A Theoretical Study on Reductive Debromination of Polybrominated Diphenyl Ethers



Abstract
:1. Introduction
2. Results and Discussion
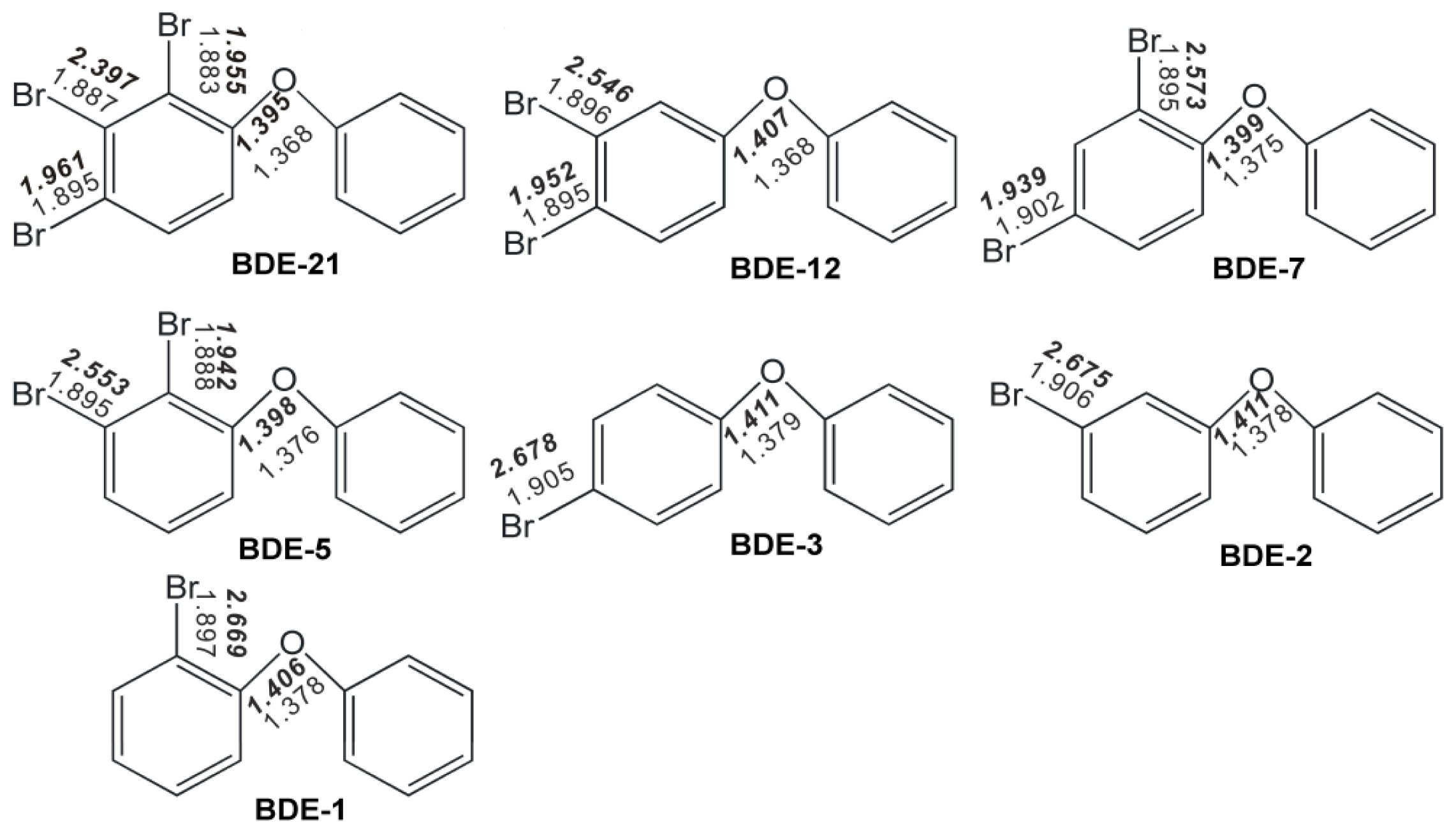
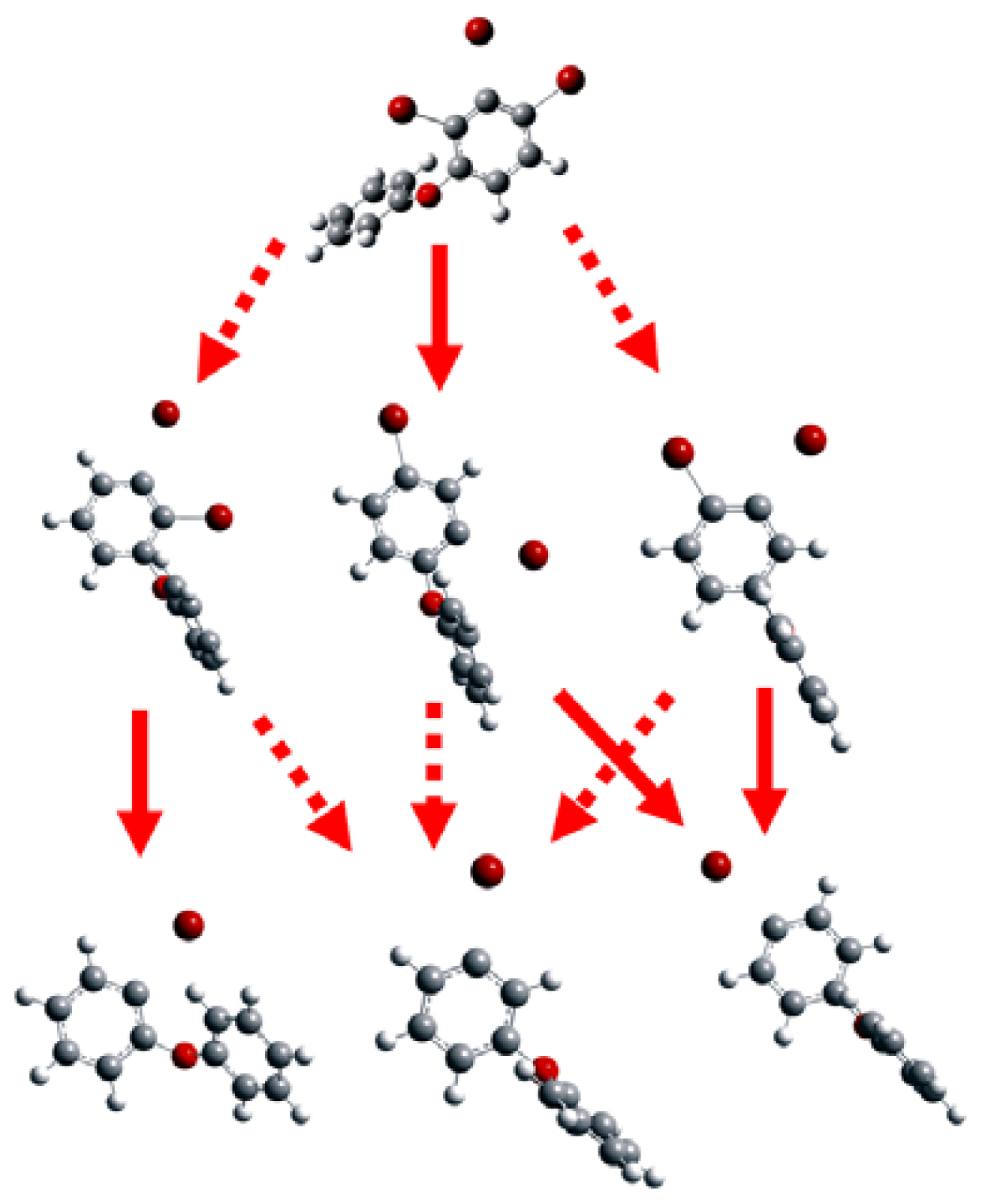
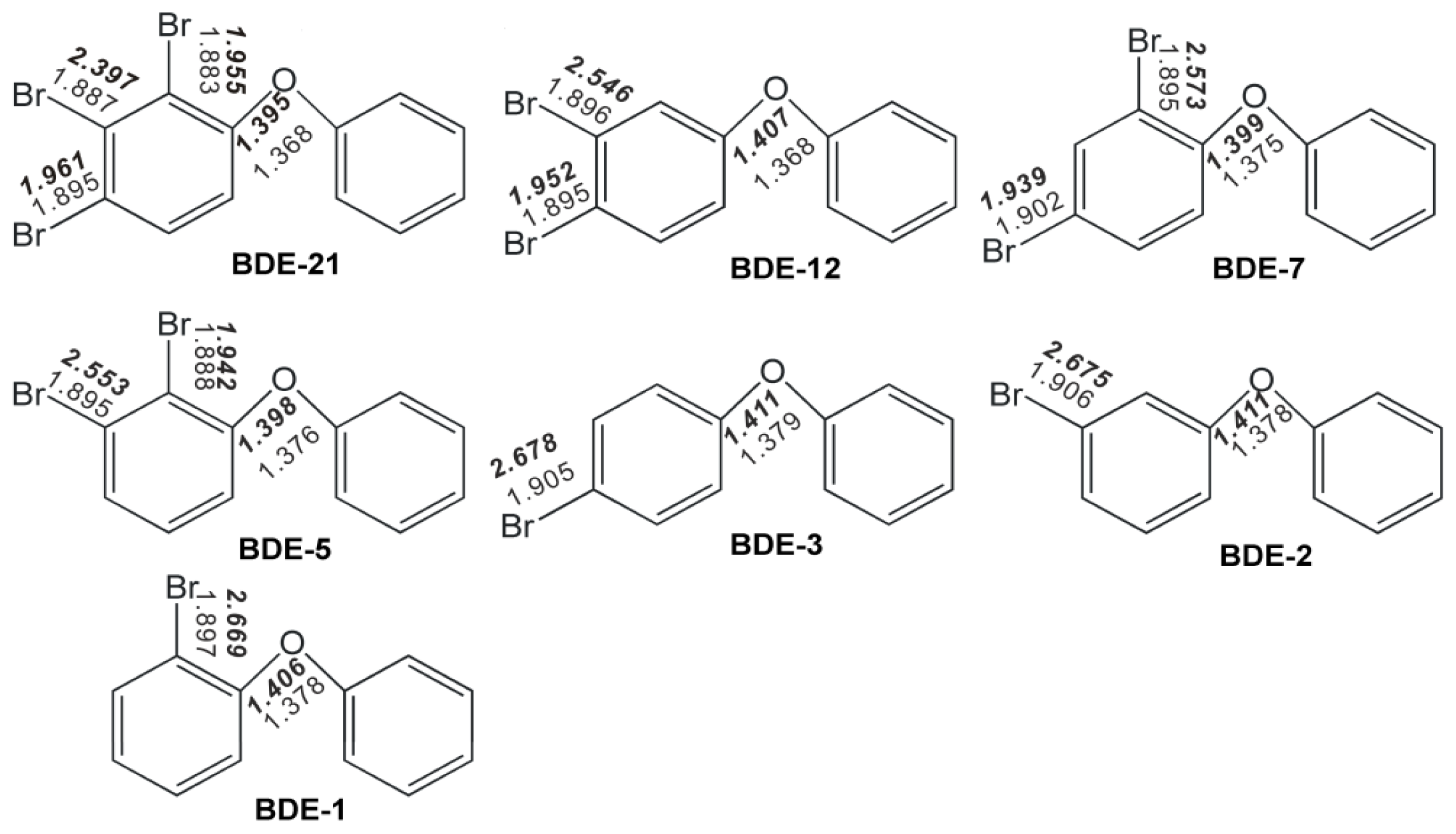
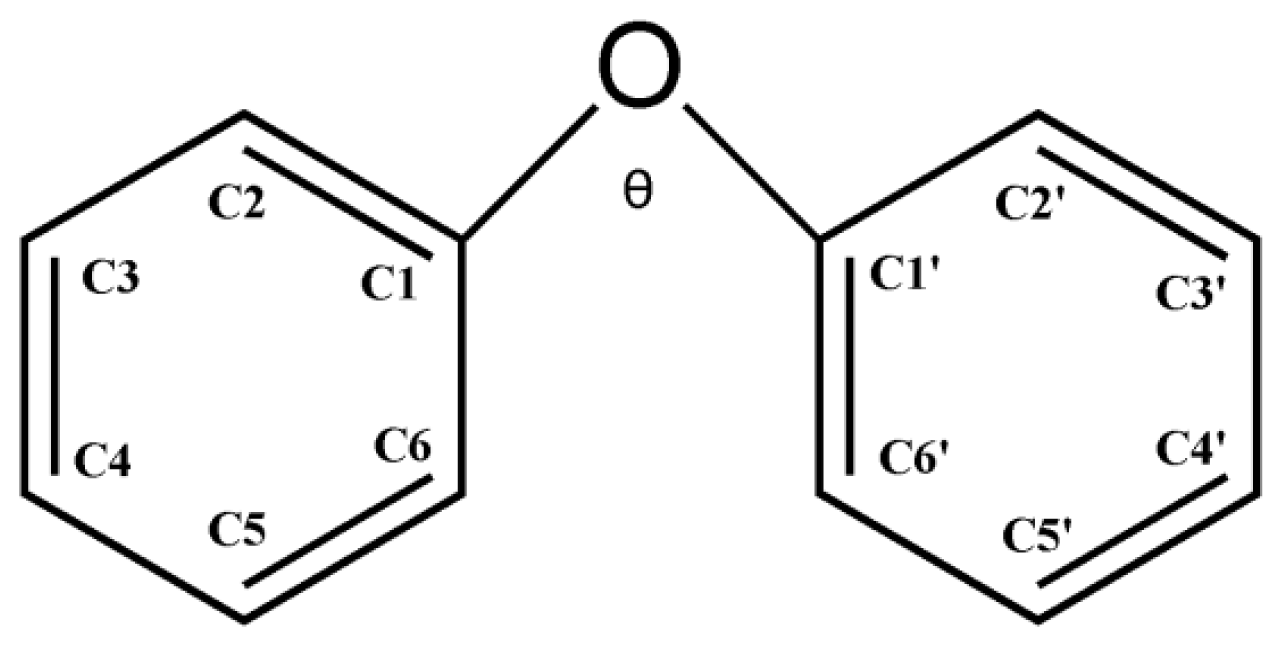
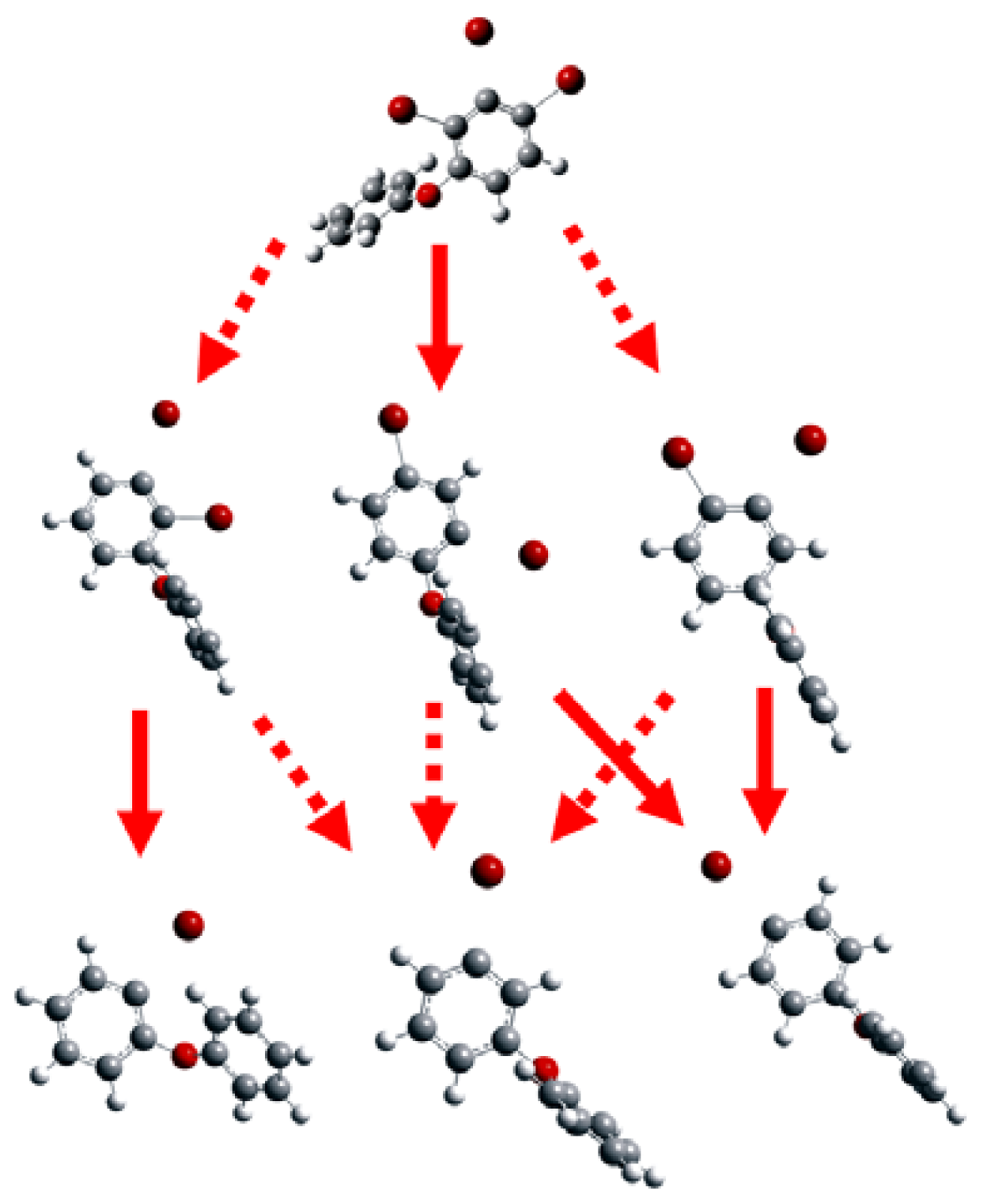
2.1. Use of Optimized Geometries of BDE Congeners for Prediction of Dominant Debromination Products by nZVI
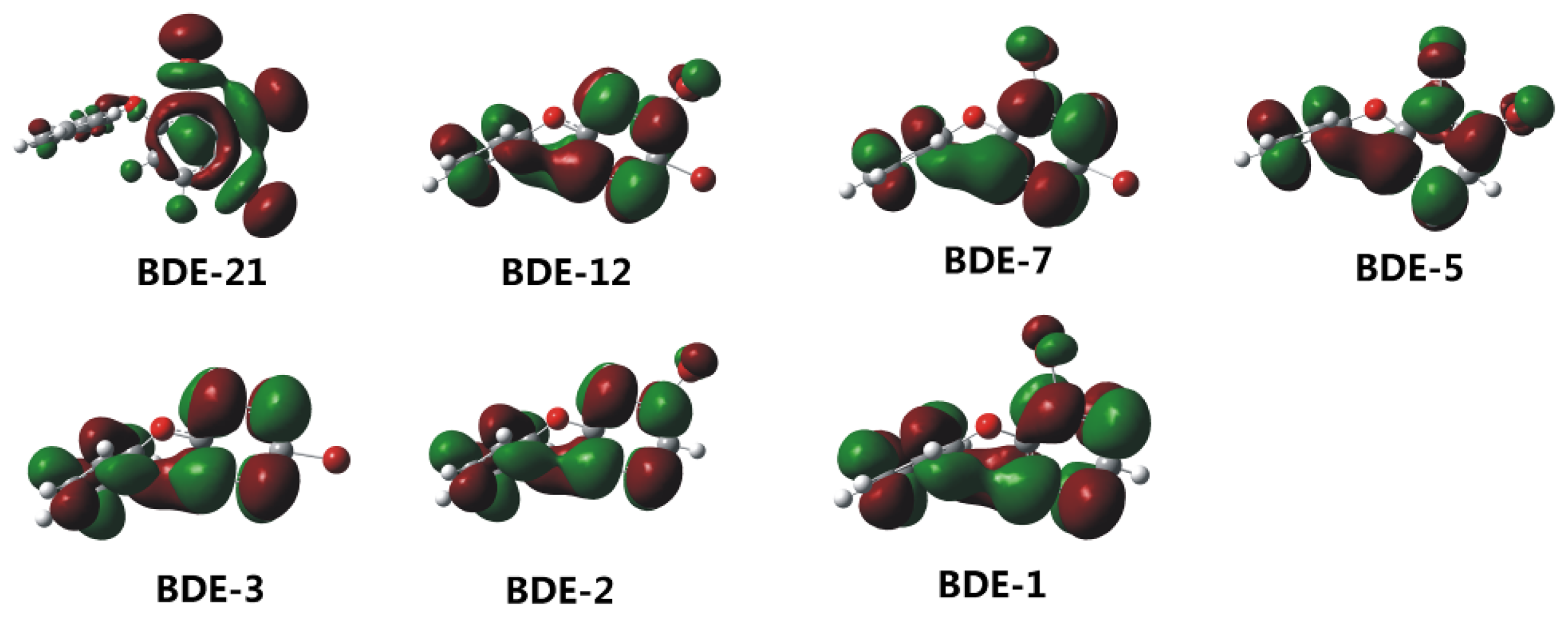
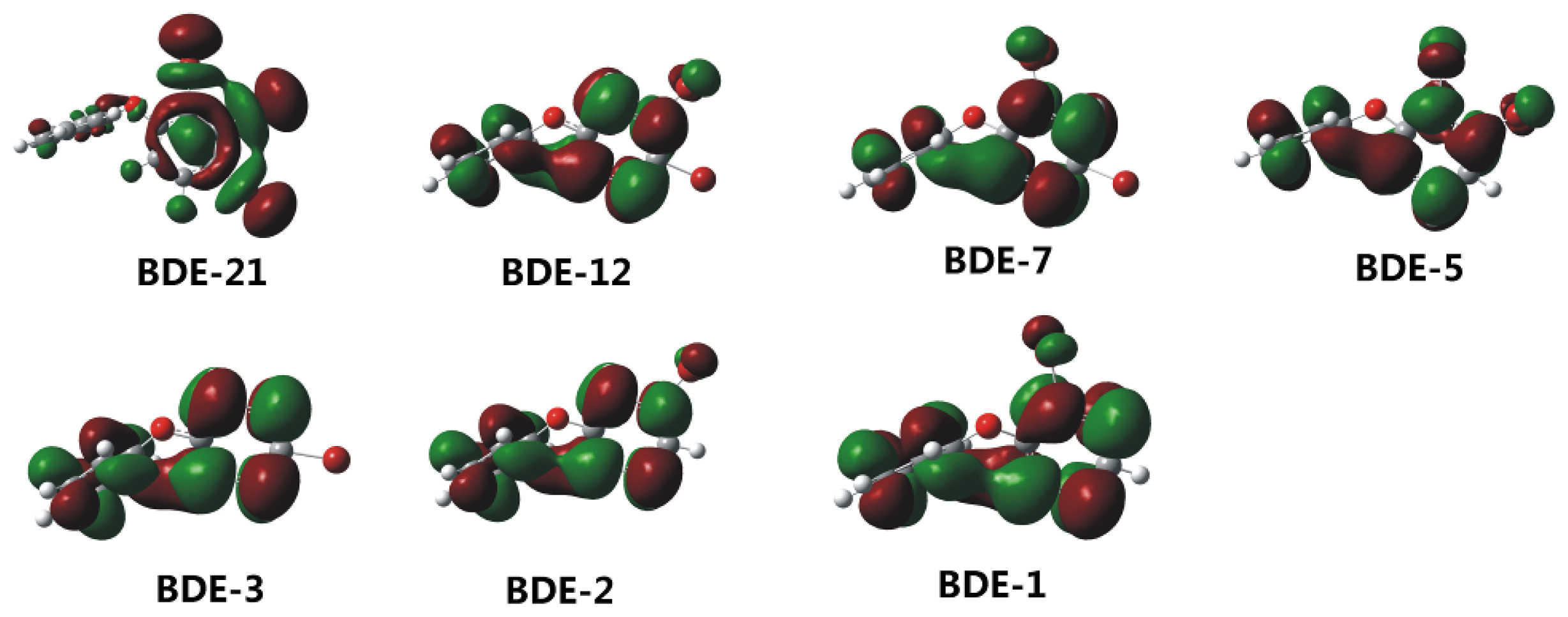
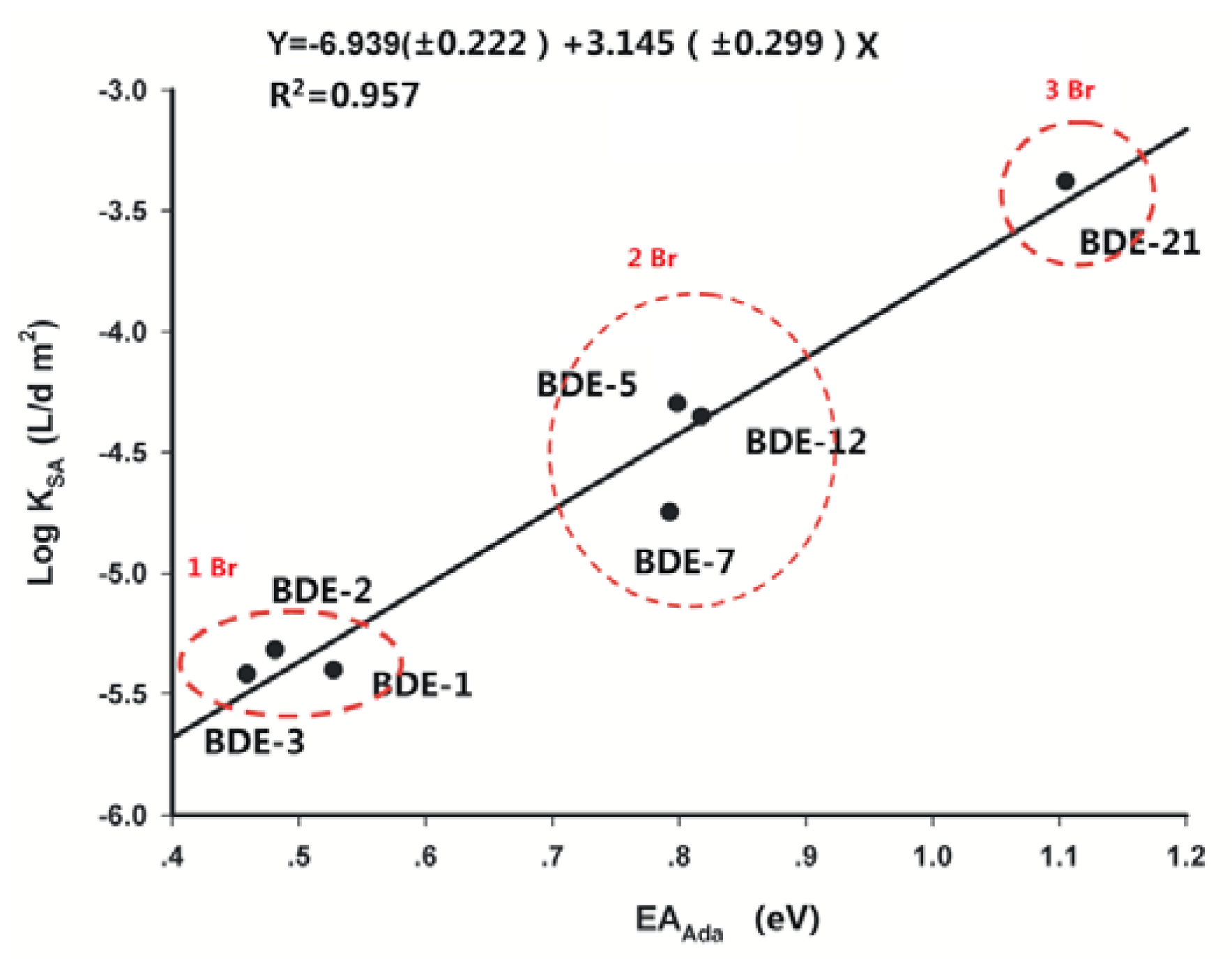
2.2. Frontier Orbital Energies, Adiabatic Electron Affinity and Their Relevance to Kinetics of Debromination Reactions by nZVI
3. Experimental Section
4. Conclusions
Supplementary Information
ijms-13-09332-s001.pdfAcknowledgments
References
- Chen, L.G.; Mai, B.X.; Bi, X.H.; Chen, S.J.; Wang, X.M.; Ran, Y.; Luo, X.J.; Sheng, G.Y.; Fu, J.M.; Zeng, E.Y. Concentration levels, compositional profiles, and gas-particle partitioning of polybrominated diphenyl ethers in the atmosphere of an urban city in South China. Environ. Sci. Technol 2006, 18, 1190–1196. [Google Scholar]
- Hites, R.A. Polybrominated diphenyl ethers in the environment and in people: A meta-analysis of concentrations. Environ. Sci. Technol 2004, 38, 945–956. [Google Scholar]
- Darnerud, P.O.; Eriksen, G.S.; Jóhannesson, T.; Larsen, P.B.; Viluksela, M. Polybrominated diphenyl ethers: occurrence, dietary exposure, and toxicology. Environ. Health Perspect 2001, 109(Suppl 1), 49–68. [Google Scholar]
- Meerts, I.A.T.M.; Letcher, R.J.; Hoving, S.; Marsh, G.; Bergman, Å.; Lemmen, J.G.; Burg, B.V.; Brouwer, A. In vitro estrogenicity of polybrominated diphenyl ethers, hydroxylated PBDEs, and polybrominated bisphenol A compounds. Environ. Health Perspect 2001, 109, 399–407. [Google Scholar]
- Sjödin, A.; Patterson, J.; Patterson, G.D.; Bergman, Å. A review on human exposure to brominated flame retardants—Particularly polybrominated diphenyl ethers. Environ. Int 2003, 29, 829–839. [Google Scholar]
- Haglund, P.S.; Zook, D.R.; Buser, H.; Hu, J.W. Identification and quantification of polybrominated diphenyl ethers and methoxy-polybrominated diphenyl ethers in baltic biota. Environ. Sci. Technol 1997, 31, 3281–3287. [Google Scholar]
- The European parliament and the council of the European Union, Directive 2002/95/EC of the European Parliament and of the Council of 27 January 2003 on the Restriction of the Use of Certain Hazardous Substances in Electrical and Electronic Equipment; OJ L37; European Union: Brussels, Belgium, 2003.
- Canton, R.F.; Scholten, D.E.; Marsh, G.; de Jong, P.C.; van den Berg, M. Inhibition of human placental aromatase activity by hydroxylated polybrominated diphenyl ethers (OH-PBDEs). Toxicol. Appl. Pharmacol 2008, 227, 68–75. [Google Scholar]
- Ward, J.; Mohapatra, S.P.; Mitchell, A. An overview of policies for managing polybrominated diphenyl ethers (PBDEs) in the Great Lakes basin. Environ. Int 2008, 34, 1148–1156. [Google Scholar]
- Li, X.Q.; Elliott, D.W.; Zhang, W.X. Zero-valent iron nanoparticles for abatement of environmental pollutants: Materials and engineering aspects. Crit. Rev. Solid State 2006, 31, 111–122. [Google Scholar]
- Shih, Y.; Tai, Y. Reaction of decabrominated diphenyl ether by zerovalent iron nanoparticles. Chemosphere 2010, 78, 1200–1206. [Google Scholar]
- Li, A.; Tai, C.; Zhao, Z.S.; Wang, Y.W.; Zhang, Q.H.; Jiang, G.B.; Hu, J.T. Debromination of decabrominated diphenyl ether by resin-bound iron nanoparticles. Environ. Sci. Technol 2007, 41, 6841–6846. [Google Scholar]
- Zhuang, Y.; Ahn, S.; Luthy, R.G. Debromination of polybrominated diphenyl ethers by nanoscale zerovalent iron: Pathways, kinetics, and reactivity. Environ. Sci. Technol 2010, 44, 8236–8242. [Google Scholar]
- Arulmozhiraja, S.; Morita, M. Electron affinities and reductive dechlorination of toxic polychlorinated dibenzofurans: A density functional theory study. J. Phys. Chem. A 2004, 108, 3499–3508. [Google Scholar]
- Zhao, Y.Y.; Tao, F.M.; Zeng, E.Y. Structures, reductive dechlorination, and electron affinities of selected polychlorinated dibenzo-p-dioxins: Density functional theory study. J. Phys. Chem. A 2007, 111, 11638–11644. [Google Scholar]
- Keum, Y.; Li, Q.X. Reductive debromination of polybrominated diphenyl ethers by zerovalent iron. Environ. Sci. Technol 2005, 39, 2280–2286. [Google Scholar]
- Zhao, Y.Y.; Tao, F.M.; Zeng, E.Y. Theoretical study on the chemical properties of polybrominated diphenyl ethers. Chemosphere 2008, 70, 901–907. [Google Scholar]
- Hu, J.W.; Eriksson, L.; Bergman, Å.; Jakobsson, E.; Kolehmainen, E.; Knuutinen, J.; Suontamo, R.; Wei, X.H. Molecular orbital studies on brominated diphenyl ethers. Part I—Conformational properties. Chemosphere 2005, 59, 1033–1041. [Google Scholar]
- Hu, J.W.; Eriksson, L.; Bergman, Å.; Jakobsson, E.; Kolehmainen, E.; Knuutinen, J.; Suontamo, R.; Wei, X.H. Molecular orbital studies on brominated diphenyl ethers. Part II—Reactivity and quantitative structure-activity (property) relationships. Chemosphere 2005, 59, 1043–1057. [Google Scholar]
- Zhou, J.; Chen, J.W.; Liang, C.; Xie, Q.; Wang, Y.N.; Zhang, S.Y.; Qiao, X.L.; Li, X.H. Quantum chemical investigation on the mechanism and kinetics of PBDE photooxidation by ·OH: A case study for BDE-15. Environ. Sci. Technol 2011, 45, 4839–4845. [Google Scholar]
- Vogel, T.M.; Criddle, C.S.; McCarty, P.L. ES Critical Reviews: Transformations of halogenated aliphatic compounds. Environ. Sci. Technol 1987, 21, 722–736. [Google Scholar]
- Rienstra-Kiracofe, J.C.; Tschumper, G.S.; Schaefer, H.F., III; Nandi, S.; Ellison, G.B. Atomic and molecular electron affinities: Photoelectron experiments and theoretical computations. Chem. Rev 2003, 102, 231–282. [Google Scholar]
- Zhuang, Y.; Ahn, S.; Seyfferth, A.L.; Masue-Slowey, Y.; Fendorf, S.; Luthy, R.G. Dehalogenation of polybrominated diphenyl ethers and polychlorinated biphenyl by bimetallic, impregnated, and nanoscale zerovalent iron. Environ. Sci. Technol 2011, 45, 4896–4903. [Google Scholar]
- Fukui, K.; Yonezawa, T.; Nagata, C.; Shingu, H. Molecular orbital theory of orientation in aromatic, heteroaromatic and other conjugated molecules. J. Chem. Phys 1954, 22, 1433–1442. [Google Scholar]
- Scherer, M.M.; Balko, B.A.; Gallagher, D.A.; Trantnyek, P.G. Correlation analysis of rate constants for dechlorination by zerov-alent iron. Environ. Sci. Technol 1998, 32, 3026–3033. [Google Scholar]
- Lowry, G.V.; Johnson, K.M. Congener-specific dechlorination of dissolved PCBs by microscale and nanoscale zerovalent iron in a water/methanol solution. Environ. Sci. Technol 2004, 38, 5208–5216. [Google Scholar]
- Agarwal, S.; Al-Abed, S.R.; Dionysiou, D.D.; Graybill, E. Reactivity of substituted chlorines and ensuing dechlorination pathways of select PCB congeners with Pd/Mg bimetallics. Environ. Sci. Technol 2009, 43, 915–921. [Google Scholar]
- Kim, J.H.; Tratnyek, P.G.; Chang, Y.S. Rapid dechlorination of polychlorinated dibenzo-p-dioxins by bimetallic and nanosized zerovalent iron. Environ. Sci. Technol 2008, 42, 4106–4112. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Montgomery, J.A., Jr.; Vreven, T.; Kudin, K.N.; Burant, J.C.; et al. Gaussian 03; Gaussian, Inc: Wallingford, CT, USA, 2004. [Google Scholar]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys 1993, 98, 5648–5652. [Google Scholar]
- Lee, C.; Yang, W.; Parr, R.G. Development of the colle–salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar]
- Jon Baker, A.S.; Andzelm, J. Spin contamination in density functional theory. Chem. Phys. Lett 1993, 216, 380–388. [Google Scholar]
- Zeng, X.; Simonich, S.L.M.; Robrock, K.R.; Korytar, P.; Alvarez-Cohen, L.; Barofsky, D.F. Application of a congener-specific debromination model to study photodebromination, anaerobic microbial debromination, and Fe0 reduction of polybrominated diphenyl ethers. Environ. Toxicol. Chem 2010, 29, 770–778. [Google Scholar]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 6-31G(d) | 6-31+G(d) | 6-31G(d,p) | 6-311G(d,p) | 6-31+G(d) a | aug-cc-pVDZ a | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| neutral | anion | neutral | anion | neutral | anion | neutral | anion | neutral | neutral | |
| C1–O | 1.368 | 1.400 | 1.375 | 1.399 | 1.368 | 1.399 | 1.365 | 1.398 | 1.375 | 1.370 |
| C1′–O | 1.388 | 1.374 | 1.388 | 1.370 | 1.388 | 1.373 | 1.39 | 1.369 | 1.388 | 1.391 |
| C1–Br | 1.902 | 2.703 | 1.895 | 2.573 | 1.902 | 2.695 | 1.908 | 2.698 | 1.895 | 1.909 |
| C3–Br | 1.910 | 1.946 | 1.902 | 1.939 | 1.910 | 1.946 | 1.916 | 1.952 | 1.902 | 1.971 |
| C1–O–C1′ | 120.6 | 123.0 | 120.4 | 120.7 | 120.6 | 123.5 | 120.1 | 123.1 | 120.4 | 120.3 |
| IUPAC no. | 6-31G(d) | 6-31+G(d) | 6-31G(d,p) | 6-311G(d,p) | Log KSA a | ||||
|---|---|---|---|---|---|---|---|---|---|
| ELUMO | EAAda | ELUMO | EAAda | ELUMO | EAAda | ELUMO | EAAda | ||
| BDE21 | −0.0404 | 0.6481 | −0.0527 | 0.9558 | −0.0404 | 0.6509 | −0.0544 | 1.1048 | −3.378 |
| BDE12 | −0.0279 | 0.3131 | −0.0439 | 0.6708 | −0.0284 | 0.3142 | −0.0400 | 0.8174 | −4.351 |
| BDE7 | −0.0286 | 0.3707 | −0.0471 | 0.7106 | −0.0291 | 0.3743 | −0.0402 | 0.7926 | −4.747 |
| BDE5 | −0.0299 | 0.3219 | −0.0437 | 0.6817 | −0.0303 | 0.3231 | −0.0375 | 0.7983 | −4.298 |
| BDE3 | −0.0196 | −0.0394 | −0.0363 | 0.3549 | −0.0204 | −0.0398 | −0.0325 | 0.4589 | −5.417 |
| BDE2 | −0.0188 | −0.0195 | −0.0358 | 0.3701 | −0.0196 | −0.0194 | −0.0315 | 0.4812 | −5.317 |
| BDE1 | −0.0198 | 0.0969 | −0.0357 | 0.4491 | −0.0204 | 0.1029 | −0.0300 | 0.5272 | −5.401 |
| R2b | 0.961 | 0.896 | 0.863 | 0.915 | 0.961 | 0.892 | 0.896 | 0.957 | - |
© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Hu, J.-W.; Zhuang, Y.; Luo, J.; Wei, X.-H.; Huang, X.-F. A Theoretical Study on Reductive Debromination of Polybrominated Diphenyl Ethers. Int. J. Mol. Sci. 2012, 13, 9332-9342. https://doi.org/10.3390/ijms13079332
Hu J-W, Zhuang Y, Luo J, Wei X-H, Huang X-F. A Theoretical Study on Reductive Debromination of Polybrominated Diphenyl Ethers. International Journal of Molecular Sciences. 2012; 13(7):9332-9342. https://doi.org/10.3390/ijms13079332
Chicago/Turabian StyleHu, Ji-Wei, Yuan Zhuang, Jin Luo, Xiong-Hui Wei, and Xian-Fei Huang. 2012. "A Theoretical Study on Reductive Debromination of Polybrominated Diphenyl Ethers" International Journal of Molecular Sciences 13, no. 7: 9332-9342. https://doi.org/10.3390/ijms13079332