The Complete Mitochondrial Genome of the Pink Stem Borer, Sesamia inferens, in Comparison with Four Other Noctuid Moths

Abstract
:1. Introduction
2. Results and Discussion
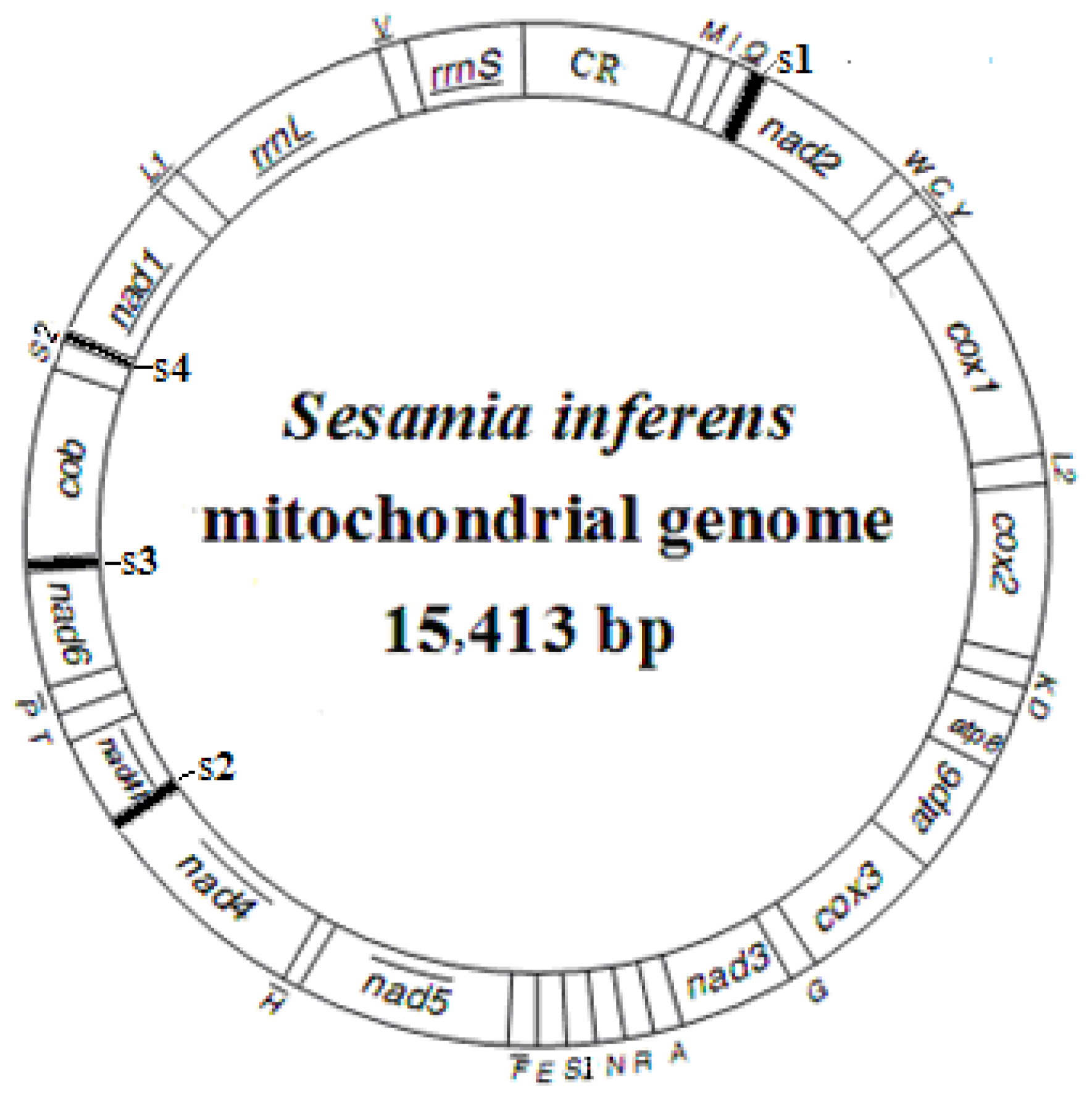
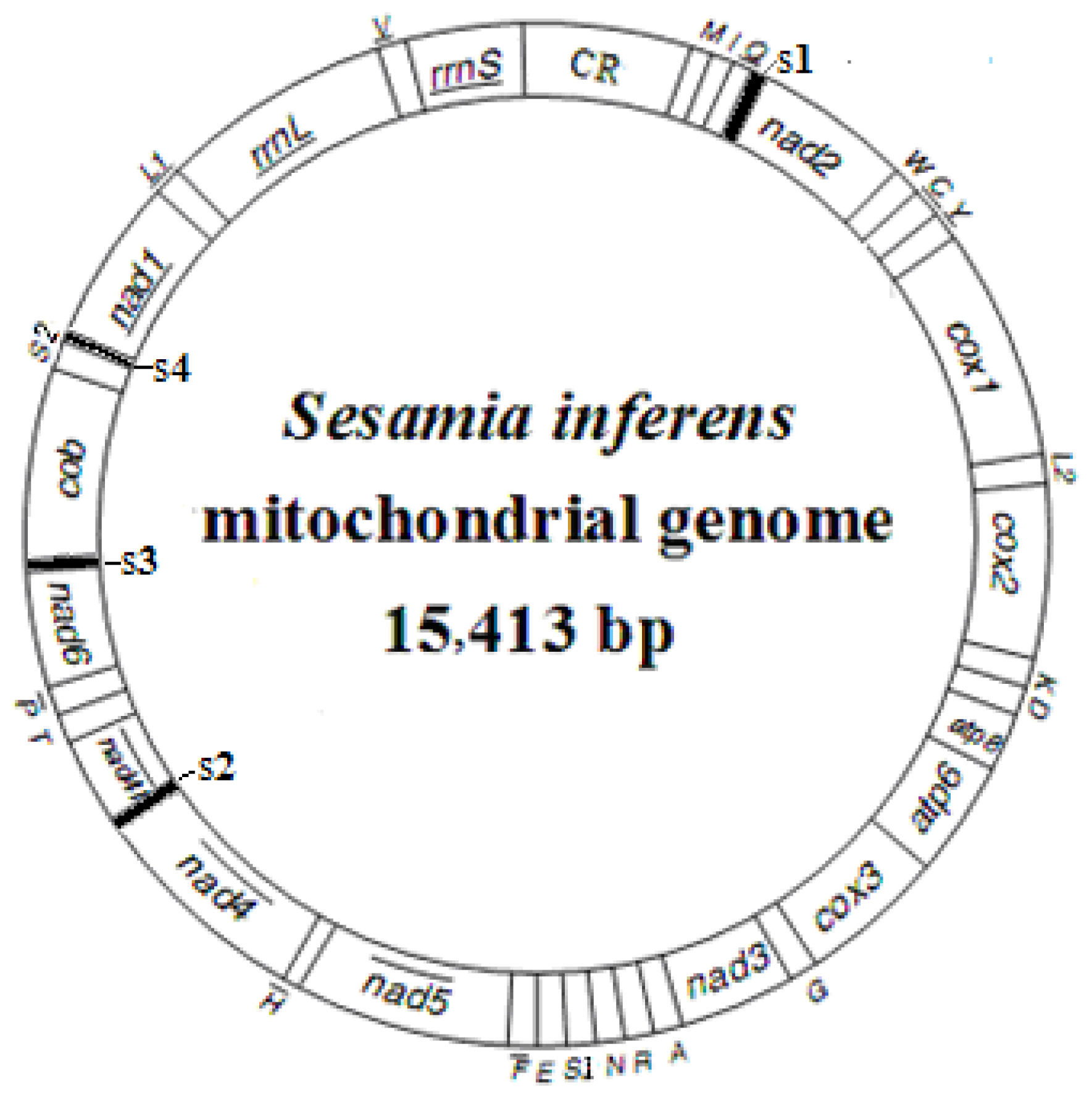
2.1. Genome Structure, Organization and Composition
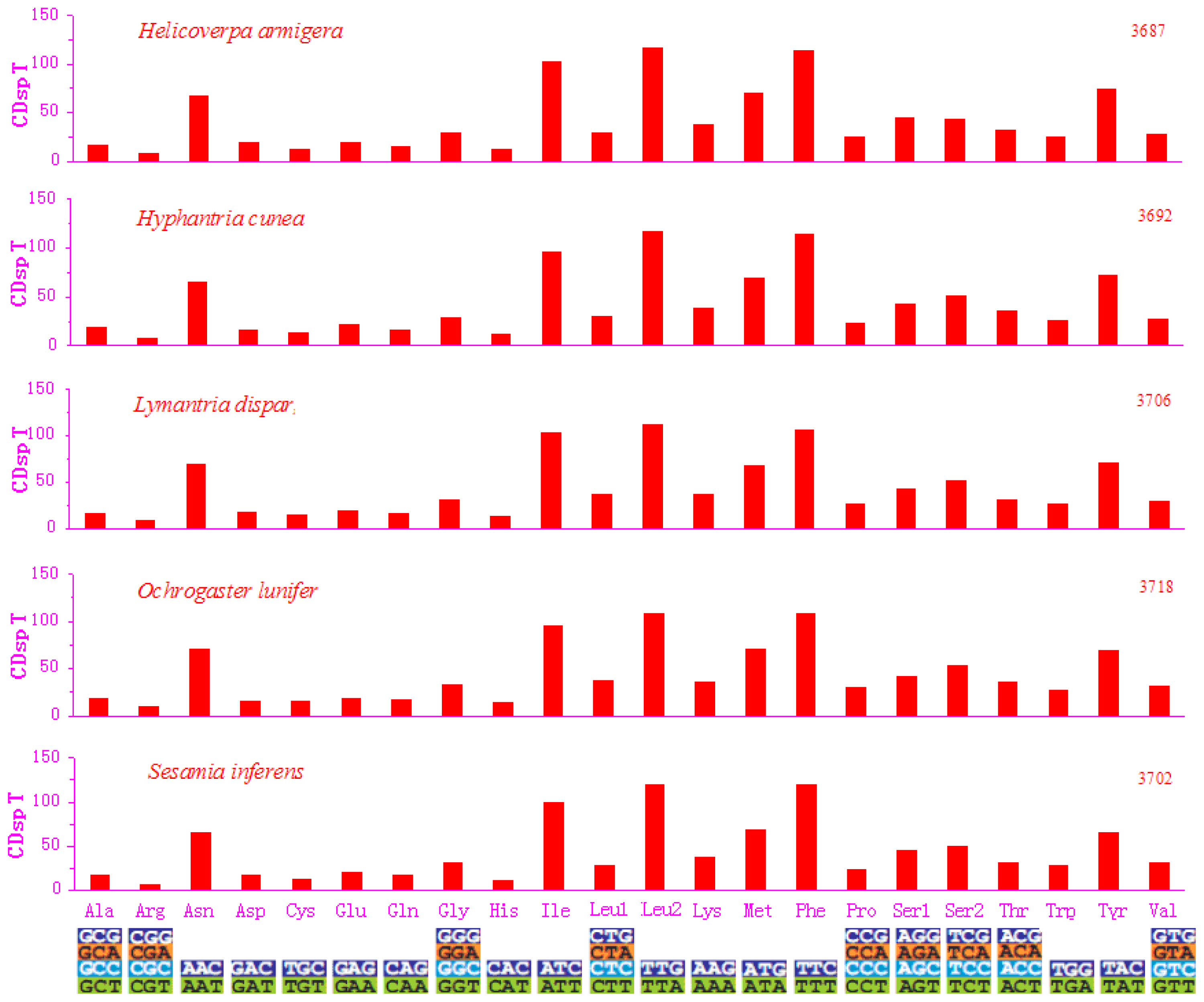
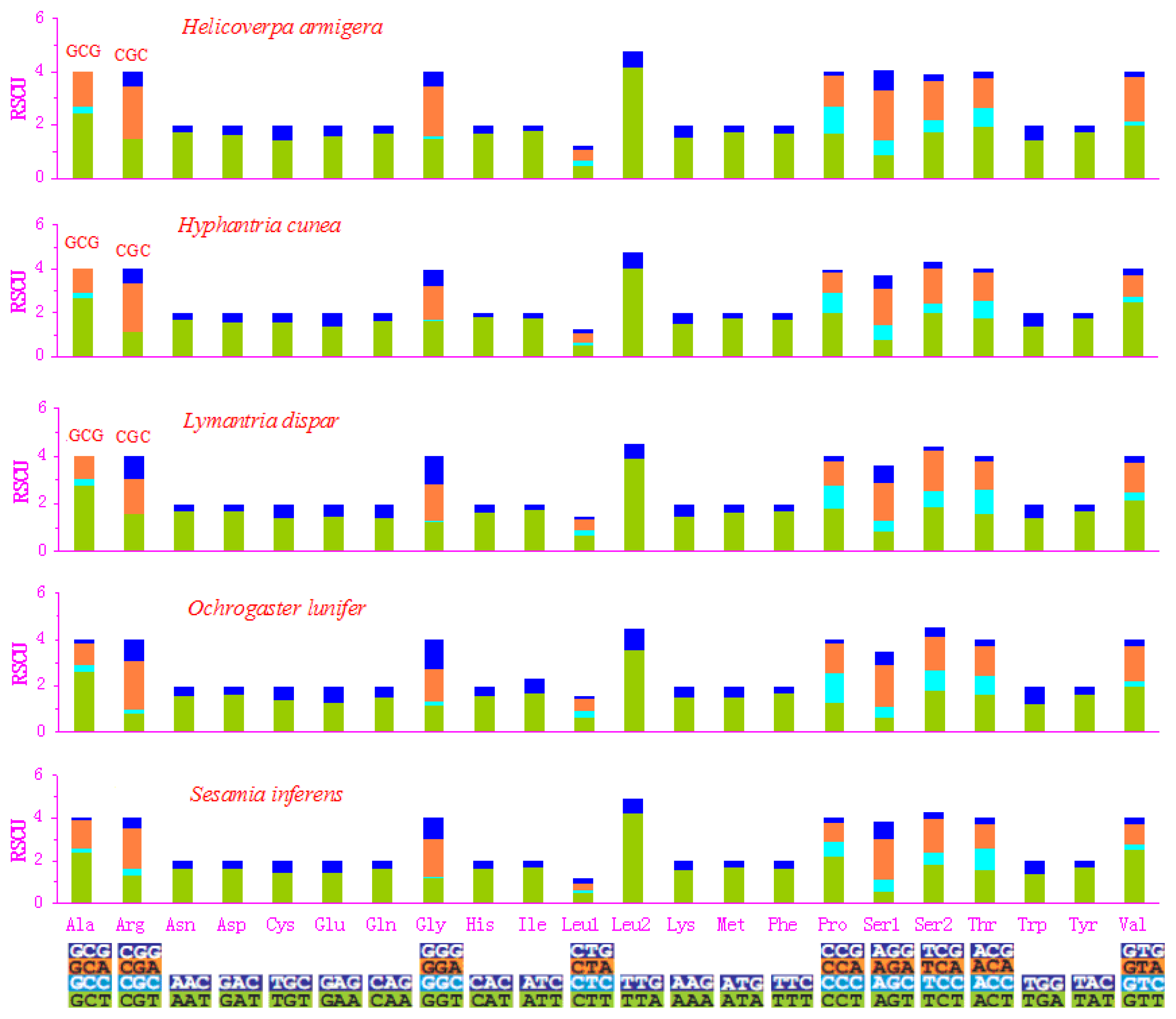
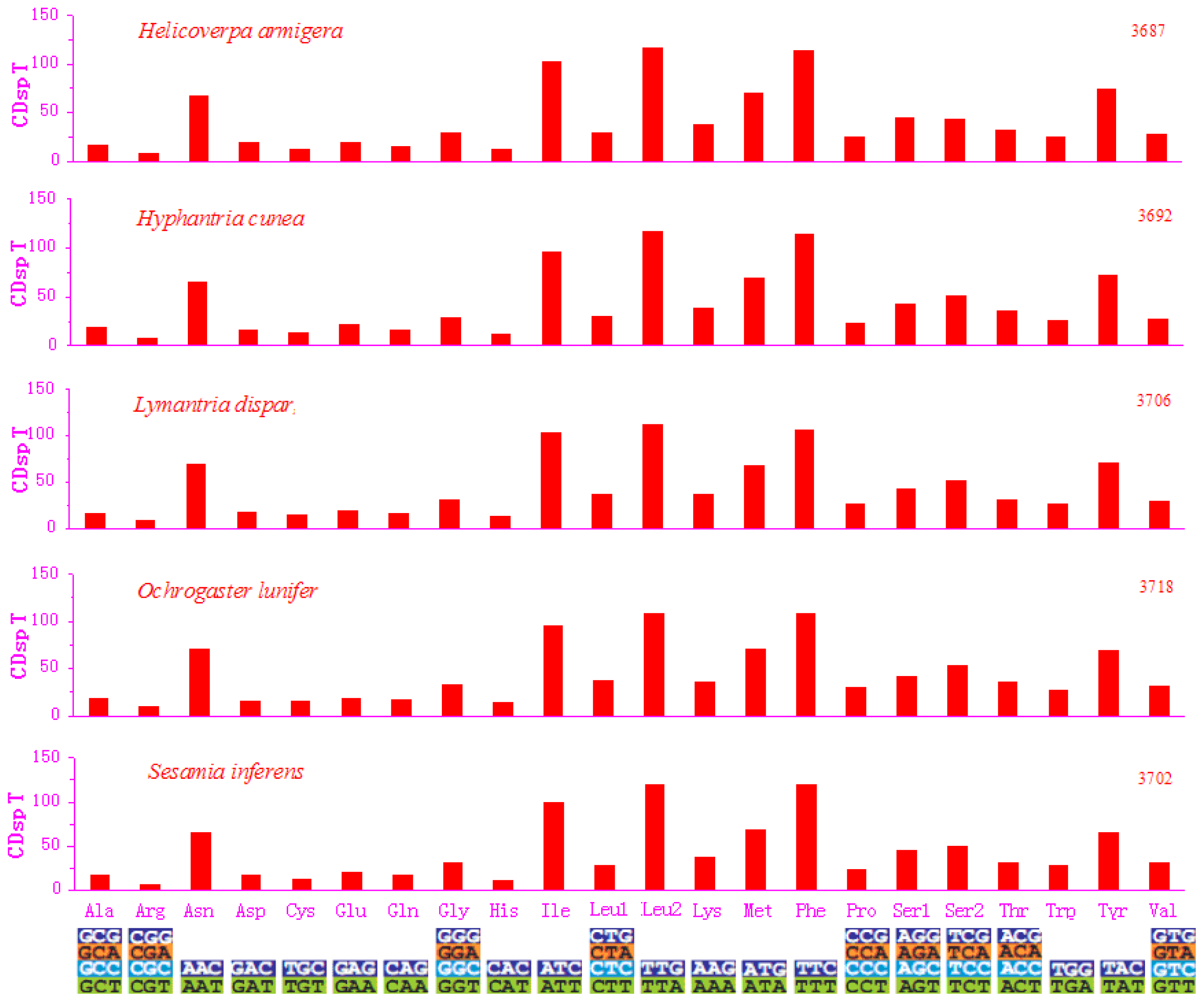
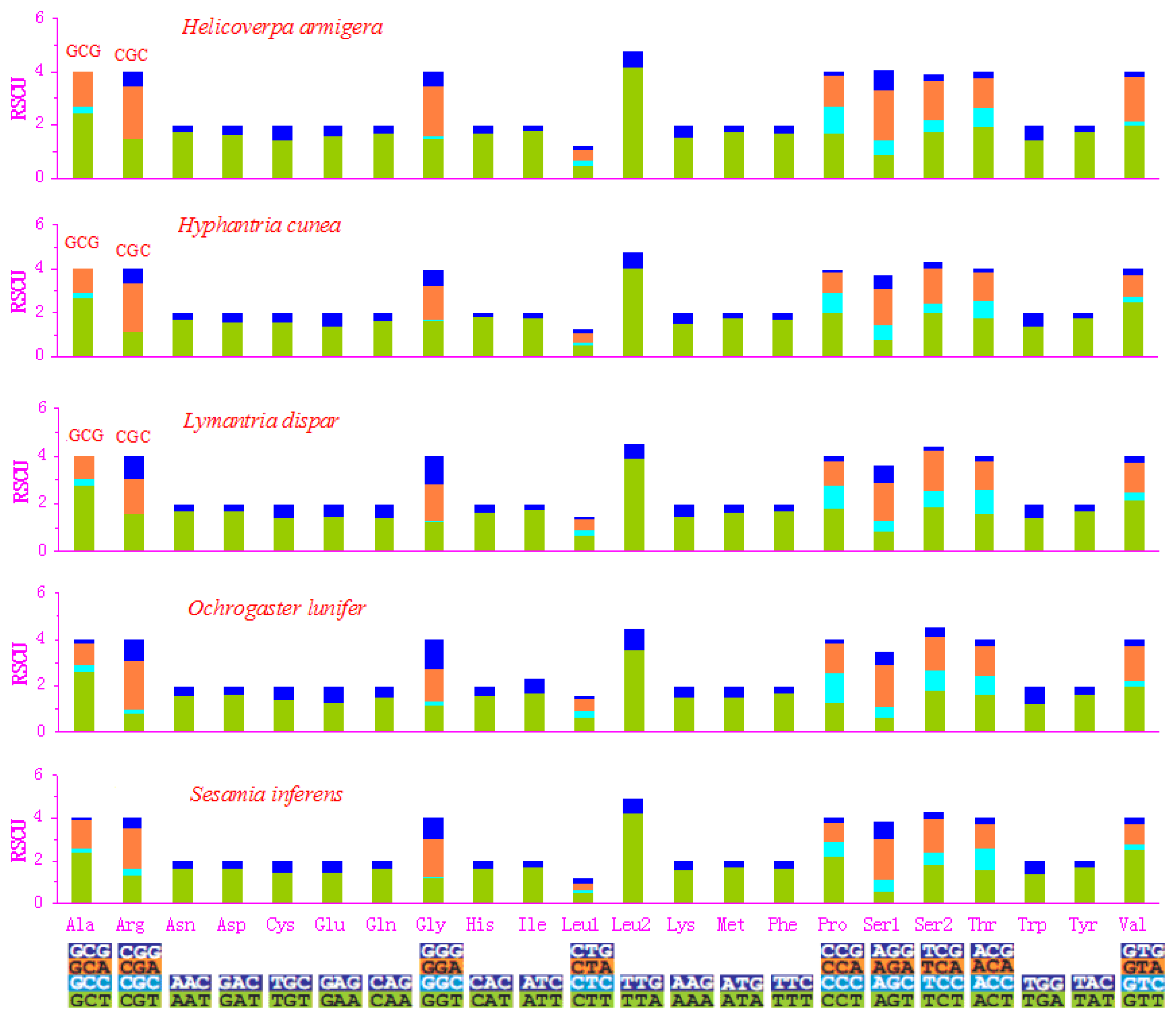
2.2. Protein-Coding Genes
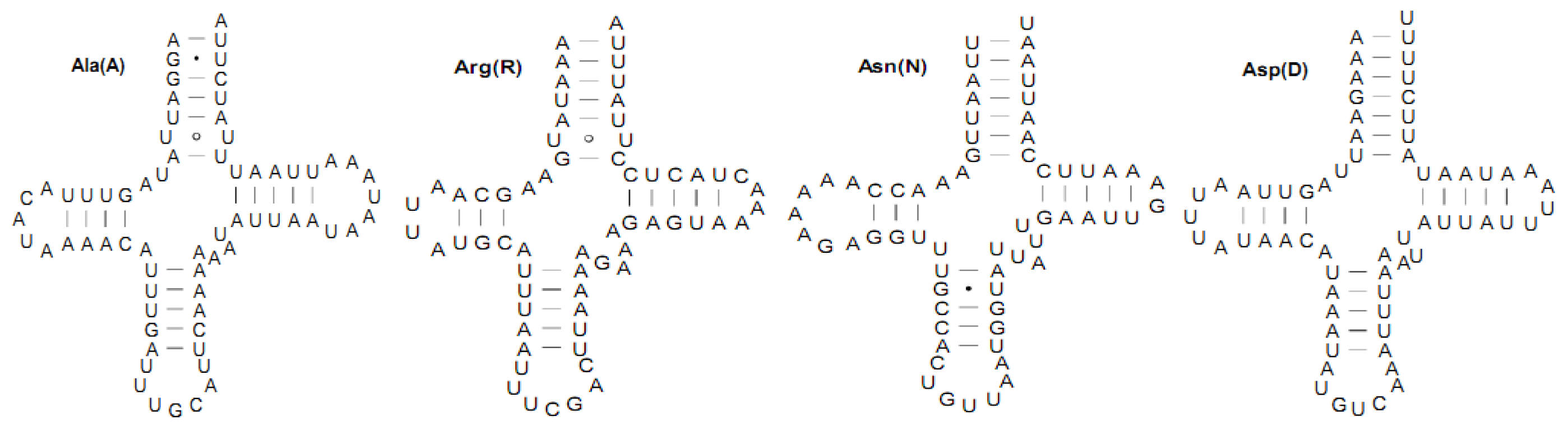
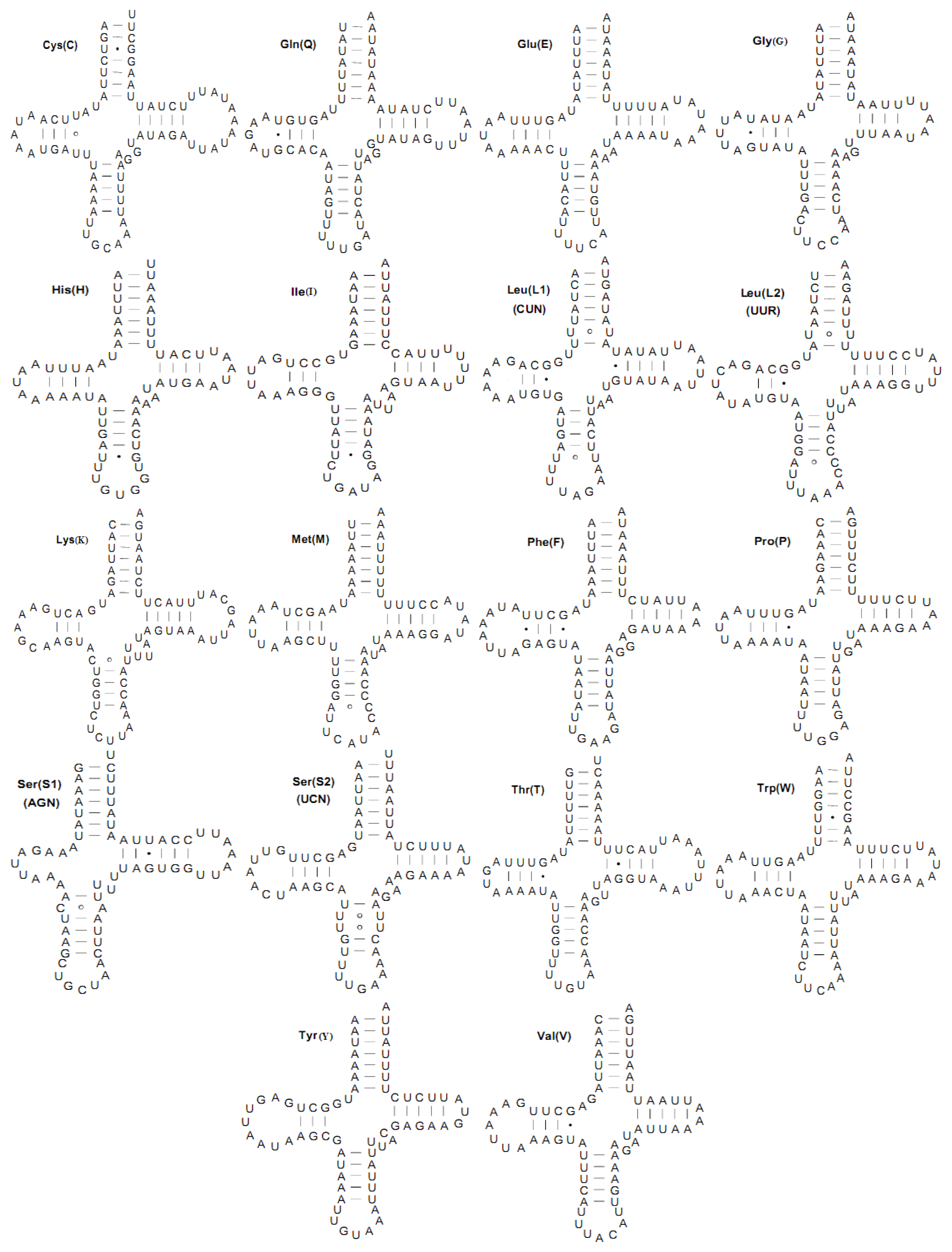
2.3. Transfer RNA Genes
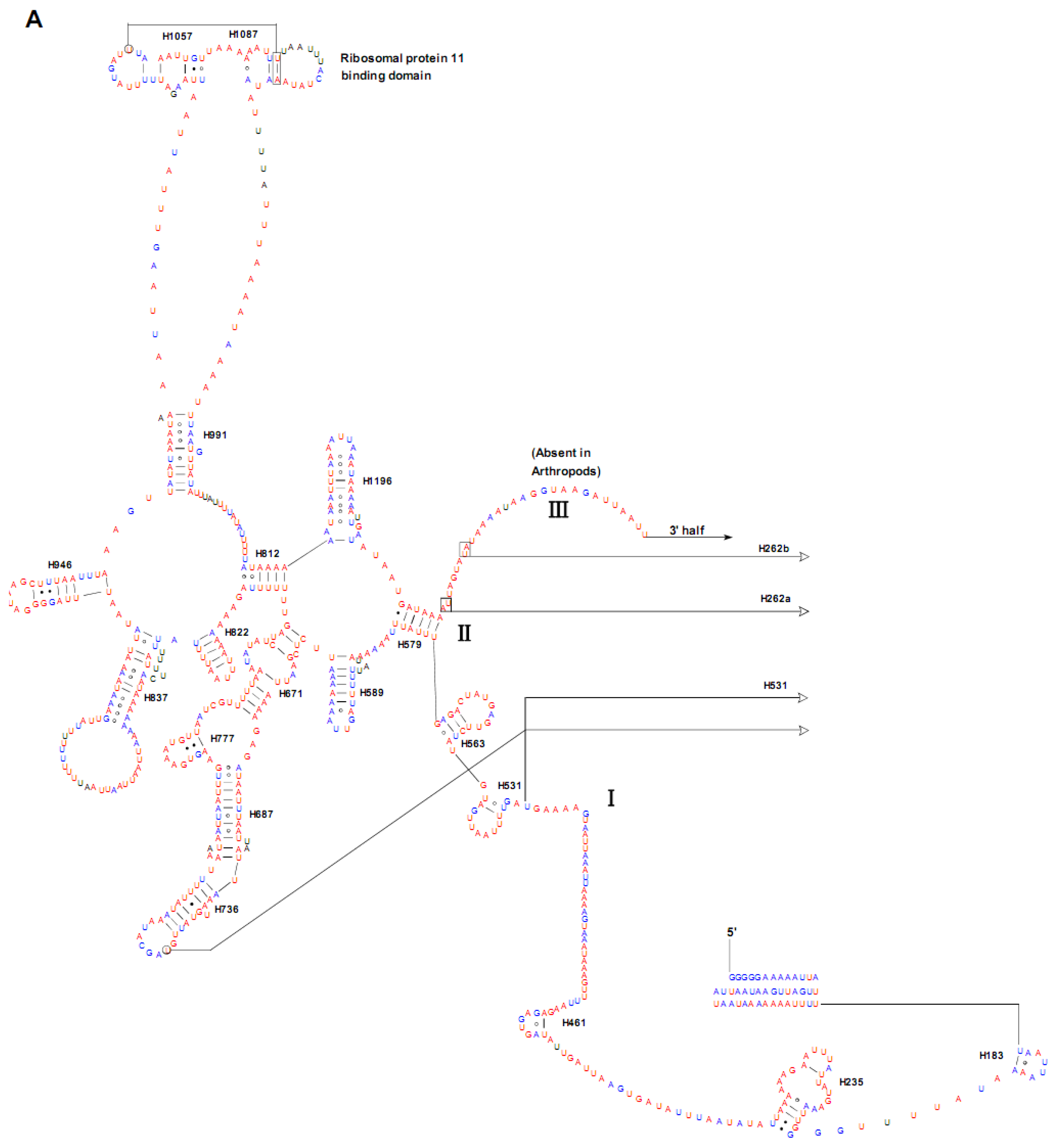
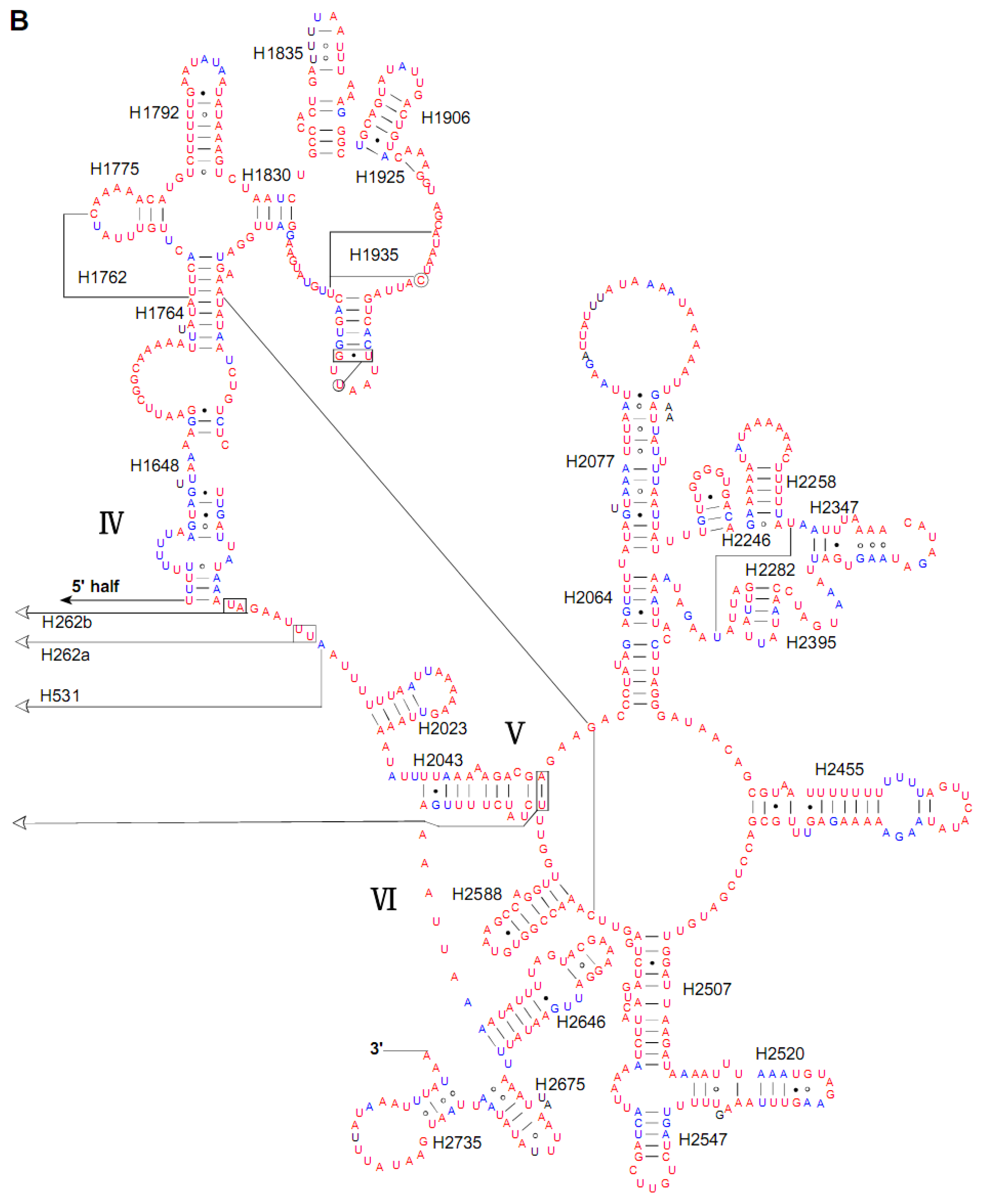
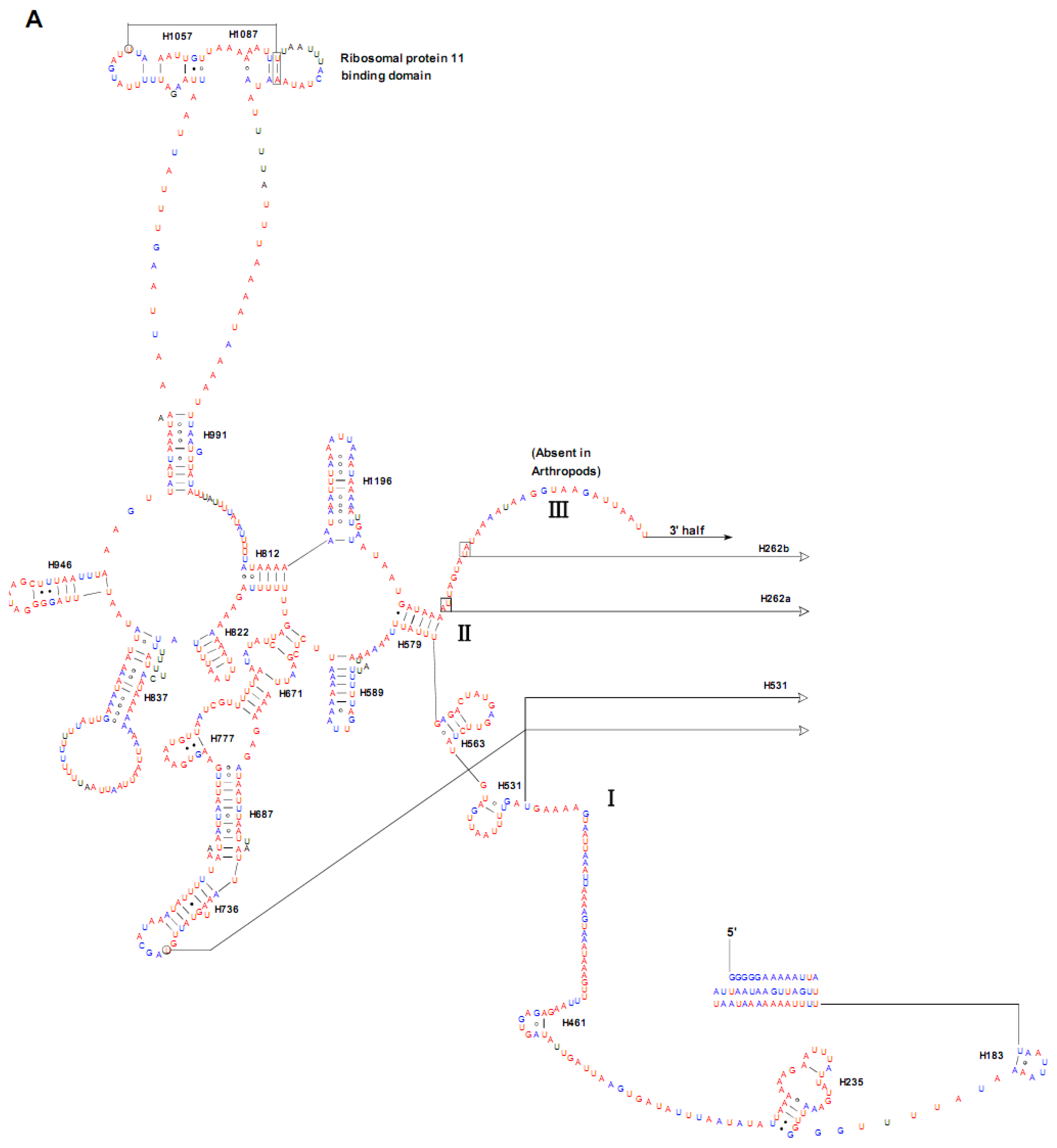
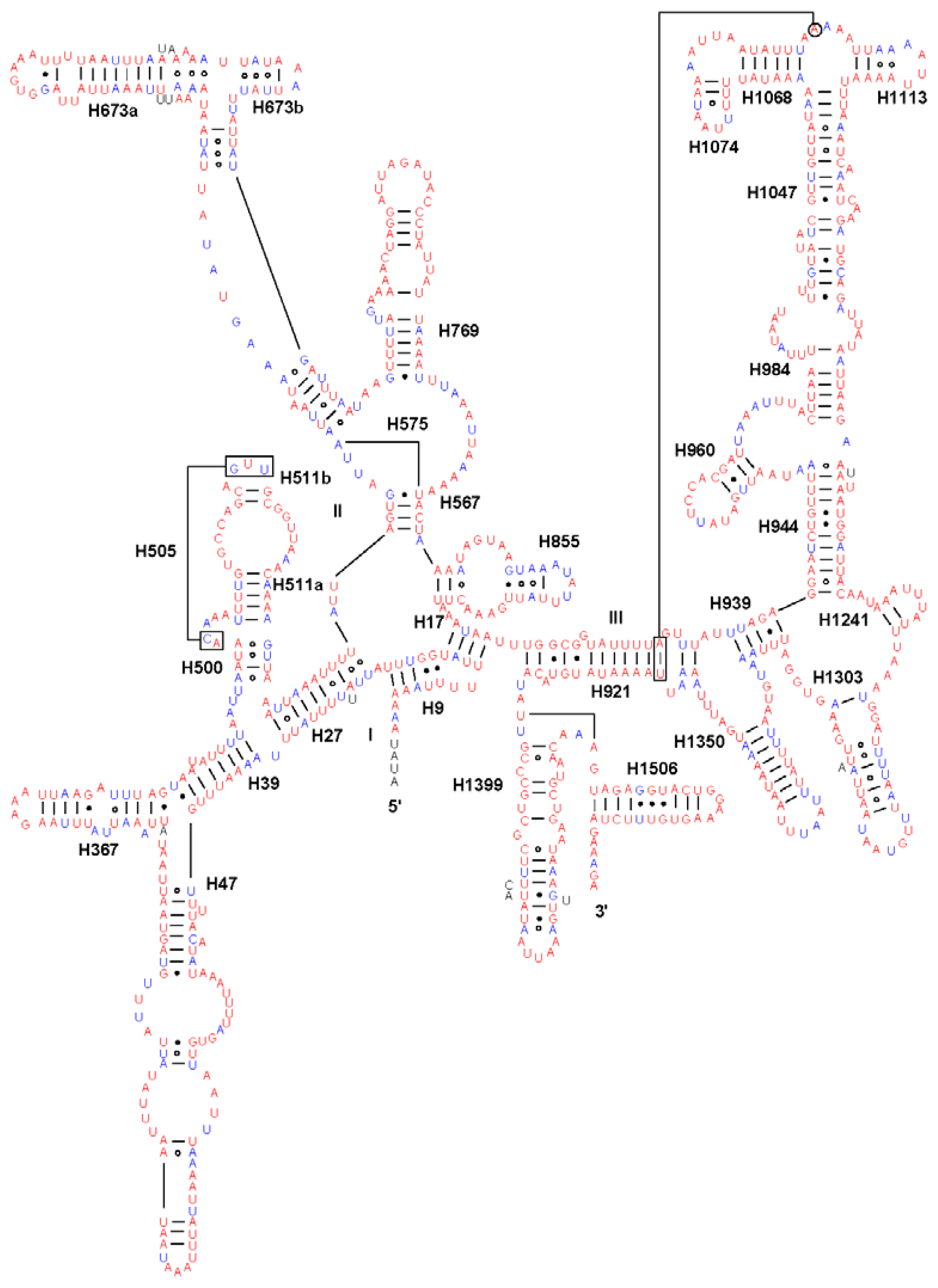
2.4. Ribosomal RNA Genes
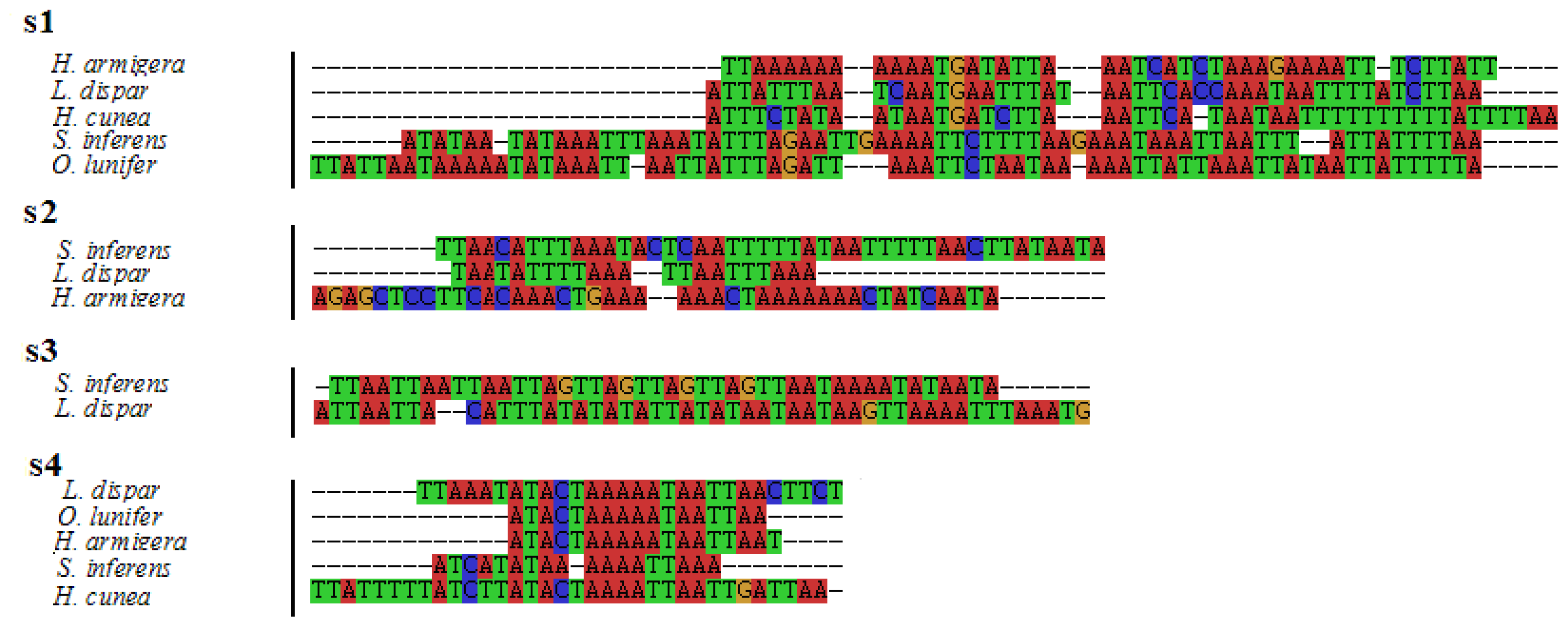
2.5. Non-Coding and Overlapping Regions
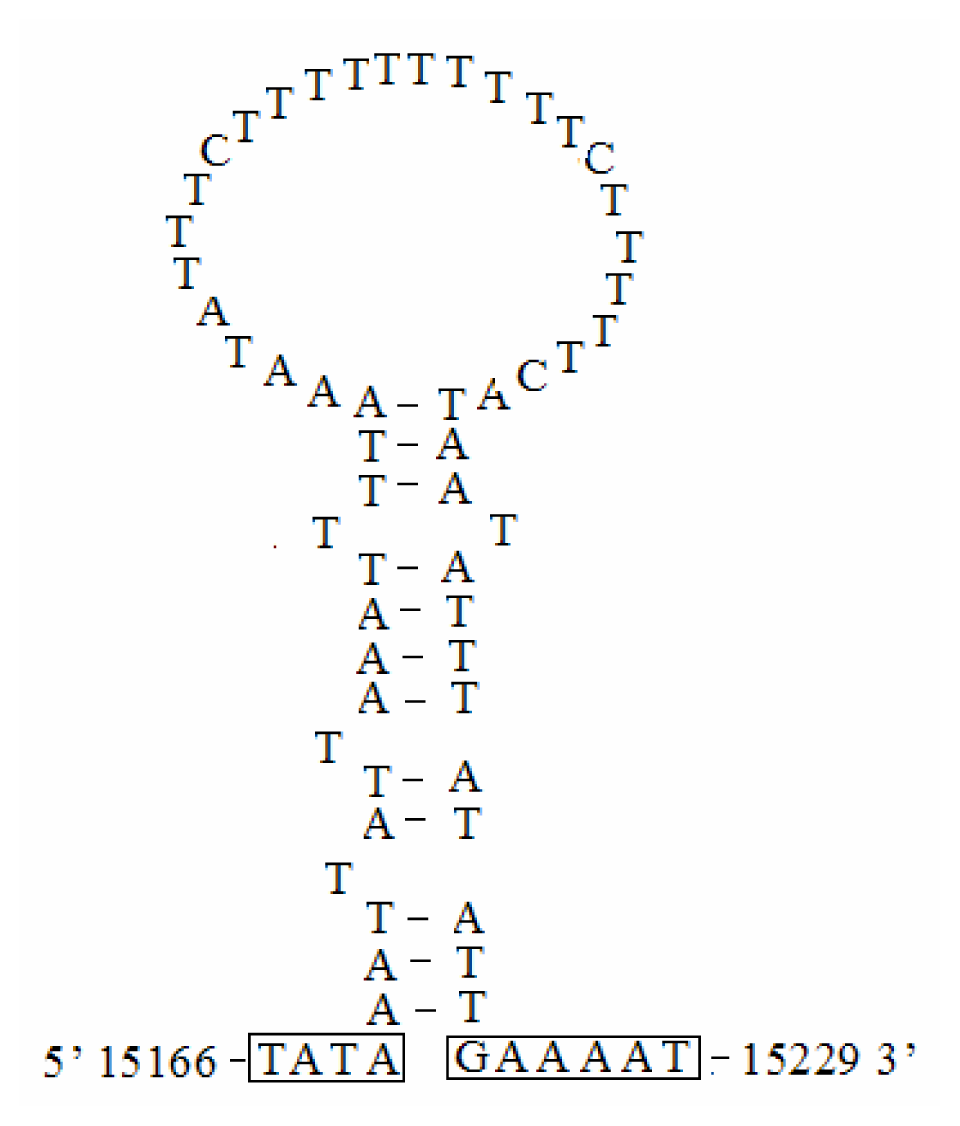
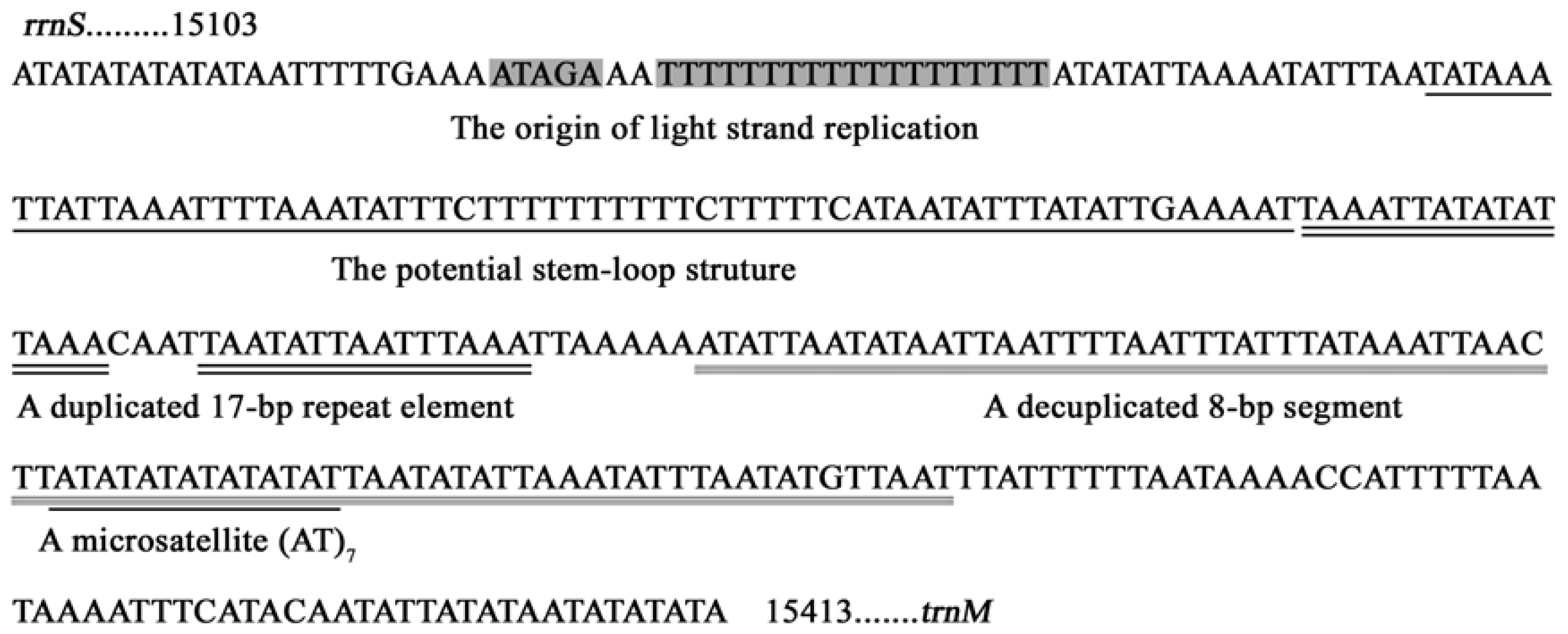
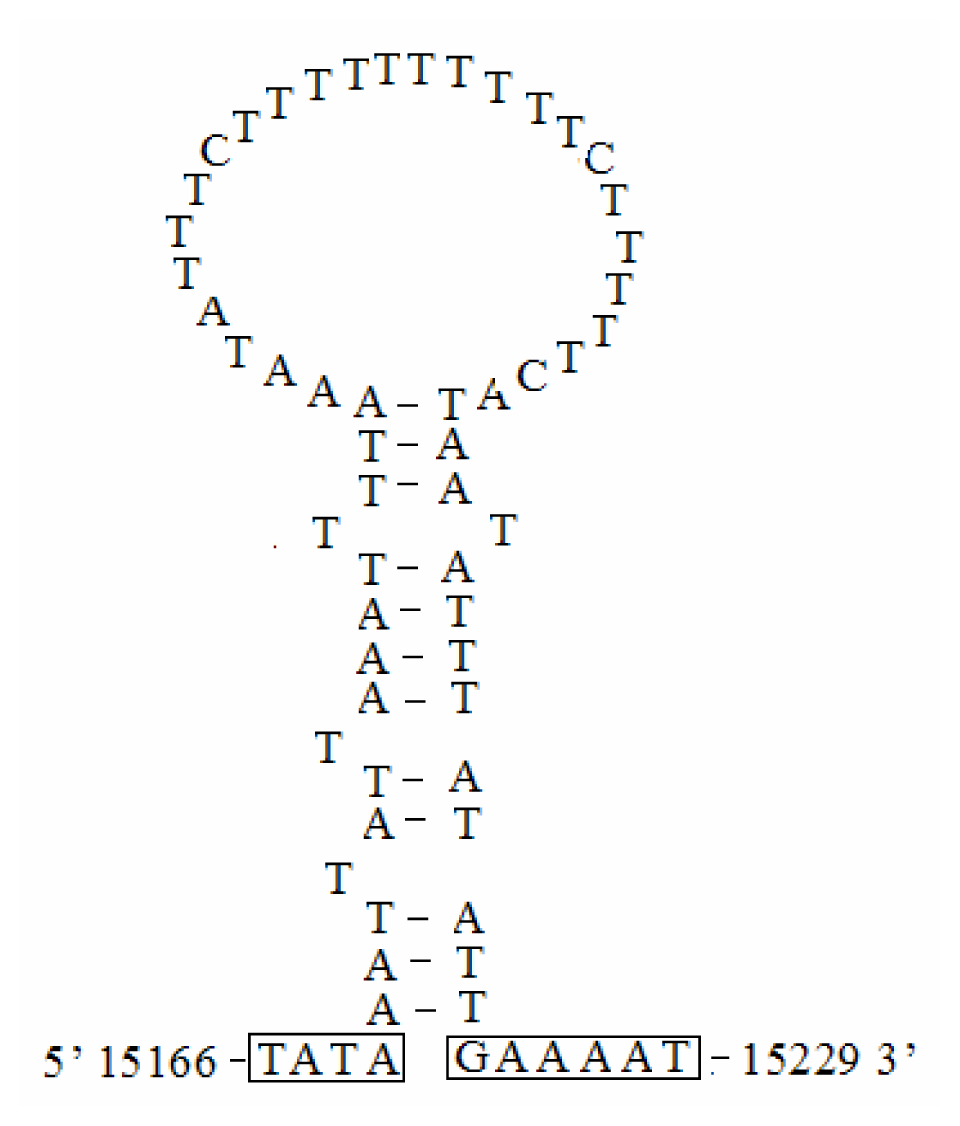
2.6. A+T-rich Region
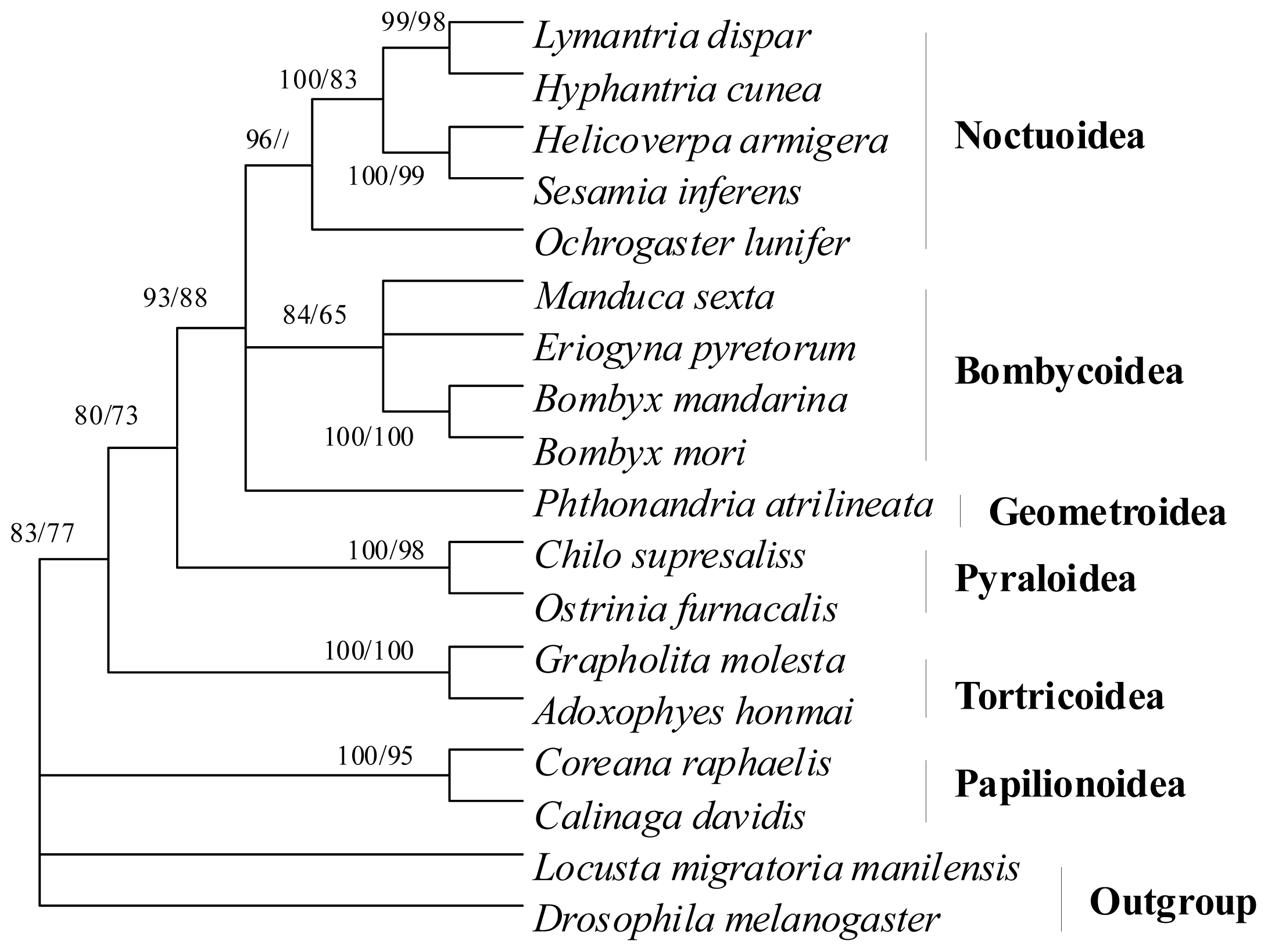
2.7. Phylogenetic Relationships
3. Experimental Section
3.1. DNA Extraction
3.2. Primers Design, PCR Amplification, Cloning and Sequencing
3.3. Sequence Analysis
3.4. Phylogenetic Analysis
4. Conclusions
Acknowledgements
References
- Savary, S.; Willocquet, L.; Elazegui, F.S.; Castilla, N.P.; Teng, P.S. Rice pest constraints in tropical Asia: Quantification of yield losses due to rice pests in a range of production situations. Plant Dis 2000, 84, 357–369. [Google Scholar]
- Singh, J.; Shera, P.S. Relative abundance of different species of rice stem borers in punjab, India. Presented at the national conference on plant protection—New horizons in the millennium. rajasthan college of agriculture, Udaipur, India; 2001. [Google Scholar]
- Sheng, C.F.; Wang, H.T.; Sheng, S.Y.; Gao, L.D.; Xuan, W.J. Pest status and loss assessment of crop damage caused by the rice borers, Chilo suppressalis and Tryporyza incertulas in China. Entomol. Knowl 2003, 40, 289–294. [Google Scholar]
- Salvato, P.; Simonato, M.; Battisti, A.; Negrisolo, E. The complete mitochondrial genome of the bag-shelter moth Ochrogaster lunifer (Lepidoptera, Notodontidae). BMC Genomics 2008, 9. [Google Scholar] [CrossRef]
- Liao, F.; Wang, L.; Wu, S.; Li, Y.P.; Zhao, L.; Huang, G.M.; Niu, C.J.; Liu, Y.Q.; Li, M.G. The complete mitochondrial genome of the fall webworm, Hyphantria cunea (Lepidoptera: Arctiidae). Int. J. Biol. Sci 2010, 6, 172–186. [Google Scholar]
- Yin, J.; Hong, G.Y.; Wang, A.M.; Cao, Y.Z.; Wei, Z.J. Mitochondrial genome of the cotton bollworm Helicoverpa armigera (Lepidoptera: Noctuidae) and comparison with other Lepidopteran. Mitochondrial DNA 2010, 21, 160–169. [Google Scholar]
- Zhu, Y.J.; Zhou, G.L.; Fang, R.; Ye, J.; Yi, J.P. The complete sequence determination and analysis of Lymantria dispar (Lepidoptera: Lymantriidae) mitochondrial genome. Plant Quar 2010, 4, 6–11. [Google Scholar]
- Wolstenholme, D.R. Animal mitochondrial DNA: Structure and evolution. Int. Rev. Cytol 1992, 141, 173–216. [Google Scholar]
- Boore, J.L. Animal mitochondrial genomes. Nucleic Acids Res 1999, 27, 1726–1780. [Google Scholar]
- Da Silva, N.M.; de Souza Dias, A.; da Silva Valente, V.L.; Victor, H.V. Characterization of mitochondrial control region, two intergenic spacers and tRNAs of Zaprionus indianus (Diptera: Drosophilidae). Genetica 2009, 137, 325–332. [Google Scholar]
- Wei, S.J.; Shi, M.; He, J.H.; Sharkey, M.J.; Achterberg, C.V.; Chen, X.X. Comparative mitogenomics of Braconidae (Insecta: Hymenoptera) and the phylogenetic utility of mitochondrial genomes with special reference to Holometabolous insects. BMC Genomics 2010, 11. [Google Scholar] [CrossRef]
- Cameron, S.L.; Yoshizawa, K.; Mizukoshi, A.; Whiting, M.F.; Johnson, K.P. Mitochondrial genome deletions and minicircles are common in lice (Insecta: Phthiraptera). BMC Genomics 2011, 12. [Google Scholar] [CrossRef] [Green Version]
- Anderson, S.; Bankier, A.T.; Barrell, B.G.; de Bruijn, M.H.; Coulson, A.R.; Drouin, J.; Eperon, I.C.; Nierlich, D.P.; Roe, B.A.; Sanger, F.; et al. Sequence and organization of the human mitochondrial genome. Nature 1981, 290, 457–465. [Google Scholar]
- Boore, J.L.; Lavrov, D.; Brown, W.M. Gene translocation links insects and crustaceans. Nature 1998, 393, 667–668. [Google Scholar]
- Kim, I.; Lee, E.M.; Seol, K.Y.; Yun, E.Y.; Lee, Y.B.; Hwang, J.S.; Jin, B.R. The mitochondrial genome of the Korean hairsteak, Coreana raphaelis (Lepidoptera: Lycaenidae). Insect Mol. Biol 2006, 15, 217–225. [Google Scholar]
- Gong, Y.J.; Shi, B.C.; Kang, Z.J.; Zhang, F.; Wei, S.J. The complete mitochondrial genome of the oriental fruit moth Grapholita molesta (Busck) (Lepidoptera: Tortricidae). Mol. Biol. Rep 2012, 39, 2893–2900. [Google Scholar]
- De Bruijn, M.H. Drosophila melanogaster mitochondrial DNA, a novel organization and genetic code. Nature 1983, 304, 234–241. [Google Scholar]
- Beard, C.B.; Hamm, D.M.; Collins, F.H. The mitochondrial genome of the mosquito Anopheles gambiae: DNA sequence, genome organization, and comparisons with mitochondrial sequences of other insects. Insect Mol. Biol 1993, 2, 103–124. [Google Scholar]
- Junqueira, A.C.; Lessinger, A.C.; Torres, T.T.; da Silva, F.R.; Vettore, A.L.; Arruda, P.; Azeredo Espin, A.M. The mitochondrial genome of the blowfly Chrysomya chloropyga (Diptera: Calliphoridae). Gene 2004, 339, 7–15. [Google Scholar]
- Krzywinski, J.; Grushko, O.G.; Besansky, N.J. Analysis of the complete mitochondrial DNA from Anopheles funestus: An improved dipteran mitochondrial genome annotation and a temporal dimension of mosquito evolution. Mol. Phylogenet. Evol 2006, 39, 417–423. [Google Scholar]
- Masta, S.E.; Boore, J.L. The complete mitochondrial genome sequence of the spider Habronattus oregonensis reveals rearranged and extremely truncated tRNAs. Mol. Biol. Evol 2004, 21, 893–902. [Google Scholar]
- Crozier, R.H.; Crozier, Y.C. The mitochondrial genome of the honeybee Apis mellifera: Complete sequence and genome organization. Genetics 1993, 133, 97–117. [Google Scholar]
- Xia, X. Maximizing transcription efficiency causes codon usage bias. Genetics 1996, 144, 1309–1320. [Google Scholar]
- Shao, R.F.; Barker, S.C. The highly rearranged mitochondrial genome of the plague thrips, Thrips imaginis (Insecta: Thysanoptera): Convergence of two novel gene boundaries and an extraordinary arrangement of rRNA genes. Mol. Biol. Evol 2003, 20, 362–370. [Google Scholar]
- Yamauchi, M.M.; Miya, M.U.; Nishida, M. Complete mitochondrial DNA sequence of the swimming crab, Portunus trituberculatus (Crustacea: Decapoda: Brachyura). Gene 2003, 311, 129–135. [Google Scholar]
- Gutell, R.R.; Lee, J.C.; Cannone, J.J. The accuracy of ribosomal RNA comparative structure models. Curr. Opin. Struct. Biol 2002, 12, 301–310. [Google Scholar]
- Gillespie, J.J.; Johnston, J.S.; Cannone, J.J.; Gutell, R.R. Characteristics of the nuclear (18S, 5.8S, 28S and 5S) and mitochondrial (12S and 16S) rRNA genes of Apis mellifera (Insecta: Hymenoptera): Structure, organization, and retrotransposable elements. Insect Mol. Biol 2006, 15, 657–686. [Google Scholar]
- Cameron, S.L.; Whiting, M.F. The complete mitochondrial genome of the tobacco hornworm, Manduca sexta (Insecta: Lepidoptera: Sphingidae), and an examination of mitochondrial gene variability within butterflies and moths. Gene 2008, 408, 112–123. [Google Scholar]
- Chai, H.N.; Du, Y.Z.; Zhai, B.P. Characterization of the complete mitochondrial genomes of Cnaphalocrocis medinalis and Chilo suppressalis (Lepidoptera: Pyralidae). Int. J. Biol. Sci 2012, 8, 561–579. [Google Scholar]
- Zhang, D.X.; Hewitt, G.M. Insect mitochondrial control region: A review of its structure, evolution and usefulness in evolutionary studies. Biochem. Syst. Ecol 1997, 25, 99–120. [Google Scholar]
- Clary, D.O.; Wolstenholme, D.R. Drosophila mitochondrial DNA: Conserved sequences in the A+T-rich region and supporting evidence for a secondary structure model of the small ribosomal RNA. J. Mol. Evol 1987, 25, 116–125. [Google Scholar]
- Zhang, D.; Szymura, J.M.; Hewitt, G.M. Evolution and structural conservation of the control region of insect mitochondrial DNA. J. Mol. Evol 1995, 40, 382–391. [Google Scholar]
- Rand, D.M.; Harrison, R.G. Molecular population genetics of mtDNA size variation in crickets. Genetics 1989, 121, 551–569. [Google Scholar]
- Schultheis, A.S.; Weigt, L.A.; Hendricks, A.C. Arrangement and structural conservation of the mitochondrial control region of two species of Plecoptera: Utility of tandem repeat-containing regions in studies of population genetics and evolutionary history. Insect Mol. Biol 2002, 11, 605–610. [Google Scholar]
- Hong, M.Y.; Lee, E.M.; Jo, Y.H.; Park, H.C.; Kim, S.R.; Hwang, J.S.; Jin, B.R.; Kang, P.D.; Kim, K.G.; Han, Y.S.; Kim, I. Complete nucleotide sequence and organization of the mitogenome of the silk moth Caligula boisduvalii (Lepidoptera: Saturniidae) and comparison with other lepidopteran insects. Gene 2008, 413, 49–57. [Google Scholar]
- Kim, S.R.; Kim, M.I.; Hong, M.Y.; Kim, K.Y.; Kang, P.D.; Hwang, J.S.; Han, Y.S.; Jin, B.R.; Kim, I. The complete mitogenome sequence of the Japanese oak silkmoth, Antheraea yamamai (Lepidoptera: Saturniidae). Mol. Biol. Rep 2009, 36, 1871–1880. [Google Scholar]
- Kim, I.; Cha, S.Y.; Yoon, M.H.; Hwang, J.S.; Lee, S.M.; Sohn, H.D.; Jin, B. The complete nucleotide sequence and gene organization of the mitochondrial genome of the oriental mole cricket, Gryllotalpa orientalis (Orthoptera: Gryllotalpidae). Gene 2005, 353, 155–168. [Google Scholar]
- Lee, E.S.; Shin, K.S.; Kim, M.S.; Park, H.; Cho, S.; Kim, C.B. The mitochondrial genome of the smaller tea tortrix Adoxophyes honmai (Lepidoptera: Tortricidae). Gene 2006, 373, 52–57. [Google Scholar]
- Hassanin, A. Phylogeny of Arthropoda inferred from mitochondrial sequences: Strategies for limiting the misleading effects of multiple changes in pattern and rates of substitution. Mol. Phylogenet. Evol 2006, 38, 100–116. [Google Scholar]
- Kristensen, N.P.; Skalski, A.W. Phylogeny and Paleontology. Lepidoptera: Moths and Butterflies. 1. Evolution, Systematics, and Biogeography. Handbook of Zoology 1999, IV(Part 35), 7–25. [Google Scholar]
- Yang, L.; Wei, Z.J.; Hong, G.Y.; Jiang, S.T.; Wen, L.P. The complete nucleotide sequence of the mitochondrial genome of Phthonandria atrilineata (Lepidoptera: Geometridae). Mol. Biol. Rep 2009, 36, 1441–1449. [Google Scholar]
- Hu, X.L.; Cao, G.L.; Xue, R.Y.; Zheng, X.J.; Zhang, X.; Duan, H.R.; Gong, C.L. The complete mitogenome and phylogenetic analysis of Bombyx mandarina strain Qingzhou. Mol. Biol. Rep 2010, 37, 2599–2608. [Google Scholar]
- Lu, C.; Liu, Y.Q.; Liao, X.S.; Li, B.; Xiang, Z.X.; Han, H.; Wang, X.G. Complete sequence determination and analysis of Bombyx mori mitochondrial genome. J. Agric. Biotechnol 2002, 10, 163–170. [Google Scholar]
- Jiang, S.T.; Hong, G.Y.; Yu, M.; Li, N.; Yang, Y.; Liu, Y.Q.; Wei, Z.J. Characterization of the complete mitochondrial genome of the giant silkworm moth, Eriogyna pyretorum (Lepidoptera: Saturniidae). Int. J. Biol. Sci 2009, 5, 351–365. [Google Scholar]
- Xia, J.; Hu, J.; Zhu, G.P.; Zhu, C.D.; Hao, J.S. Sequencing and analysis of the complete mitochondrial genome of Calinaga davidis Oberthür (Lepidoptera: Nymphalidae). Acta Entomol. Sin 2011, 54, 555–565. [Google Scholar]
- Coates, B.S.; Sumerford, D.V.; Hellmich, R.L.; Lewis, L.C. Partial mitochondrial genome sequences of Ostrinia nubilalis and Ostrinia furnicalis. Int. J. Biol. Sci 2005, 1, 13–18. [Google Scholar]
- Lewis, D.L.; Farr, C.L.; Kaguni, L.S. Drosophila melanogaster mitochondrial DNA: Completion of the nucleotide sequence and evolutionary comparisons. Insect Mol. Biol 1995, 4, 263–278. [Google Scholar]
- Hwang, U.W.; Park, C.J.; Yong, T.S.; Kim, W. One-step PCR amplification of complete arthropod mitochondrial genomes. Mol. Phylogenet. Evol 2001, 19, 345–352. [Google Scholar]
- Simon, C.; Frati, F.; Beckenbach, A.T.; Crespi, B.; Liu, H.; Flook, P. Evolution, weighting and phylogenetics utility of mitochondrial gene sequences and compilation of conserved polymerase chain reaction primers. Ann. Entomol. Soc. Am 1994, 87, 651–701. [Google Scholar]
- Wei, Z.J.; Hong, G.Y.; Jiang, S.T.; Tong, Z.X.; Lu, C. Characters and expression of the gene encoding DH, PBAN and other FXPRLamide family neuropeptides in Antheraea pernyi. J. Appl. Entomol 2008, 132, 59–67. [Google Scholar]
- Staden Package. Available online: http://staden.sourceforge.net accessed on 5 December 2011.
- Larkin, M.A.; Blackshields, G.; Brown, N.P.; Chenna, R.; McGettigan, P.A.; McWilliam, H.; Valentin, F.; Wallace, I.M.; Wilm, A.; Lopez, R. Clustal W and Clustal X version 2.0. Bioinformatics 2007, 23, 2947–2948. [Google Scholar]
- Perna, N.T.; Kocher, T.D. Patterns of nucleotide composition at fourfold degenerate sites of animal mitochondrial genomes. J. Mol. Evol 1995, 41, 353–358. [Google Scholar]
- Lowe, T.M.; Eddy, S.R. tRNAscan-SE: A program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res 1997, 25, 955–964. [Google Scholar]
- Cannone, J.J.; Subramanian, S.; Schnare, M.N.; Collett, J.R.; D’Souza, L.M.; Du, Y.; Feng, B.; Lin, N.; Madabusi, L.V.; Müller, K.M.; et al. The comparative RNA web (CRW) site: An online database of comparative sequence and structure information for ribosomal, intron, and other RNAs. BMC Bioinform 2002, 3. [Google Scholar] [CrossRef]
- Gutell, R.R.; Larsen, N.; Woese, C.R. Lessons from an evolving rRNA: 16S and 23S rRNA structures from a comparative perspective. Microbiol. Rev 1994, 58, 10–26. [Google Scholar]
- Buckley, T.R.; Simon, C.; Flook, P.K.; Misof, B. Secondary structure and conserved motifs of the frequently sequenced domains IV and V of the insect mitochondrial large subunit rRNA gene. Insect Mol. Biol 2000, 9, 565–580. [Google Scholar]
- Benson, G. Tandem repeats finder: A program to analyze DNA sequences. Nucleic Acids Res 1999, 27, 573–580. [Google Scholar]
- Bachtrog, D. Reduced selection for codon usage bias in Drosophila Miranda. J. Mol. Evol 2007, 64, 586–590. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Direction | Site | Size (bp) | Intergenic spacer | Anticodon | Start codon | Stop codon |
|---|---|---|---|---|---|---|---|
| trnM | F | 1..68 | 68 | 0 | 32..34 CAT | ||
| trnI | F | 69..133 | 65 | −3 | 98..100 GAT | ||
| trnQ | R | 131..199 | 69 | 68 | 167..169 TTG | ||
| nad2 | F | 268..1284 | 1017 | 6 | ATT | TAA | |
| trnW | F | 1291..1358 | 68 | −8 | 1322..1324 TCA | ||
| trnC | R | 1351..1423 | 73 | 4 | 1391..1393 GCA | ||
| trnY | R | 1428..1494 | 67 | 5 | 1460..1462 GTA | ||
| cox1 | F | 1500..3030 | 1531 | 3 | CGA | T- | |
| trnL2(UUR) | F | 3034..3100 | 67 | 0 | 3064..3066 TAA | ||
| cox2 | F | 3101..3782 | 682 | 0 | ATT | T- | |
| trnK | F | 3783..3853 | 71 | 0 | 3813..3815 CTT | ||
| trnD | F | 3854..3919 | 66 | 0 | 3884..3886 GTC | ||
| atp8 | F | 3920..4087 | 168 | −11 | ATT | TAA | |
| atp6 | F | 4077..4754 | 696 | −1 | ATG | TAA | |
| cox3 | F | 4754..5542 | 789 | 2 | ATG | TAA | |
| trnG | F | 5545..5609 | 65 | 0 | 5575..5577 TCC | ||
| nad3 | F | 5610..5963 | 354 | 16 | ATT | TAA | |
| trnA) | F | 5980..6048 | 69 | 2 | 6010..6012 TGC | ||
| trnR | F | 6051..6114 | 64 | 9 | 6078..6080 TCG | ||
| trnN | F | 6124..6188 | 65 | 2 | 6154..6156 GTT | ||
| trnS1(AGN) | F | 6191..6256 | 66 | 0 | 6216..6218 GCT | ||
| trnE | F | 6257..6324 | 68 | 8 | 6287..6289 TTC | ||
| trnF | R | 6333..6399 | 67 | −1 | 6364..6366 GAA | ||
| nad5 | R | 6399..8150 | 1752 | 0 | ATT | TAA | |
| trnH | R | 8151..8215 | 65 | 0 | 8183..8185 GTG | ||
| nad4 | R | 8216..9554 | 1339 | 44 | ATG | T- | |
| nad4L | R | 9599..9892 | 294 | 7 | ATG | TAA | |
| trnT | F | 9900..9969 | 70 | 0 | 9929..9931 TGT | ||
| trnP | R | 9970..10034 | 65 | 7 | 10002..10004 TGG | ||
| nad6 | F | 10042..10575 | 534 | 44 | ATC | TAA | |
| cob | F | 10620..11768 | 1149 | 1 | ATG | TAA | |
| trnS2(UCN) | F | 11770..11836 | 67 | 18 | 11802..11804 TGA | ||
| nad1 | R | 11855..12799 | 945 | 1 | ATG | TAA | |
| trnL1(CUN) | R | 12801..12867 | 67 | −1 | 12836..12838 TAG | ||
| rrnL | R | 12867..14251 | 1385 | 0 | - | ||
| trnV | R | 14253..14318 | 66 | 0 | 14284..14286 TAC | ||
| rrnS | R | 14319..15102 | 784 | 0 | |||
| A+T-rich region | 15103..15413 | 311 | 0 |
| Region | Whole mitogenome | PCGs | |||||
|---|---|---|---|---|---|---|---|
| Species | Length (bp) | AT (%) | Length (bp) | AT (%) | 1st position (%) | 2nd position (%) | 3rd position (%) |
| H. armigera | 15347 | 81.0 | 11,133 | 79.4 | 70.2 | 94.1 | 73.8 |
| H. cunea | 15481 | 80.4 | 11,136 | 78.5 | 73.2 | 70.3 | 92.0 |
| L. dispar | 15569 | 79.9 | 11,169 | 77.7 | 90.2 | 72.9 | 70.1 |
| O. lunifer | 15593 | 77.8 | 11,196 | 75.7 | 72.0 | 70.0 | 85.1 |
| S. inferens | 15413 | 80.3 | 11,160 | 78.6 | 87.7 | 72.9 | 75.2 |
© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Chai, H.-N.; Du, Y.-Z. The Complete Mitochondrial Genome of the Pink Stem Borer, Sesamia inferens, in Comparison with Four Other Noctuid Moths. Int. J. Mol. Sci. 2012, 13, 10236-10256. https://doi.org/10.3390/ijms130810236
Chai H-N, Du Y-Z. The Complete Mitochondrial Genome of the Pink Stem Borer, Sesamia inferens, in Comparison with Four Other Noctuid Moths. International Journal of Molecular Sciences. 2012; 13(8):10236-10256. https://doi.org/10.3390/ijms130810236
Chicago/Turabian StyleChai, Huan-Na, and Yu-Zhou Du. 2012. "The Complete Mitochondrial Genome of the Pink Stem Borer, Sesamia inferens, in Comparison with Four Other Noctuid Moths" International Journal of Molecular Sciences 13, no. 8: 10236-10256. https://doi.org/10.3390/ijms130810236