’s disease? CNS Drug 1997, 8, 376–393. [Google Scholar]
- Smith, T.S.; Parker, W.D., Jr; Bennett, J.P., Jr. l-dopa increases nigral production of hydroxyl radicals in vivo: Potential l-dopa toxicity? Neuroreport 1994, 5, 1009–1011. [Google Scholar]
- Ogawa, N.; Asanuma, M.; Kondo, Y.; Kawada, Y.; Yamamoto, M.; Mori, A. Differential effects of chronic l-dopa treatment on lipid peroxidation in the mouse brain with or without pretreatment with 6-hydroxydopamine. Neurosci. Lett 1994, 171, 55–58. [Google Scholar]
- Spencer, J.P.; Jenner, A.; Aruoma, O.I.; Evans, P.J.; Kaur, H.; Dexter, D.T.; Jenner, P.; Lees, A.J.; Marsden, D.C.; Halliwell, B. Intense oxidative DNA damage promoted by l-dopa and its metabolites. Implications for neurodegenerative disease. FEBS Lett 1994, 353, 246–250. [Google Scholar]
- Uttara, B.; Singh, A.V.; Zamboni, P.; Mahajan, R.T. Oxidative stress and neurodegenerative diseases: A review of upstream and downstream antioxidant therapeutic options. Curr. Neuropharmacol 2009, 7, 65–74. [Google Scholar]
- Ebadi, M.; Srinivasan, S.K.; Baxi, M.D. Oxidative stress and antioxidant therapy in Parkinson’s disease. Prog. Neurobiol 1996, 48, 1–19. [Google Scholar]
- Floyd, R.A.; Carney, J.M. Free radical damage to protein and DNA: Mechanisms involved and relevant observations on brain undergoing oxidative stress. Ann. Neurol 1992, 32, S22–S27. [Google Scholar]
- Hald, A.; Lotharius, J. Oxidative stress and inflammation in Parkinson’s disease: Is there a causal link? Exp. Neurol 2005, 193, 279–290. [Google Scholar]
- Gutteridge, J.M.; Halliwell, B. Iron toxicity and oxygen radicals. Baillieres Clin. Haematol 1989, 2, 195–256. [Google Scholar]
- Halliwell, B.; Gutteridge, J.M. Oxygen toxicity, oxygen radicals, transition metals and disease. Biochem. J 1984, 219, 1–14. [Google Scholar]
- Schapira, A.H. Mitochondria in the aetiology and pathogenesis of Parkinson’s disease. Lancet Neurol 2008, 7, 97–109. [Google Scholar]
- Schapira, A.H.V.; Cooper, J.M.; Dexter, D.; Jenner, P.; Clark, J.B.; Marsden, C.D. Mitochondrial complex I deficiency in Parkinson’s disease. Lancet 1989, 333, 1269. [Google Scholar]
- Uéda, K.; Fukushima, H.; Masliah, E.; Xia, Y.; Iwai, A.; Yoshimoto, M.; Otero, D.A.; Kondo, J.; Ihara, Y.; Saitoh, T. Molecular cloning of cDNA encoding an unrecognized component of amyloid in Alzheimer disease. Proc. Natl. Acad. Sci. USA 1993, 90, 11282–11286. [Google Scholar]
- Trojanowski, J.Q.; Lee, V.M.-Y. Parkinson’s disease and related α-synucleinopathies are brain amyloidoses. Ann. N. Y. Acad. Sci 2003, 991, 107–110. [Google Scholar]
- Xia, Y.; Saitoh, T.; Uéda, K.; Tanaka, S.; Chen, X.; Hashimoto, M.; Hsu, L.; Conrad, C.; Sundsmo, M.; Yoshimoto, M.; Thal, L.; Katzman, R.; Masliah, E. Characterization of the human alpha-synuclein gene: Genomic structure, transcription start site, promoter region and polymorphisms. J. Alzheimers Dis 2001, 3, 485–494. [Google Scholar]
- McGeer, P.L.; Itagaki, S.; Boyes, B.E.; McGeer, E.G. Reactive microglia are positive for HLA-DR in the substantia nigra of Parkinson’s and Alzheimer Parkinson’s disease brains. Neurology 1988, 38, 1285–1291. [Google Scholar]
- Jonsson, G.; Sachs, C. Actions of 6-hydroxydopamine quinones on catecholamine neurons. J. Neurochem 1975, 25, 509–516. [Google Scholar]
- Ungerstedt, U. 6-Hydroxy-dopamine induced degeneration of central monoamine neurons. Eur. J. Pharmacol 1968, 5, 107–110. [Google Scholar]
- Nicklas, W.J.; Vyas, I.; Heikkila, R.E. Inhibition of NADH-linked oxidation in brain mitochondria by 1-methyl-4-phenyl-pyridine, a metabolite of the neurotoxin, 1-methyl-4-phenyl-1,2,5,6-tetrahydropyridine. Life Sci 1985, 36, 2503–2508. [Google Scholar]
- Hasegawa, E.; Takeshige, K.; Oishi, T.; Murai, Y.; Minakami, S. 1-Methyl-4-phenylpyridinium (MPP+) induces NADH-dependent superoxide formation and enhances NADH-dependent lipid peroxidation in bovine heart submitochondrial particles. Biochem. Biophys. Res. Commun 1990, 170, 1049–1055. [Google Scholar]
- Cleeter, M.W.; Cooper, J.M.; Schapira, A.H. Irreversible inhibition of mitochondrial complex I by 1-methyl-4-phenylpyridinium: Evidence for free radical involvement. J. Neurochem 1992, 58, 786–789. [Google Scholar]
- Liou, H.H.; Tsai, M.C.; Chen, C.J.; Jeng, J.S.; Chang, Y.C.; Chen, S.Y.; Chen, R.C. Environmental risk factors and Parkinson’s disease: A case-control study in Taiwan. Neurology 1997, 48, 1583–1588. [Google Scholar]
- Meco, G.; Bonifati, V.; Vanacore, N.; Fabrizio, E. Parkinsonism after chronic exposure to the fungicide maneb (manganese ethylene-bis-dithiocarbamate). Scand. J. Work Environ. Health 1994, 20, 301–305. [Google Scholar]
- Hertzman, C.; Wiens, M.; Bowering, D.; Snow, B.; Calne, D. Parkinson’s disease: A case-control study of occupational and environmental risk factors. Am. J. Ind. Med 1990, 17, 349–355. [Google Scholar]
- Savitt, J.M.; Dawson, V.L.; Dawson, T.M. Diagnosis and treatment of Parkinson disease: Molecules to medicine. J. Clin. Invest 2006, 116, 1744–1754. [Google Scholar]
- Thomas, B.; Beal, M.F. Parkinson’s disease. Hum. Mol. Genet 2007, 16, R183–R194. [Google Scholar]
- Banerjee, R.; Starkov, A.A.; Beal, M.F.; Thomas, B. Mitochondrial dysfunction in the limelight of Parkinson’s disease pathogenesis. Biochim. Biophys. Acta 2009, 1792, 651–663. [Google Scholar]
- Moreira, E.L.G.; Rial, D.; Aguiar, A.S., Jr; Figueiredo, C.P.; Siqueira, J.M.; DalBó, S.; Horst, H.; de Oliveira, J.; Mancini, G.; dos Santos, T.S.; et al. Proanthocyanidin-rich fraction from Croton celtidifolius Baill confers neuroprotection in the intranasal 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine rat model of Parkinson’s disease. J. Neural Transm. 2010, 117, 1337–1351. [Google Scholar]
- Zhou, C.; Huang, Y.; Przedborski, S. Oxidative stress in Parkinson’s disease: A mechanism of pathogenic and therapeutic significance. Ann. N. Y. Acad. Sci 2008, 1147, 93–104. [Google Scholar]
- Fukae, J.; Mizuno, Y.; Hattori, N. Mitochondrial dysfunction in Parkinson’s disease. Mitochondrion 2007, 7, 58–62. [Google Scholar]
- Zhu, B.T. CNS dopamine oxidation and catechol-O-methyltransferase: Importance in the etiology, pharmacotherapy, and dietary prevention of Parkinson’s disease. Int. J. Mol. Med 2004, 13, 343–353. [Google Scholar]
- Chaturvedi, R.K.; Shukla, S.; Seth, K.; Chauhan, S.; Sinha, C.; Shukla, Y.; Agrawal, A.K. Neuroprotective and neurorescue effect of black tea extract in 6-hydroxydopamine-lesioned rat model of Parkinson’s disease. Neurobiol. Dis 2006, 22, 421–434. [Google Scholar]
- Paraskevas, G.P.; Kapaki, E.; Petropoulou, O.; Anagnostouli, M.; Vagenas, V.; Papageorgiou, C. Plasma levels of antioxidant vitamins C and E are decreased in vascular parkinsonism. J. Neurol. Sci 2003, 215, 51–55. [Google Scholar]
- Kelloff, G.J.; Crowell, J.A.; Steele, V.E.; Lubet, R.A.; Malone, W.A.; Boone, C.W.; Kopelovich, L.; Hawk, E.T.; Lieberman, R.; Lawrence, J.A.; et al. Progress in cancer chemoprevention: Development of diet-derived chemopreventive agents. J. Nutr 2000, 130, 467S–471S. [Google Scholar]
- Goel, A.; Kunnumakkara, A.B.; Aggarwal, B.B. Curcumin as “Curecumin”: From kitchen to clinic. Biochem. Pharmacol 2008, 75, 787–809. [Google Scholar]
- Liu, F.; Fung, M.C.; Ooi, V.E.C.; Chang, S.T. Induction in the mouse of gene expression of immunomodulating cytokines by mushroom polysaccharide-protein complexes. Life Sci 1996, 58, 1795–1803. [Google Scholar]
- Sreejayana, N.; Rao, M.N.A.; Priyadarsini, K.I.; Devasagayam, T.P.A. Inhibition of radiation-induced lipid peroxidation by curcumin. Int. J. Pharm 1997, 151, 127–130. [Google Scholar]
- Wei, Q.Y.; Chen, W.F.; Zhou, B.; Yang, L.; Liu, Z.L. Inhibition of lipid peroxidation and protein oxidation in rat liver mitochondria by curcumin and its analogues. Biochim. Biophys. Acta 2006, 1760, 70–77. [Google Scholar]
- Cole, G.M.; Teter, B.; Frautschy, S.A. Neuroprotective effects of curcumin. Adv. Exp. Med. Biol 2007, 595, 197–212. [Google Scholar]
- Thomas, P.; Wang, Y.J.; Zhong, J.-H.; Kosaraju, S.; O’Callaghan, N.J.; Zhou, X.-F.; Fenech, M. Grape seed polyphenols and curcumin reduce genomic instability events in a transgenic mouse model for Alzheimer’s disease. Mutat. Res.-Fund Mol. Mech 2009, 661, 25–34. [Google Scholar]
- Ma, Q.L.; Yang, F.; Rosario, E.R.; Ubeda, O.J.; Beech, W.; Gant, D.J.; Chen, P.P.; Hudspeth, B.; Chen, C.; Zhao, Y.; et al. β-amyloid oligomers induce phosphorylation of tau and inactivation of insulin receptor substrate via c-Jun N-terminal kinase signaling: Suppression by omega-3 fatty acids and curcumin. J. Neurosci 2009, 29, 9078–9089. [Google Scholar]
- Rajeswari, A.; Sabesan, M. Inhibition of monoamine oxidase-B by the polyphenolic compound, curcumin and its metabolite tetrahydrocurcumin, in a model of Parkinson’s disease induced by MPTP neurodegeneration in mice. Inflammopharmacology 2008, 16, 96–99. [Google Scholar]
- Jagatha, B.; Mythri, R.B.; Vali, S.; Bharath, M.M. Curcumin treatment alleviates the effects of glutathione depletion in vitro and in vivo: Therapeutic implications for Parkinson’s disease explained via in silico studies. Free Radic. Biol. Med 2008, 44, 907–917. [Google Scholar]
- Mythri, R.B.; Veena, J.; Harish, G.; Rao, B.S.S.; Bharath, M.M.S. Chronic dietary supplementation with turmeric protects against 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-mediated neurotoxicity in vivo: Implications for Parkinson’s disease. Br. J. Nutr 2011, 106, 63–72. [Google Scholar]
- Liu, Z.; Yu, Y.; Li, X.; Ross, C.A.; Smith, W.W. Curcumin protects against A53T alpha-synuclein-induced toxicity in a PC12 inducible cell model for Parkinsonism. Pharmacol. Res 2011, 63, 439–444. [Google Scholar]
- Kawanishi, S.; Oikawa, S.; Murata, M. Evaluation for safety of antioxidant chemopreventive agents. Antioxid. Redox Signal 2005, 7, 1728–1739. [Google Scholar]
- Shan, B.E.; Wang, M.X.; Li, R.Q. Quercetin inhibit human SW480 colon cancer growth in association with inhibition of cyclin D1 and survivin expression through Wnt/β-catenin signaling pathway. Cancer Invest 2009, 27, 604–612. [Google Scholar]
- Choi, E.J.; Lee, B.H.; Lee, K.; Chee, K.M. Long-term combined administration of quercetin and daidzein inhibits quercetin-induced suppression of glutathione antioxidant defenses. Food Chem. Toxicol 2005, 43, 793–798. [Google Scholar]
- Kaur, R.; Chopra, K.; Singh, D. Role of α2 receptors in quercetin-induced behavioral despair in mice. J. Med. Food 2007, 10, 165–168. [Google Scholar]
- Ruiz, P.A.; Braune, A.; Holzlwimmer, G.; Quintanilla-Fend, L.; Haller, D. Quercetin inhibits TNF-induced NF-κB transcription factor recruitment to proinflammatory gene promoters in murine intestinal epithelial cells. J. Nutr 2007, 137, 1208–1215. [Google Scholar]
- Zhang, Z.-J.; Cheang, L.C.V.; Wang, M.-W.; Li, G.H.; Chu, I.K.; Lin, Z.-X.; Lee, S.M.Y. Ethanolic extract of fructus Alpinia oxyphylla protects against 6-hydroxydopamine-induced damage of PC12 cells in vitro and dopaminergic neurons in zebrafish. Cell. Mol. Neurobiol 2012, 32, 27–40. [Google Scholar]
- Pandey, A.K.; Verma, S.; Bhattacharya, P.; Paul, S.; Mishra, A.; Patnaik, R. An in-silico strategy to explore neuroprotection by quercetin in cerebral ischemia: A novel hypothesis based on inhibition of matrix metalloproteinase (MMPs) and acid sensing ion channel 1a (ASIC1a). Med. Hypotheses 2012, 79, 76–81. [Google Scholar]
- Hilliard, J.J.; Krause, H.M.; Bernstein, J.I.; Fernandez, J.A.; Nguyen, V.; Ohemeng, K.A.; Barrett, J.F. A comparison of active site binding of 4-quinolones and novel flavone gyrase inhibitors to DNA gyrase. Adv. Exp. Med. Biol 1995, 390, 59–69. [Google Scholar]
- Noack, H.; Kube, U.; Augustin, W. Relations between tocopherol depletion and coenzyme Q during lipid peroxidation in rat liver mitochondria. Free Radic. Res 1994, 20, 375–386. [Google Scholar]
- Forsmark-Andrée, P.; Lee, C.P.; Dallner, G.; Ernster, L. Lipid peroxidation and changes in the ubiquinone content and the respiratory chain enzymes of submitochondrial particles. Free Radic. Biol. Med 1997, 22, 391–400. [Google Scholar]
- Battino, M.; Littarru, G.P.; Gorini, A.; Villa, R.F. Coenzyme Q, peroxidation and cytochrome oxidase features after Parkinson’s-like disease by MPTP toxicity in intra-synaptic and non-synaptic mitochondria from Macaca fascicularis cerebral cortex and hippocampus: Action of dihydroergocriptine. Neurochem. Res 1996, 21, 1505–1514. [Google Scholar]
- Chaturvedi, R.K.; Beal, M.F. Mitochondrial approaches for neuroprotection. Ann. N. Y. Acad. Sci 2008, 1147, 395–412. [Google Scholar]
- Mancuso, M.; Orsucci, D.; Volpi, L.; Calsolaro, V.; Siciliano, G. Coenzyme Q10 in neuromuscular and neurodegenerative disorders. Curr. Drug Targets 2010, 11, 111–121. [Google Scholar]
- Young, A.J.; Johnson, S.; Steffens, D.C.; Doraiswamy, P.M. Coenzyme Q10: A review of its promise as a neuroprotectant. CNS Spectr 2007, 12, 62–68. [Google Scholar]
- Ostrowski, R.P. Effect of coenzyme Q(10) on biochemical and morphological changes in experimental ischemia in the rat brain. Brain Res. Bull 2000, 53, 399–407. [Google Scholar]
- Yoshida, Y.; Hayakawa, M.; Habuchi, Y.; Niki, E. Evaluation of the dietary effects of coenzyme Q in vivo by the oxidative stress marker, hydroxyoctadecadienoic acid and its stereoisomer ratio. Biochim. Biophys. Acta 2006, 1760, 1558–1568. [Google Scholar]
- Cleren, C.; Yang, L.; Lorenzo, B.; Calingasan, N.Y.; Schomer, A.; Sireci, A.; Wille, E.J.; Beal, M.F. Therapeutic effects of coenzyme Q10 (CoQ10) and reduced CoQ10 in the MPTP model of Parkinsonism. J. Neurochem 2008, 104, 1613–1621. [Google Scholar]
- Shults, C.W.; Oakes, D.; Kieburtz, K.; Beal, M.F.; Haas, R.; Plumb, S.; Juncos, J.L.; Nutt, J.; Shoulson, I.; Carter, J.; et al. Effects of coenzyme Q10 in early Parkinson disease: Evidence of slowing of the functional decline. Arch. Neurol 2002, 59, 1541–1550. [Google Scholar]
- Yang, L.; Calingasan, N.Y.; Wille, E.J.; Cormier, K.; Smith, K.; Ferrante, R.J.; Beal, M.F. Combination therapy with coenzyme Q10 and creatine produces additive neuroprotective effects in models of Parkinson’s and Huntington’s diseases. J. Neurochem 2009, 109, 1427–1439. [Google Scholar]
- Beal, M.F.; Shults, C.W. Effects of Coenzyme Q10 in Huntington’s disease and early Parkinson’s disease. Biofactors 2003, 18, 153–161. [Google Scholar]
- Jang, M.; Cai, L.; Udeani, G.O.; Slowing, K.V.; Thomas, C.F.; Beecher, C.W.; Fong, H.H.; Farnsworth, N.R.; Kinghorn, A.D.; Mehta, R.G.; et al. Cancer chemopreventive activity of resveratrol, a natural product derived from grapes. Science 1997, 275, 218–220. [Google Scholar]
- Soleas, G.J.; Diamandis, E.P.; Goldberg, D.M. Resveratrol: A molecule whose time has come? And gone? Clin. Biochem 1997, 30, 91–113. [Google Scholar]
- Surh, Y. Molecular mechanisms of chemopreventive effects of selected dietary and medicinal phenolic substances. Mutat. Res 1999, 428, 305–327. [Google Scholar]
- Wang, Y.; Xu, H.; Fu, Q.; Ma, R.; Xiang, J. Protective effect of resveratrol derived from Polygonum cuspidatum and its liposomal form on nigral cells in parkinsonian rats. J. Neurol. Sci 2011, 304, 29–34. [Google Scholar]
- Boocock, D.J.; Faust, G.E.S.; Patel, K.R.; Schinas, A.M.; Brown, V.A.; Ducharme, M.P.; Booth, T.D.; Crowell, J.A.; Perloff, M.; Gescher, A.J.; et al. Phase I dose escalation pharmacokinetic study in healthy volunteers of resveratrol, a potential cancer chemopreventive agent. Cancer Epidemiol. Biomarker. Prev 2007, 16, 1246–1252. [Google Scholar]
- Van Zanden, J.J.; Geraets, L.; Wortelboer, H.M.; van Bladeren, P.J.; Rietjens, I.M.C.M.; Cnubben, N.H.P. Structural requirements for the flavonoid-mediated modulation of glutathione S-transferase P1-1 and GS-X pump activity in MCF7 breast cancer cells. Biochem. Pharmacol 2004, 67, 1607–1617. [Google Scholar]
- Zhao, G.; Yao-Yue, C.; Qin, G.W.; Guo, L.-H. Luteolin from Purple Perilla mitigates ROS insult particularly in primary neurons. Neurobiol. Aging 2012, 33, 176–186. [Google Scholar]
- Park, K.-H.; Yokota, T.; Sakurai, A.; Takahashi, N. Occurence of castasterone, brassinolide and methyl 4-chloroindole-3-acetate in immature Vicia faba seeds. Agric. Biol. Chem 1987, 51, 3081–3086. [Google Scholar]
- Ikekawa, N.; Nishiyama, F.; Fujimoto, Y. Identification of 24-epibrassinolide in bee pollen of the broad bean, Vicia faba L. Chem. Pharm. Bull 1988, 36, 405–407. [Google Scholar]
- Mazorra, L.M.; Núňez, M.; Hechavarria, M.; Coll, F.; Sánchez-Blanco, M.J. Influence of brassinosteroids on antioxidant enzymes activity in tomato under different temperatures. Biol. Plant 2002, 45, 593–596. [Google Scholar]
- Ali, B.; Hasan, S.A.; Hayat, S.; Hayat, Q.; Yadav, S.; Fariduddin, Q.; Ahmad, A. A role for brassinosteroids in the amelioration of aluminium stress through antioxidant system in mung bean (Vigna radiata L. Wilczek). Environ. Exp. Bot 2008, 62, 153–159. [Google Scholar]
- Arora, N.; Bhardwaj, R.; Sharma, P.; Arora, H.K. Effect of 28-homobrassinolide on growth, lipid peroxidation and antioxidative enzyme activities in seedlings of Zea mays L. under salinity stress. Acta Physiol. Plant 2008, 30, 833–839. [Google Scholar]
- Ismaili, J.; Boisvert, M.; Longpre, F.; Carange, J.; Le Gall, C.; Martinoli, M.-G.; Daoust, B. Brassinosteroids and analogs as neuroprotectors: Synthesis and structure-activity relationships. Steroids 2012, 77, 91–99. [Google Scholar]
- Geromel, V.; Darin, N.; Chrétien, D.; Bénit, P.; DeLonlay, P.; Rötig, A.; Munnich, A.; Rustin, P. Coenzyme Q(10) and idebenone in the therapy of respiratory chain diseases: Rationale and comparative benefits. Mol. Genet. Metab 2002, 77, 21–30. [Google Scholar]
- Gerhardt, E.; Gräber, S.; Szegő, É.M.; Moisoi, N.; Martins, L.M.; Outeiro, T.F.; Kermer, P. Idebenone and resveratrol extend lifespan and improve motor function of HtrA2 knockout mice. PLoS One 2011, 6, e28855. [Google Scholar]
- Koo, U.; Nam, K.-W.; Ham, A.; Lyu, D.; Kim, B.; Lee, S.-J.; Kim, K.H.; Oh, K.-B.; Mar, W.; Shin, J. Neuroprotective effects of 3alpha-acetoxyeudesma-1,4(15),11(13)-trien-12,6alpha-olide against dopamine-induced apoptosis in the human neuroblastoma SH-SY5Y cell line. Neurochem. Res 2011, 36, 1991–2001. [Google Scholar]
- Rojas, P.; Serrano-García, N.; Medina-Campos, O.N.; Pedraza-Chaverri, J.; Maldonado, P.D.; Ruiz-Sánchez, E. S-Allylcysteine, a garlic compound, protects against oxidative stress in 1-methyl-4-phenylpyridinium-induced parkinsonism in mice. J. Nutr. Biochem 2011, 22, 937–944. [Google Scholar]
- Lopes, F.M.; Londero, G.F.; de Medeiros, L.M.; da Motta, L.L.; Behr, G.A.; de Oliveira, V.A.; Ibrahim, M.; Moreira, J.C.F.; de Oliveira Porciuncula, L.; da Rocha, J.B.; et al. Evaluation of the neurotoxic/neuroprotective role of organoselenides using differentiated human neuroblastoma SH-SY5Y cell line challenged with 6-hydroxydopamine. Neurotox. Res 2012, 22, 138–149. [Google Scholar]
- Müller, A.; Cadenas, E.; Graf, P.; Sies, H. A novel biologically active seleno-organic compound-I. Glutathione peroxidase-like activity in vitro and antioxidant capacity of PZ 51 (Ebselen). Biochem. Pharmacol 1984, 33, 3235–3239. [Google Scholar]
- Bhabak, K.P.; Mugesh, G. Synthesis, characterization, and antioxidant activity of some ebselen analogues. Chemistry 2007, 13, 4594–4601. [Google Scholar]
- Porciuncúla, L.O.; Rocha, J.B.T.; Cimarosti, H.; Vinadé, L.; Ghisleni, G.; Salbego, C.G.; Souza, D.O. Neuroprotective effect of ebselen on rat hippocampal slices submitted to oxygen-glucose deprivation: Correlation with immunocontent of inducible nitric oxide synthase. Neurosci. Lett 2003, 346, 101–104. [Google Scholar]
- Ardais, A.P.; Viola, G.G.; Costa, M.S.; Nunes, F.; Behr, G.A.; Klamt, F.; Moreira, J.C.F.; Souza, D.O.; Rocha, J.B.T; Porciuncula, L.O. Acute treatment with diphenyl diselenide inhibits glutamate uptake into rat hippocampal slices and modifies glutamate transporters, SNAP-25, and GFAP immunocontent. Toxicol. Sci. 2010, 113, 434–443. [Google Scholar]
- Posser, T.; Franco, J.L.; dos Santos, D.A.; Rigon, A.P.; Farina, M.; Dafré, A.L.; Teixeira Rocha, J.B.; Leal, R.B. Diphenyl diselenide confers neuroprotection against hydrogen peroxide toxicity in hippocampal slices. Brain Res 2008, 1199, 138–147. [Google Scholar]
- Nakaso, K.; Nakamura, C.; Sato, H.; Imamura, K.; Takeshima, T.; Nakashima, K. Novel cytoprotective mechanism of anti-parkinsonian drug deprenyl: PI3K and Nrf2-derived induction of antioxidative proteins. Biochem. Biophys. Res. Commun 2006, 339, 915–922. [Google Scholar]
- Carrillo, M.-C.; Kanai, S.; Nokubo, M.; Kitani, K. (−) deprenyl induces activities of both superoxide dismutase and catalase but not of glutathione peroxidase in the striatum of young male rats. Life Sci. 1991, 48, 517–521. [Google Scholar]
- Xiao, H.; Lv, F.; Xu, W.; Zhang, L.; Jing, P.; Cao, X. Deprenyl prevents MPP+-induced oxidative damage in PC12 cells by the upregulation of Nrf2-mediated NQO1 expression through the activation of PI3K/Akt and Erk. Toxicology 2011, 290, 287–295. [Google Scholar]
- Liu, X.-H.; Xin, H.; Hou, A.-J.; Zhu, Y.-Z. Protective effects of leonurine in neonatal rat hypoxic cardiomyocytes and rat infarcted heart. Clin. Exp. Pharmacol. Physiol 2009, 36, 696–703. [Google Scholar]
- Loh, K.P.; Huang, S.H.; Tan, B.K.H.; Zhu, Y.Z. Cerebral protection of purified Herba Leonuri extract on middle cerebral artery occluded rats. J. Ethnopharmacol 2009, 125, 337–343. [Google Scholar]
- Loh, K.P.; Qi, J.; Tan, B.K.H.; Liu, X.H.; Wei, B.G.; Zhu, Y.Z. Leonurine protects middle cerebral artery occluded rats through antioxidant effect and regulation of mitochondrial function. Stroke 2010, 41, 2661–2668. [Google Scholar]
- Sun, J.; Huang, S.H.; Zhu, Y.C.; Whiteman, M.; Wang, M.J.; Tan, B.K.-H.; Zhu, Y.Z. Anti-oxidative stress effects of Herba leonuri on ischemic rat hearts. Life Sci 2005, 76, 3043–3056. [Google Scholar]
- Shi, X.R.; Hong, Z.Y.; Liu, H.R.; Zhang, Y.C.; Zhu, Y.Z. Neuroprotective effects of SCM198 on 6-hydroxydopamine-induced behavioral deficit in rats and cytotoxicity in neuronal SH-SY5Y cells. Neurochem. Int 2011, 58, 851–860. [Google Scholar]
- Jeding, I.; Evans, P.J.; Akanmu, D.; Dexter, D.; Spencer, J.D.; Aruoma, O.I.; Jenner, P.; Halliwell, B. Characterization of the potential antioxidant and pro-oxidant actions of some neuroleptic drugs. Biochem. Pharmacol 1995, 49, 359–365. [Google Scholar]
- Hajieva, P.; Mocko, J.B.; Moosmann, B.; Behl, C. Novel imine antioxidants at low nanomolar concentrations protect dopaminergic cells from oxidative neurotoxicity. J. Neurochem 2009, 110, 118–132. [Google Scholar]
- Mocko, J.B.; Kern, A.; Moosmann, B.; Behl, C.; Hajieva, P. Phenothiazines interfere with dopaminergic neurodegeneration in Caenorhabditis elegans models of Parkinson’s disease. Neurobiol. Dis 2010, 40, 120–129. [Google Scholar]
- Martin, I.C.A. Implications of phenothiazine side effects: A study of antiparkinsonian agents in an older population. Acta Psychiatr. Scand 1975, 51, 110–118. [Google Scholar]
- Terry, P.M. Side-effects of phenothiazines. Br. Med. J 1967, 2, 55. [Google Scholar]
- Huang, J.-Z.; Chen, Y.-Z.; Su, M.; Zheng, H.-F.; Yang, Y.-P.; Chen, J.; Liu, C.-F. dl-3-n-Butylphthalide prevents oxidative damage and reduces mitochondrial dysfunction in an MPP(+)-induced cellular model of Parkinson’s disease. Neurosci. Lett 2010, 475, 89–94. [Google Scholar]
- Annoura, H.; Nakanishi, K.; Toba, T.; Takemoto, N.; Imajo, S.; Miyajima, A.; Tamura-Horikawa, Y.; Tamura, S. Discovery of (2S)-1-(4-amino-2,3,5-trimethylphenoxy)-3-[4-[4-(4-fluorobenzyl)phenyl]-1-piperazinyl]-2-propanol dimethanesulfonate (SUN N8075): A dual Na(+) and Ca(2+) channel blocker with antioxidant activity. J. Med. Chem 2000, 43, 3372–3376. [Google Scholar]
- Kotani, Y.; Morimoto, N.; Oida, Y.; Tamura, Y.; Tamura, S.; Inoue, T.; Shimazawa, M.; Yoshimura, S.; Iwama, T.; Hara, H. Prevention of in vitro and in vivo acute ischemic neuronal damage by (2S)-1-(4-amino-2,3,5-trimethylphenoxy)-3-{4-[4-(4-fluorobenzyl) phenyl]-1-piperazinyl}-2-propanol dimethanesulfonate (SUN N8075), a novel neuroprotective agent with antioxidant properties. Neuroscience 2007, 149, 779–788. [Google Scholar]
- Oyagi, A.; Oida, Y.; Hara, H.; Izuta, H.; Shimazawa, M.; Matsunaga, N.; Adachi, T. Protective effects of SUN N8075, a novel agent with antioxidant properties, in in vitro and in vivo models of Parkinson’s disease. Brain Res 2008, 1214, 169–176. [Google Scholar]
- Xu, J.; Kao, S.-Y.; Lee, F.J.S.; Song, W.; Jin, L.-W.; Yankner, B.A. Dopamine-dependent neurotoxicity of alpha-synuclein: A mechanism for selective neurodegeneration in Parkinson disease. Nat. Med 2002, 8, 600–606. [Google Scholar]
- Clark, J.; Clore, E.L.; Zheng, K.; Adame, A.; Masliah, E.; Simon, D.K. Oral N-acetyl-cysteine attenuates loss of dopaminergic terminals in alpha-synuclein overexpressing mice. PLoS One 2010, 5, e12333. [Google Scholar]
- Yang, L.; Zhao, K.; Calingasan, N.Y.; Luo, G.; Szeto, H.H.; Beal, M.F. Mitochondria targeted peptides protect against 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine neurotoxicity. Antioxid. Redox Signal 2009, 11, 2095–2104. [Google Scholar]
- García-Palomero, E.; Usán, P.; Pérez-Baz, J.; Fernández, R.I.; Martínez, A.; Fernández, A.; Medina, M. Therapeutic Potential of Potent Marine Neuroprotectants. In New Trends in Alzheimer and Parkinson Related Disorders: ADPD; Hanin, I., Windisch, M., Poewe, W., Fisher, A., Eds.; Medimond S.r.l-Monduzzi Editore International Proceedings Division: Salzburg, Austria, 2007; pp. 289–294. [Google Scholar]
- Mena, M.A.; Casarejos, M.J.; Solano, R.; Rodríguez-Navarro, J.A.; Gómez, A.; Rodal, I.; Medina, M.; de Yebenes, J.G. NP7 protects from cell death induced by oxidative stress in neuronal and glial midbrain cultures from parkin null mice. FEBS Lett 2009, 583, 168–174. [Google Scholar]
- Radad, K.; Gabriele, G.; Rausch, W.D. Short review on dopamine agonists: Insight into clinical and research studies relevant to Parkinson’s disease. Pharmacological. Rep 2005, 57, 701–712. [Google Scholar]
- Schapira, A.H.V. The clinical relevance of levodopa toxicity in the treatment of Parkinson’s disease. Mov. Disord 2008, 23, S515–S520. [Google Scholar]
- Yoshikawa, T.; Minamiyama, Y.; Naito, Y.; Kondo, M. Antioxidant properties of bromocriptine, a dopamine agonist. J. Neurochem 1994, 62, 1034–1038. [Google Scholar]
- Lim, J.H.; Kim, K.-M.; Kim, S.W.; Hwang, O.; Choi, H.J. Bromocriptine activates NQO1 via Nrf2-PI3K/Akt signaling: Novel cytoprotective mechanism against oxidative damage. Pharmacol. Res 2008, 57, 325–331. [Google Scholar]
- Aslam, F.; Khan, A.; Khan, M.Z.; Sharaf, S.; Gul, S.T.; Saleemi, M.K. Toxico-pathological changes induced by cypermethrin in broiler chicks: Their attenuation with Vitamin E and selenium. Exp. Toxicol. Pathol 2010, 62, 441–450. [Google Scholar]
- Zhang, H.; Jia, H.; Liu, J.; Ao, N.; Yan, B.; Shen, W.; Wang, X.; Li, X.; Luo, C. Combined R-alpha-lipoic acid and acetyl-l-carnitine exerts efficient preventative effects in a cellular model of Parkinson’s disease. J. Cell. Mol. Med 2010, 14, 215–225. [Google Scholar]
- Du, T.; Li, L.; Song, N.; Xie, J.; Jiang, H. Rosmarinic acid antagonized 1-methyl-4-phenylpyridinium (MPP+)-induced neurotoxicity in MES23.5 dopaminergic cells. Int. J. Toxicol 2010, 29, 625–633. [Google Scholar]
- Kabuto, H.; Tada, M.; Kohno, M. Eugenol [2-methoxy-4-(2-propenyl)phenol] prevents 6-hydroxydopamine-induced dopamine depression and lipid peroxidation inductivity in mouse striatum. Biol. Pharm. Bull 2007, 30, 423–427. [Google Scholar]
- Tian, L.-L.; Zhou, Z.; Zhang, Q.; Sun, Y.-N.; Li, C.R.; Cheng, C.-H.; Zhong, Z.-Y.; Wang, S.-Q. Protective effect of (+/−) isoborneol against 6-OHDA-induced apoptosis in SH-SY5Y cells. Cell Physiol. Biochem 2007, 20, 1019–1032. [Google Scholar]
- Suzen, S. Recent developments of melatonin related antioxidant compounds. Comb. Chem. High Throughput Screen 2006, 9, 409–419. [Google Scholar]
- Hague, T.; Andrews, P.L.R.; Barker, J.; Naughton, D.P. Dietary chelators as antioxidant enzyme mimetics: Implications for dietary intervention in neurodegenerative diseases. Behav. Pharmacol 2006, 17, 425–430. [Google Scholar]
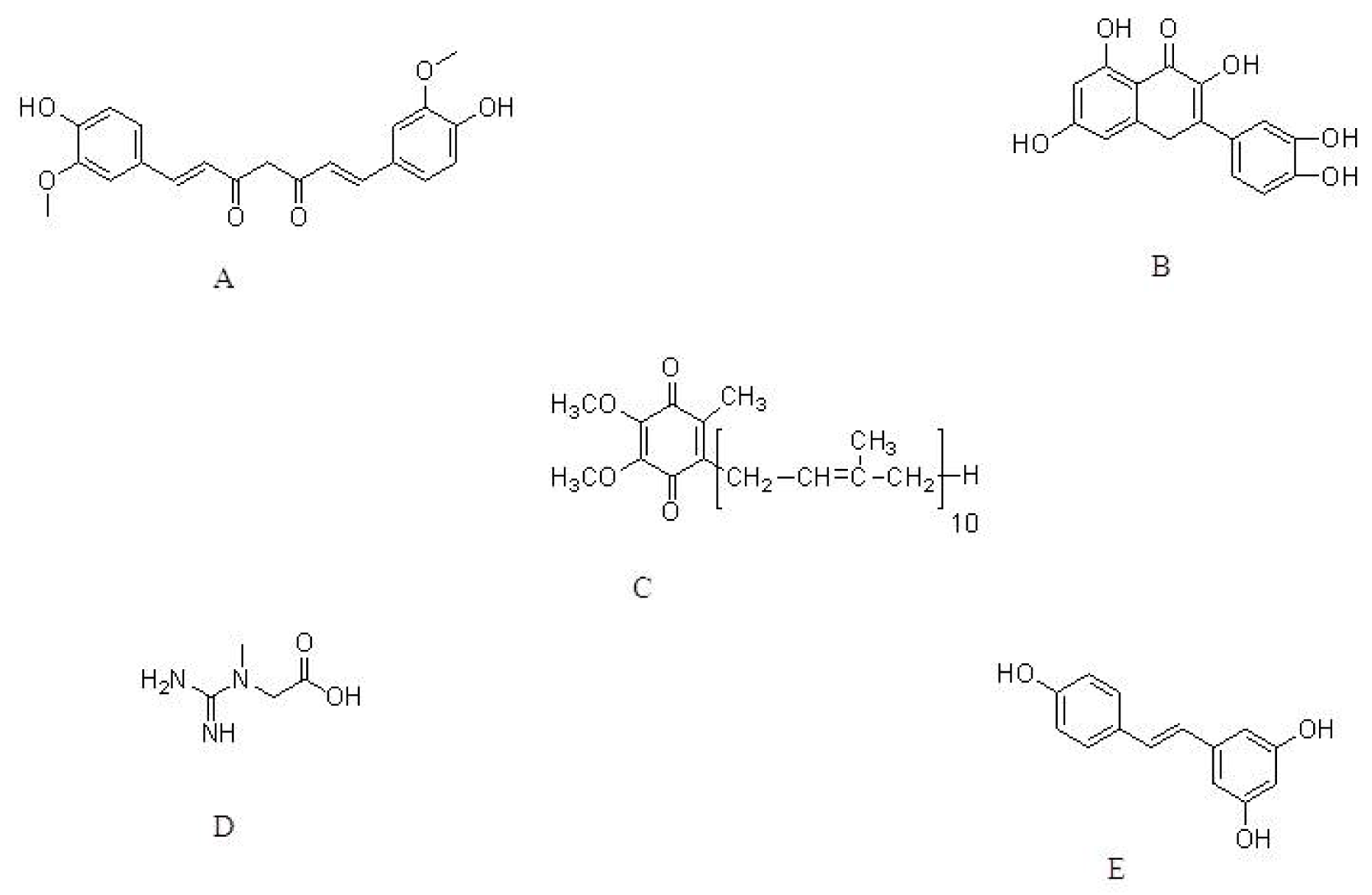
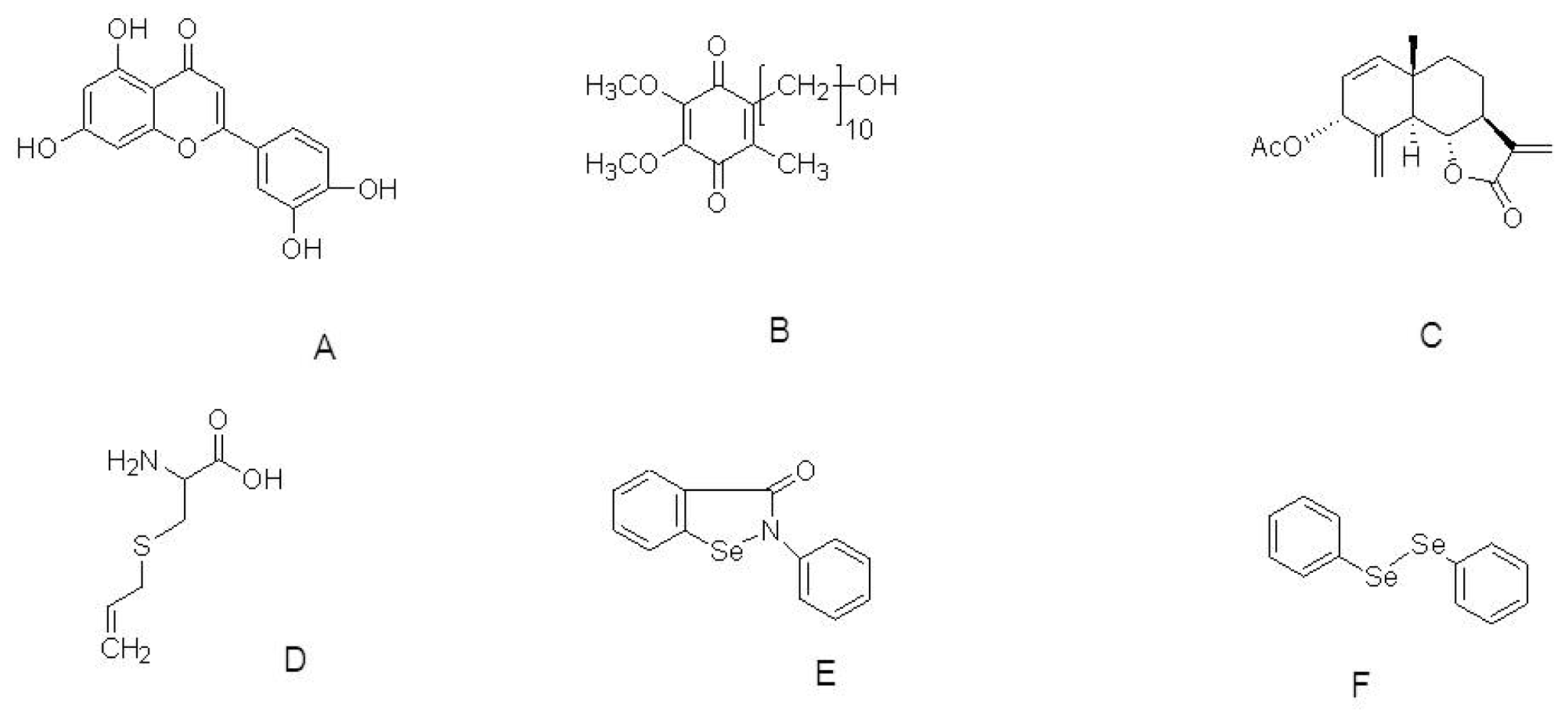
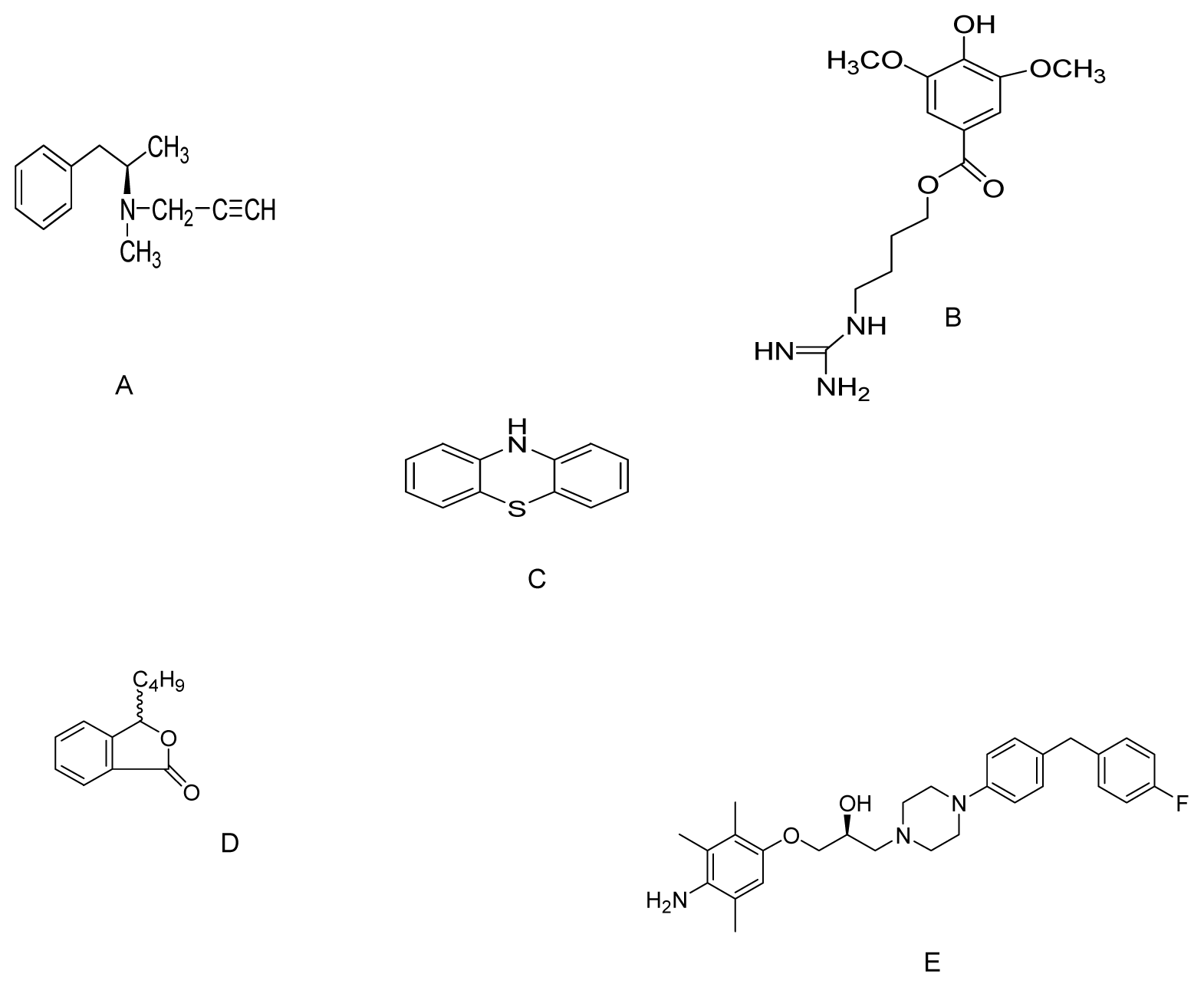
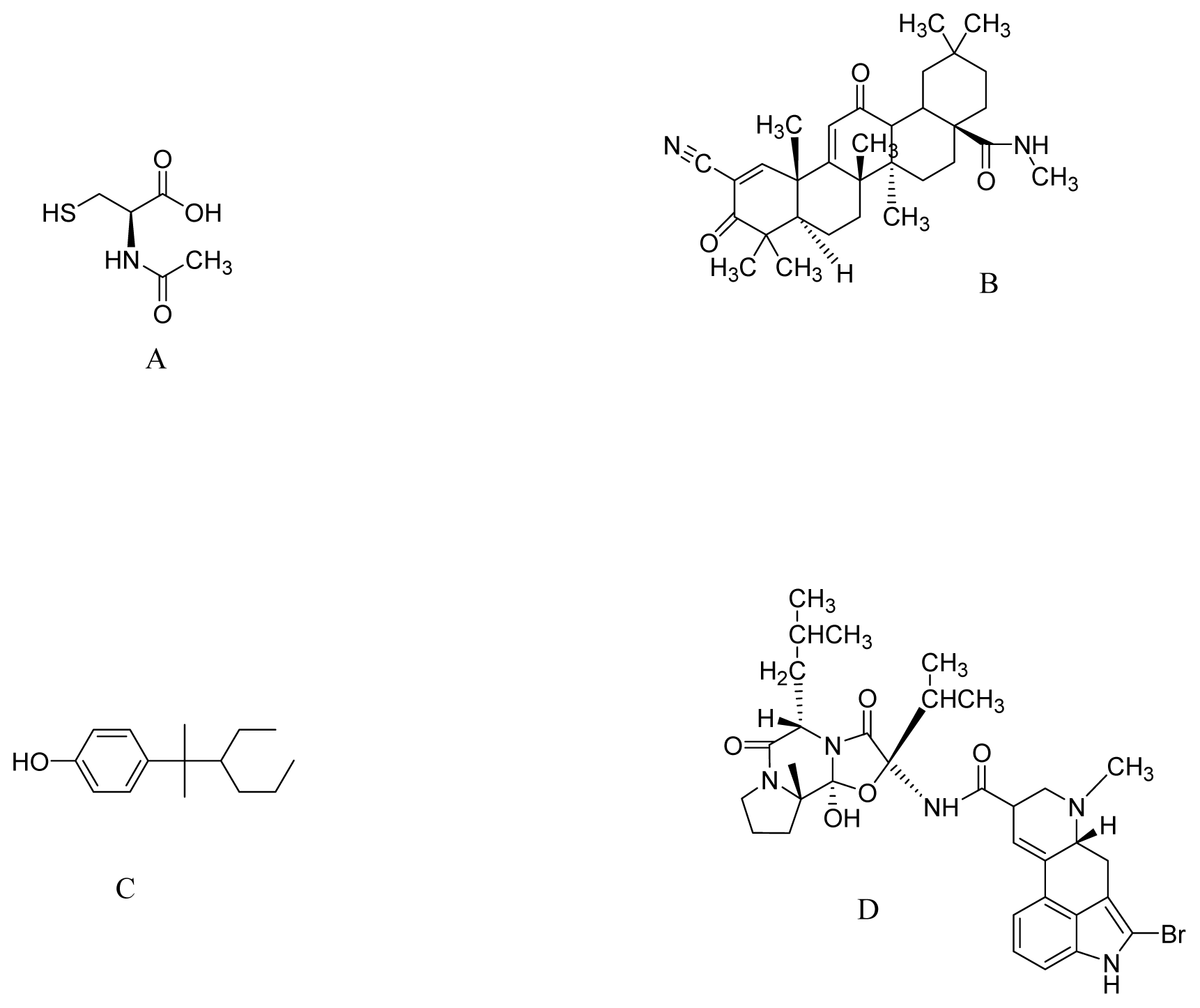

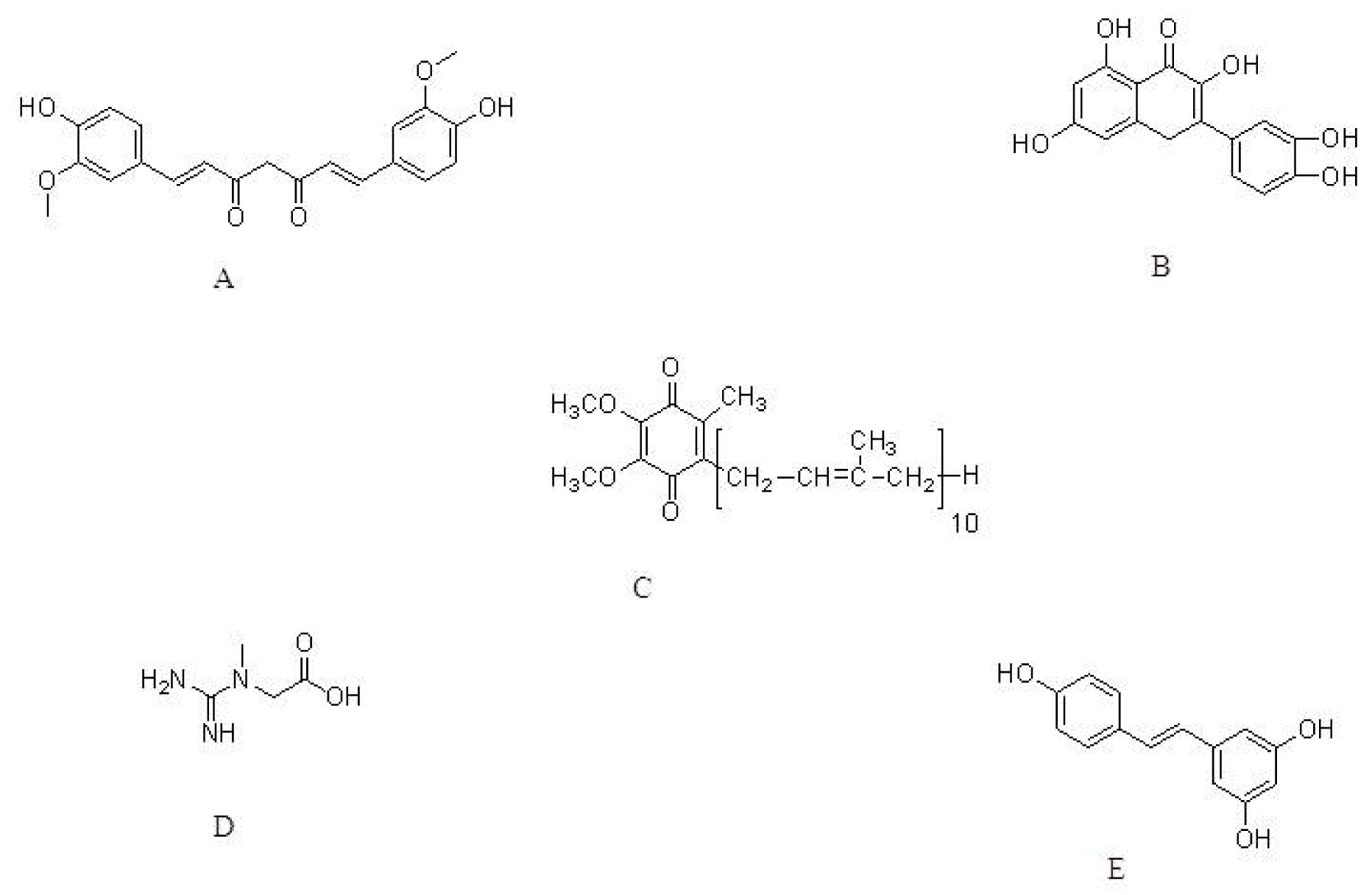{kind=link}
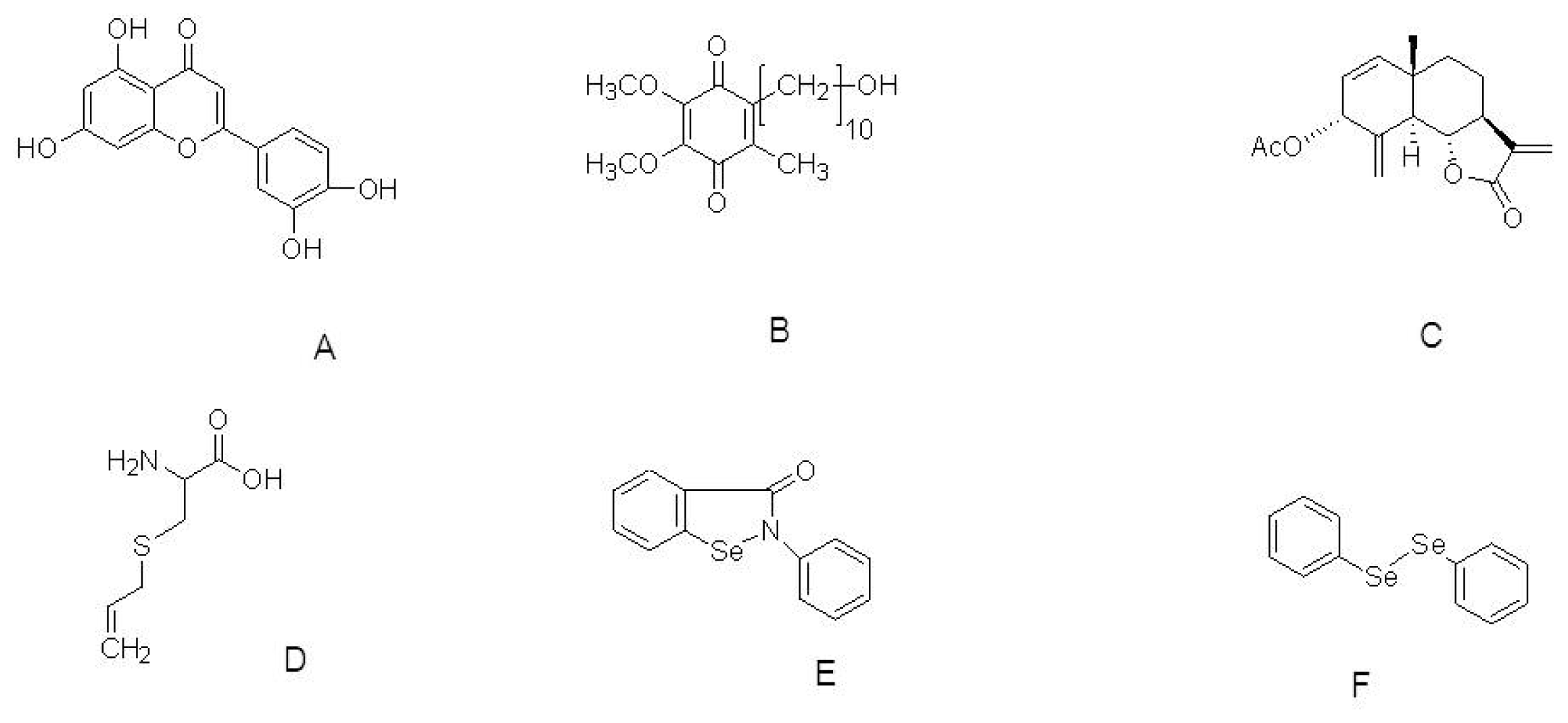{kind=link}
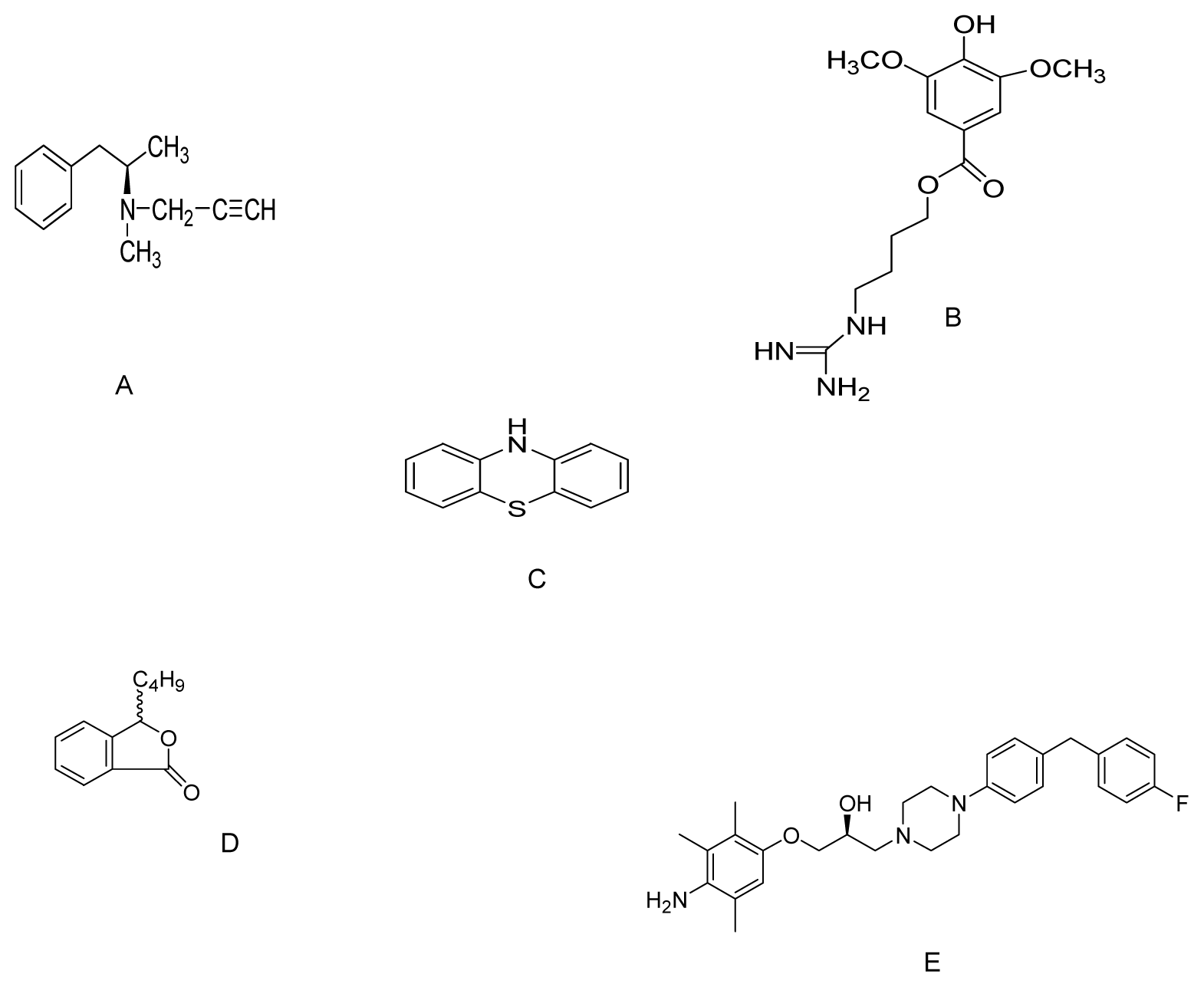{kind=link}
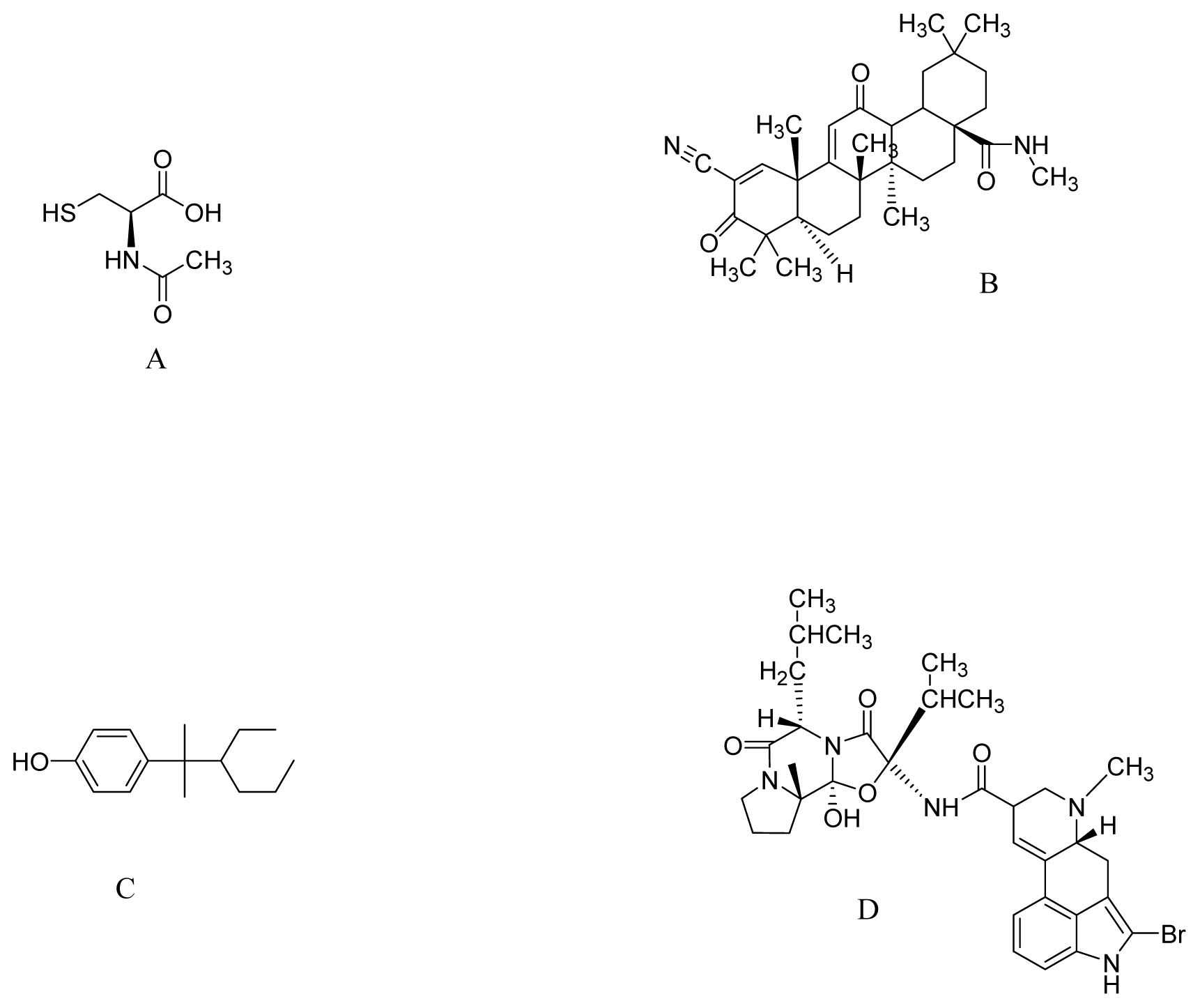{kind=link}
| Compound | Model | Effective dose | Antioxidant activity | Ref. |
|---|---|---|---|---|
| Curcumin | MPTP mouse model | Dietary supplementation | Increase in GSH levels and protected against peroxynitrite-mediated inhibition of brain mitochondrial complex I. | [57] |
| A53T SNCA-induced toxicity in PC12 cells | 0.1, 0.5 and 1.0 μM | Decrease in oxidative stress and apoptosis | [58] | |
| Quercetin | 6-OHDA-induced toxicity to PC12 cell | 25, 50 and 100 μM | Suppression of oxidative stress. | [54] |
| 6-OHDA-induced toxicity to zebrafish | 6 and 12 μM | Protect against 6-OHDA-induced apoptosis. Decrease in dopaminergic neuron loss. | [65] | |
| Coenzyme Q10 | Acute MPTP model | 1600 mg/kg/day | Protection against dopamine loss. | [75] |
| Chronic MPTP model | 1600 mg/kg/day, via diet | Increase in CoQ10 plasma concentration | ||
| Creatine with CoQ10 | MPTP mouse model | 2% Creatine & 1% CoQ10 in diet | Reduced lipid peroxidation and alpha-synuclein accumulation. | [77] |
| Resveratrol | 6-OHDA model | 20 mg/kg per day | Decrease in ROS. Increase in antioxidant capability of nigral tissues. | [82] |
| Luteolin | ROS insult to neural cells | 5, 10, and 20 μM | Decrease in ROS production and increase the activities of catalase and glutathione. | [85] |
| Idebenone | HtrA2 knockout mice | 500 mg/kg body weight/day orally | Extends lifespan and improves motor symptoms. Regulation of apoptotic pathway | [93] |
| AETO | SH-SY5Y cells | 0.4, 2, and 10 μM | Suppression of ROS & DA-induced apoptosis | [94] |
| S-allylcysteine | MPTP mouse Model | 125 mg/kg; i.p. | Blocks lipid peroxidation and reduction of superoxide production | [95] |
| Ebselen & Diphenyl diselenide | 6-OHDA-induced toxicity to SH-SY5Y cell | 3 μM each | Peroxyl radical scavenging. Increase the GPx activity and SOD activity. | [96] |
| Deprenyl | MPP+ treated PC12 cells | 10, 20, 50 and 100 μM | Nrf2/ARE pathway | [104] |
| SCM198 | 6-OHDA-induced toxicity to SH-SY5Y cells | 0.1, 1, and 10 mM | Increase in SOD activity. Suppression of apoptosis | [109] |
| Phenothiazine | MPP+ and rotenone toxicity to C. elegans | 500 nM | Increase free radical scavenging effects | [112] |
| dl-3n-Butylphthalide | MPP+ treated PC12 cells | 0.1, 1.0 and 10 μM | Reducing oxidative stress & increasing cellular GSH content | [115] |
| SUN N8075 | MPTP mouse model | 10 and 30 mg/kg i.p | Inhibited lipid peroxidation and H2O2-induced ROS. | [118] |
| N-acetyl-l-cysteine | Transgenic mice overexpressing α-synuclein | Drinking water supplemented with 40 mM | Increase of GSH levels in SN. | [120] |
| CDDO-methyl amide | MPTP and 3-nitropropionic acid induced neurotoxicity | 800 mg/kg of diet | Nrf2/ARE pathway | [77] |
| NP7 | Parkin null mice | 5–10 μM | Inhibits H2O2-induced apoptosis | [123] |
| Bromocriptine | H2O2-treated PC12 cells | 5 μM | Increase activity of NQO1 and Nrf2 signaling | [127] |
© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Koppula, S.; Kumar, H.; More, S.V.; Kim, B.W.; Kim, I.S.; Choi, D.-K. Recent Advances on the Neuroprotective Potential of Antioxidants in Experimental Models of Parkinson’s Disease. Int. J. Mol. Sci. 2012, 13, 10608-10629. https://doi.org/10.3390/ijms130810608
Koppula S, Kumar H, More SV, Kim BW, Kim IS, Choi D-K. Recent Advances on the Neuroprotective Potential of Antioxidants in Experimental Models of Parkinson’s Disease. International Journal of Molecular Sciences. 2012; 13(8):10608-10629. https://doi.org/10.3390/ijms130810608
Chicago/Turabian StyleKoppula, Sushruta, Hemant Kumar, Sandeep Vasant More, Byung Wook Kim, In Su Kim, and Dong-Kug Choi. 2012. "Recent Advances on the Neuroprotective Potential of Antioxidants in Experimental Models of Parkinson’s Disease" International Journal of Molecular Sciences 13, no. 8: 10608-10629. https://doi.org/10.3390/ijms130810608