TRAIL and Paclitaxel Synergize to Kill U87 Cells and U87-Derived Stem-Like Cells in Vitro

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
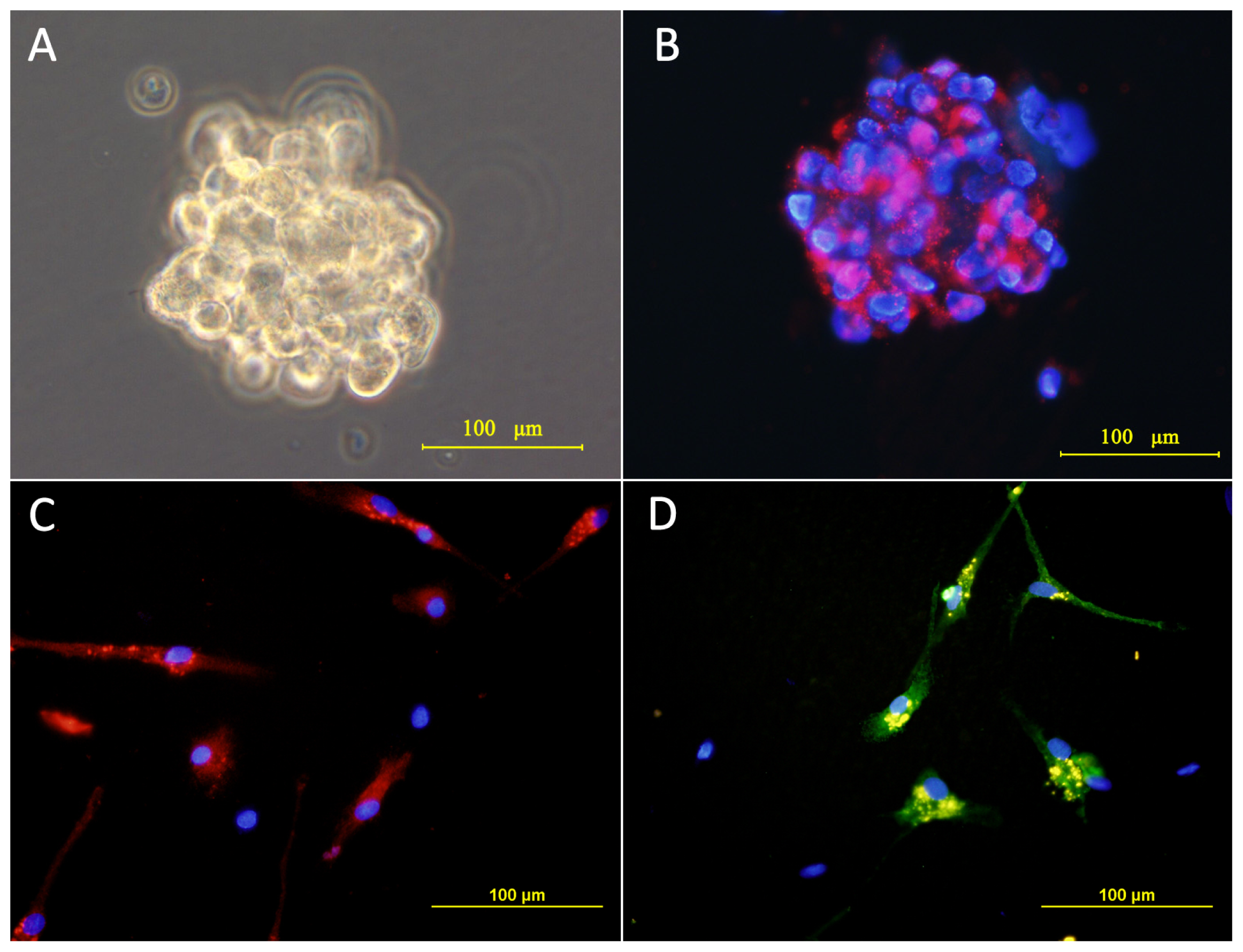
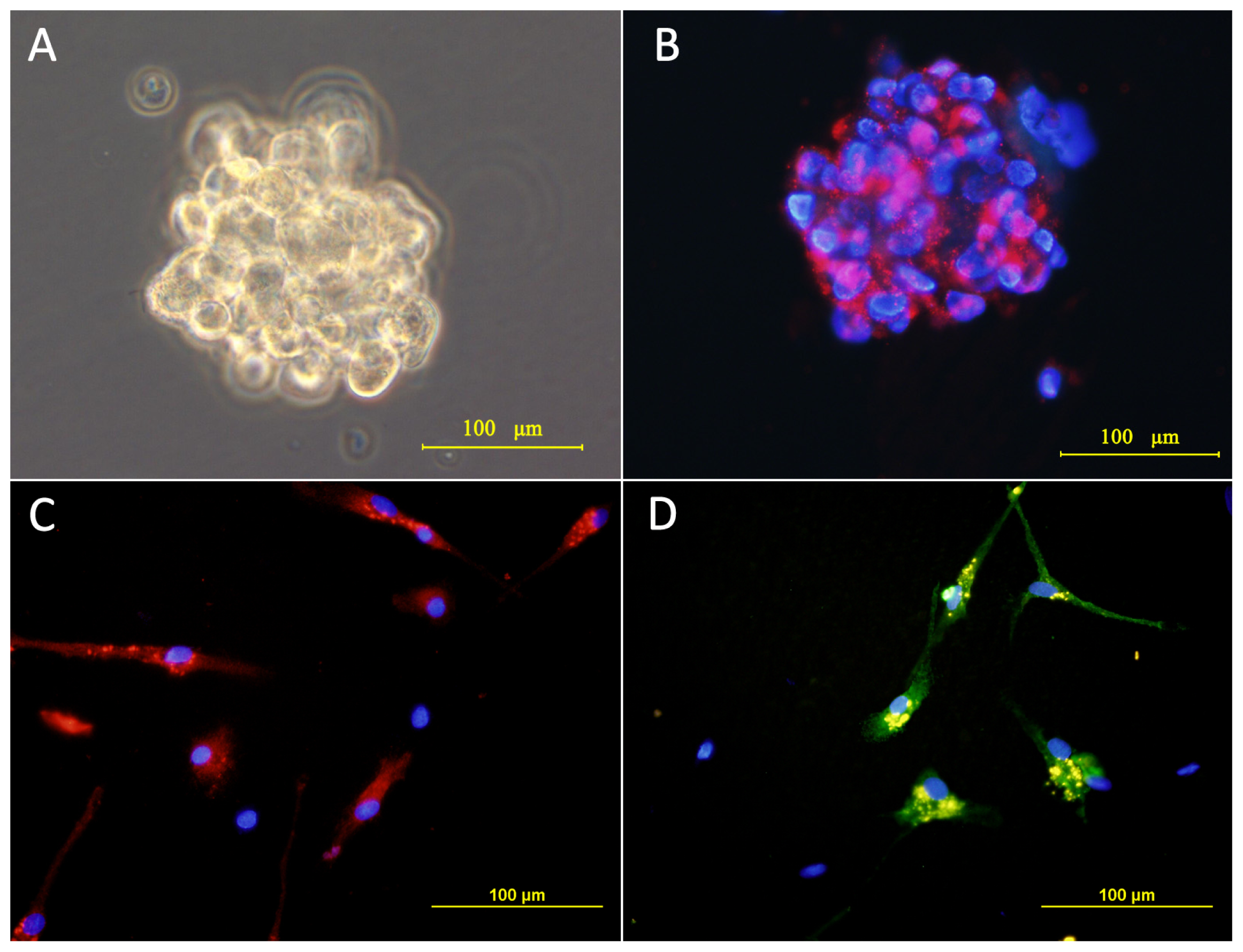
2.1. Culture and Identification of Cells
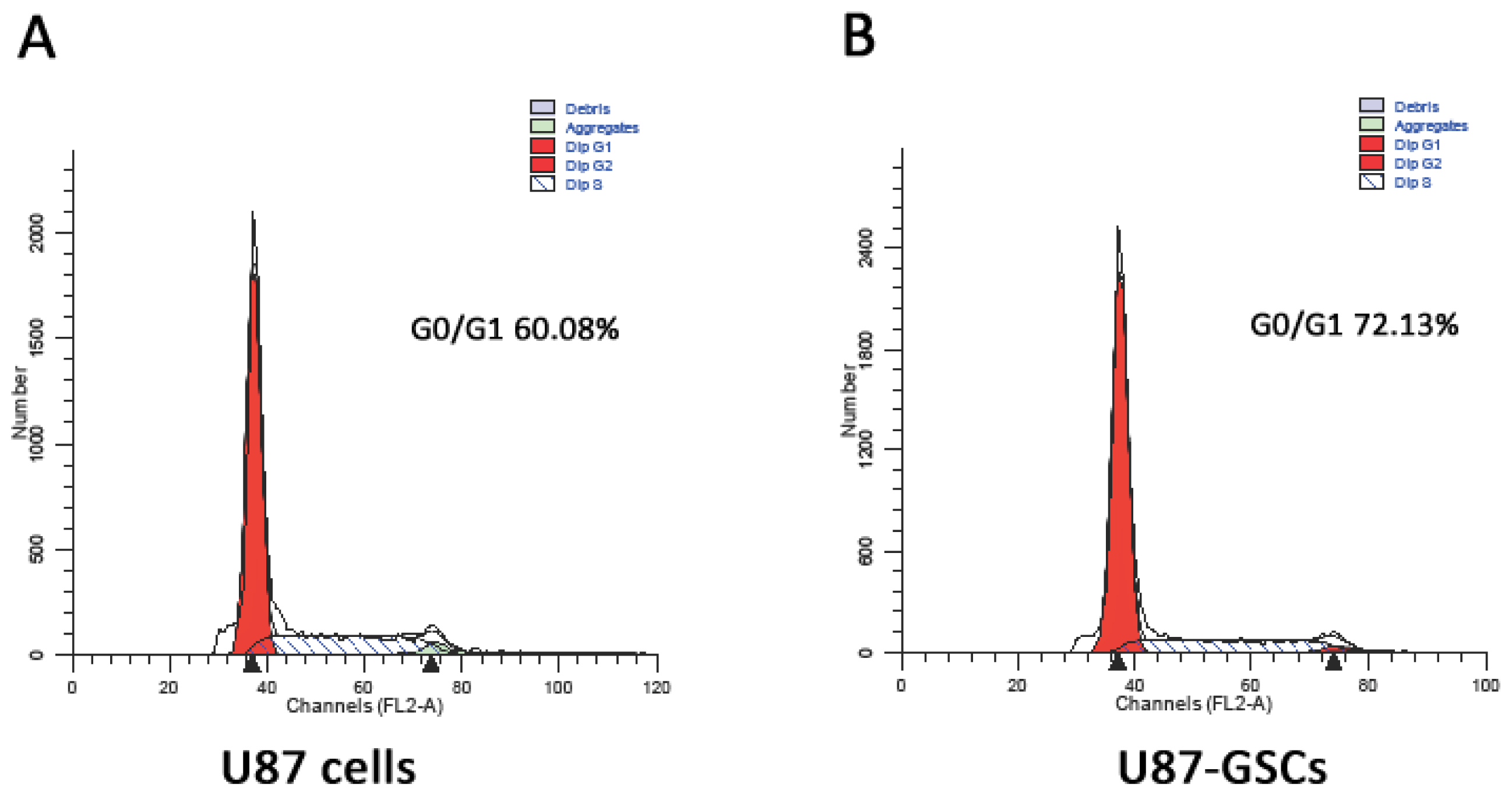
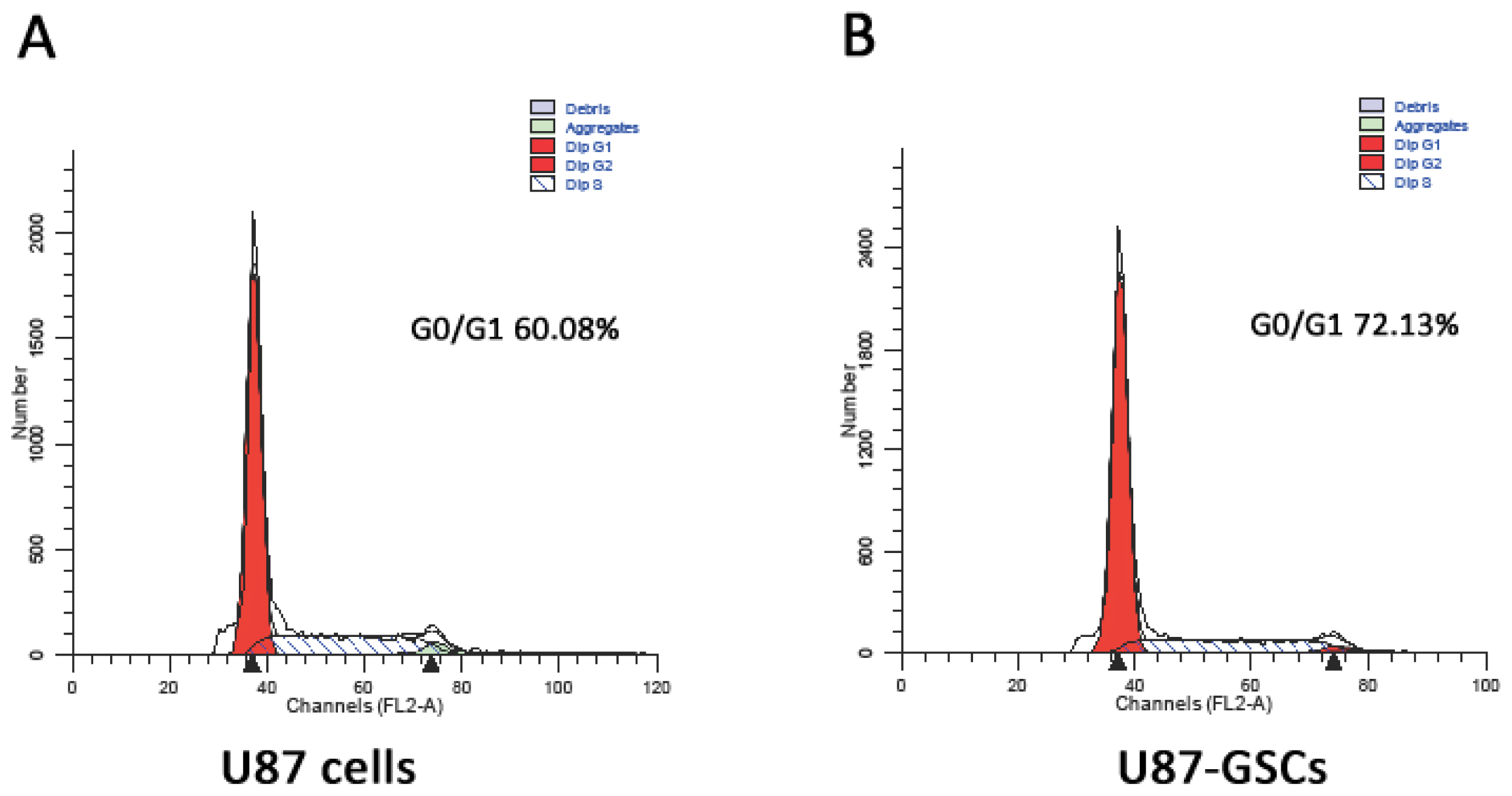
2.2. Cell Cycle Analysis
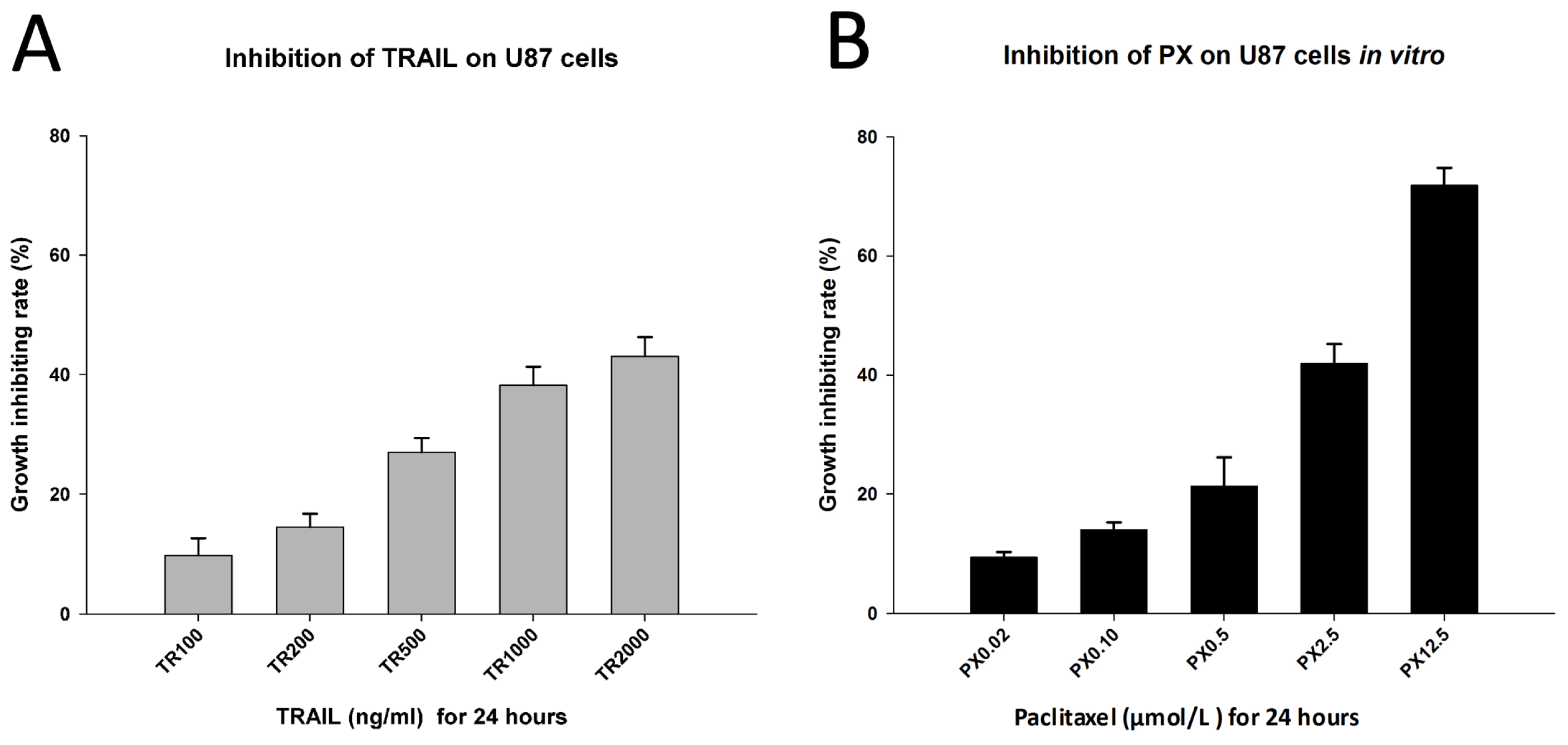
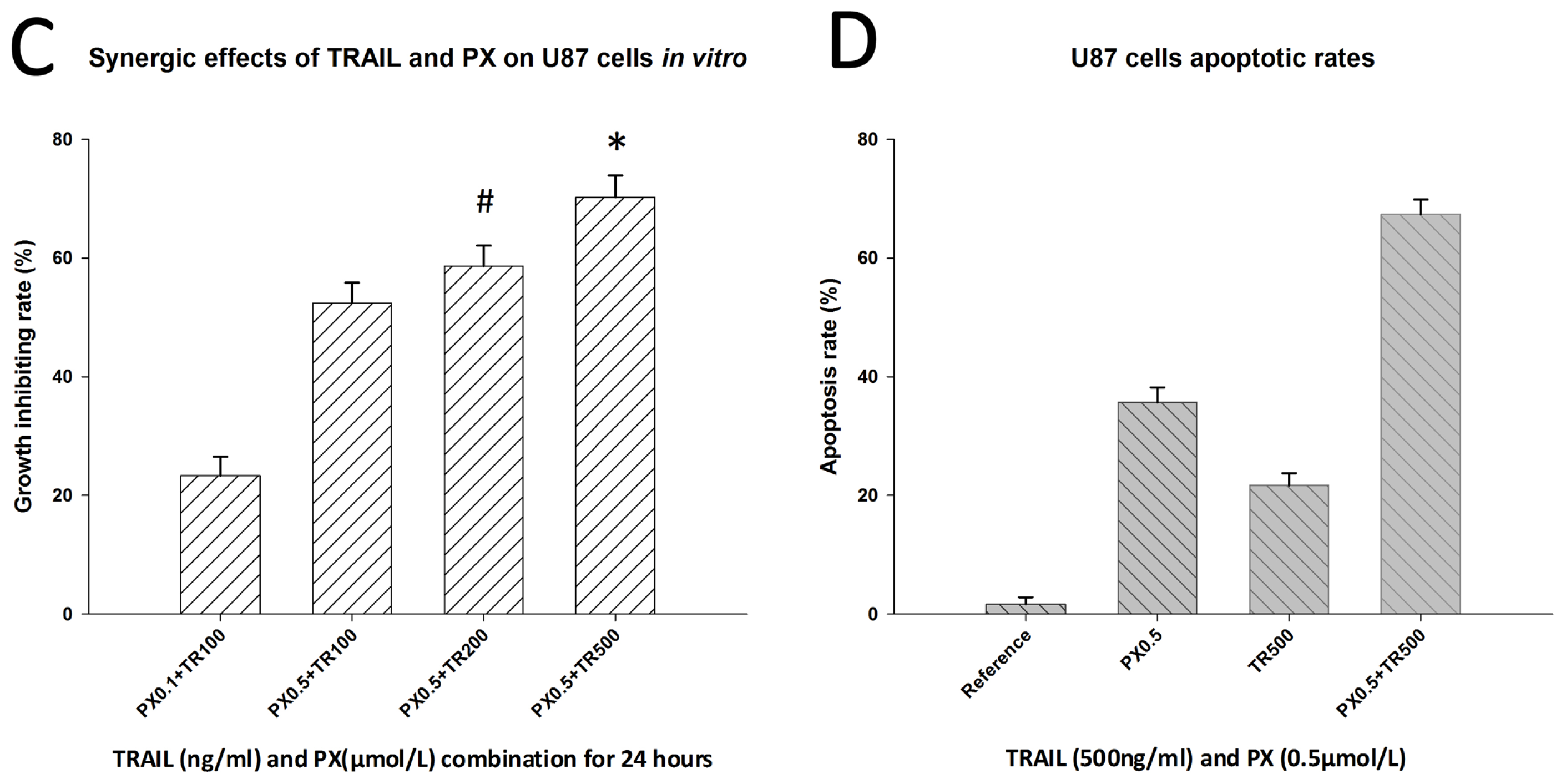
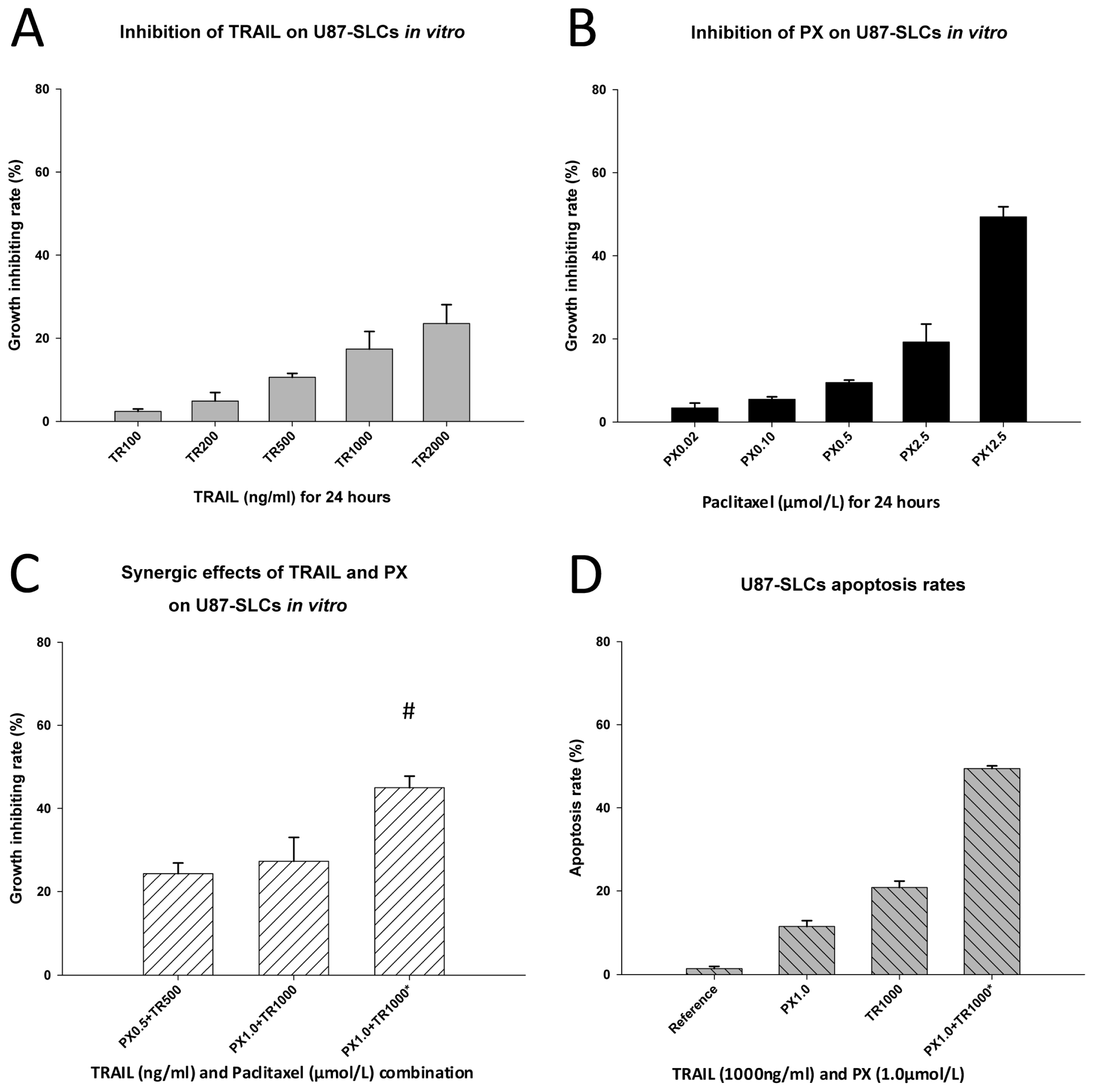
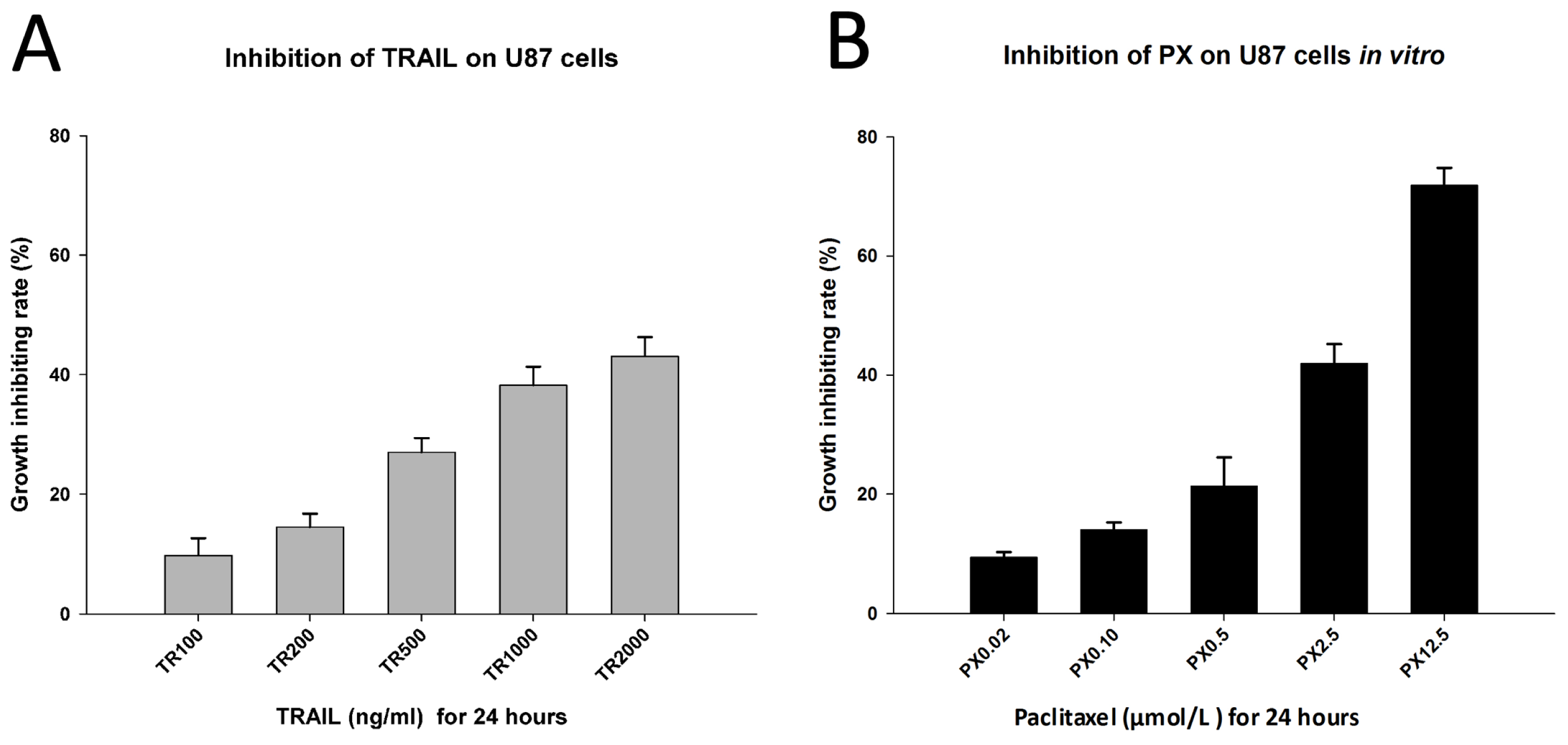
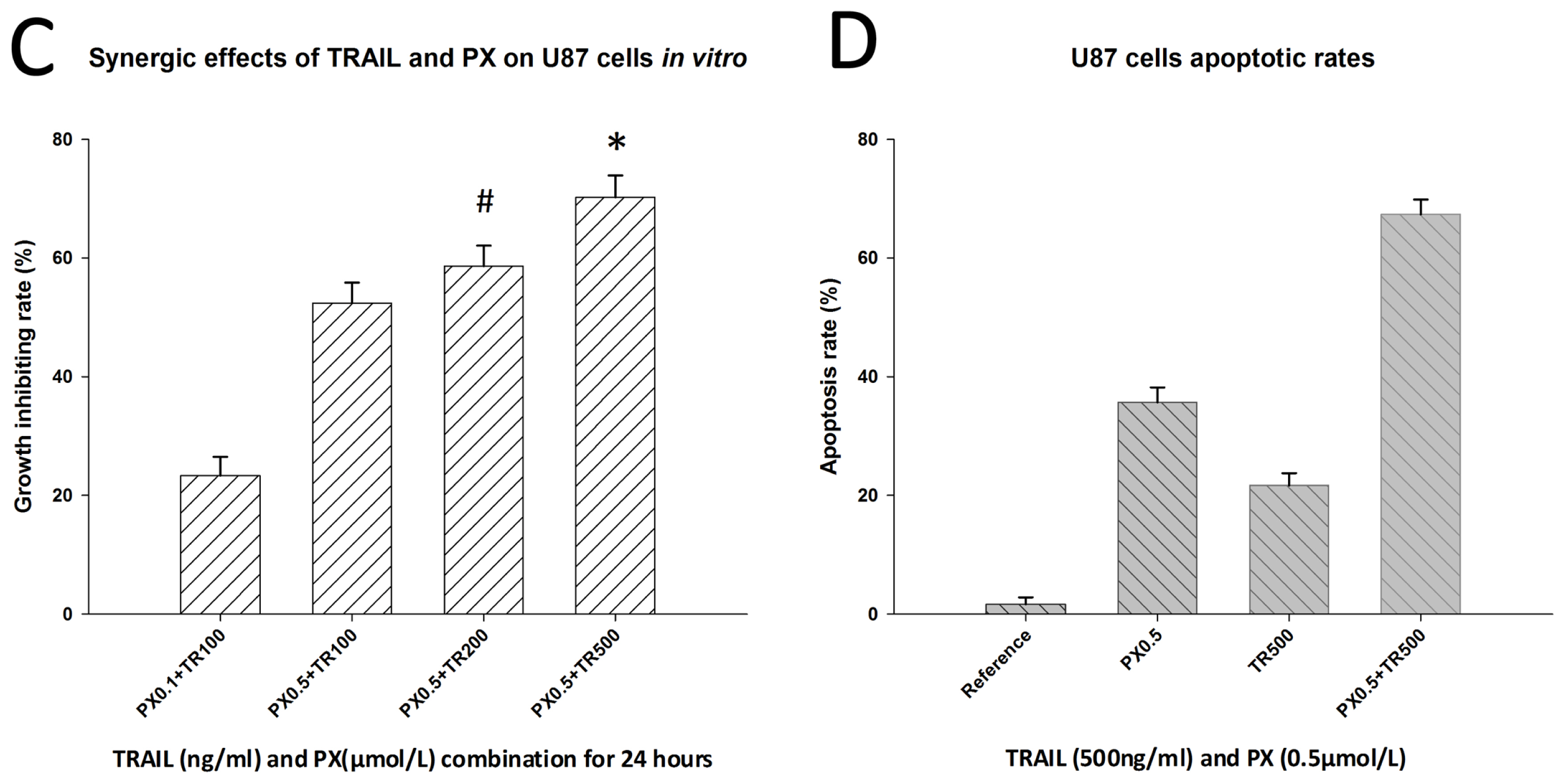
2.3. Chemotherapeutic Effects of TRAIL and PX on U87 Cells
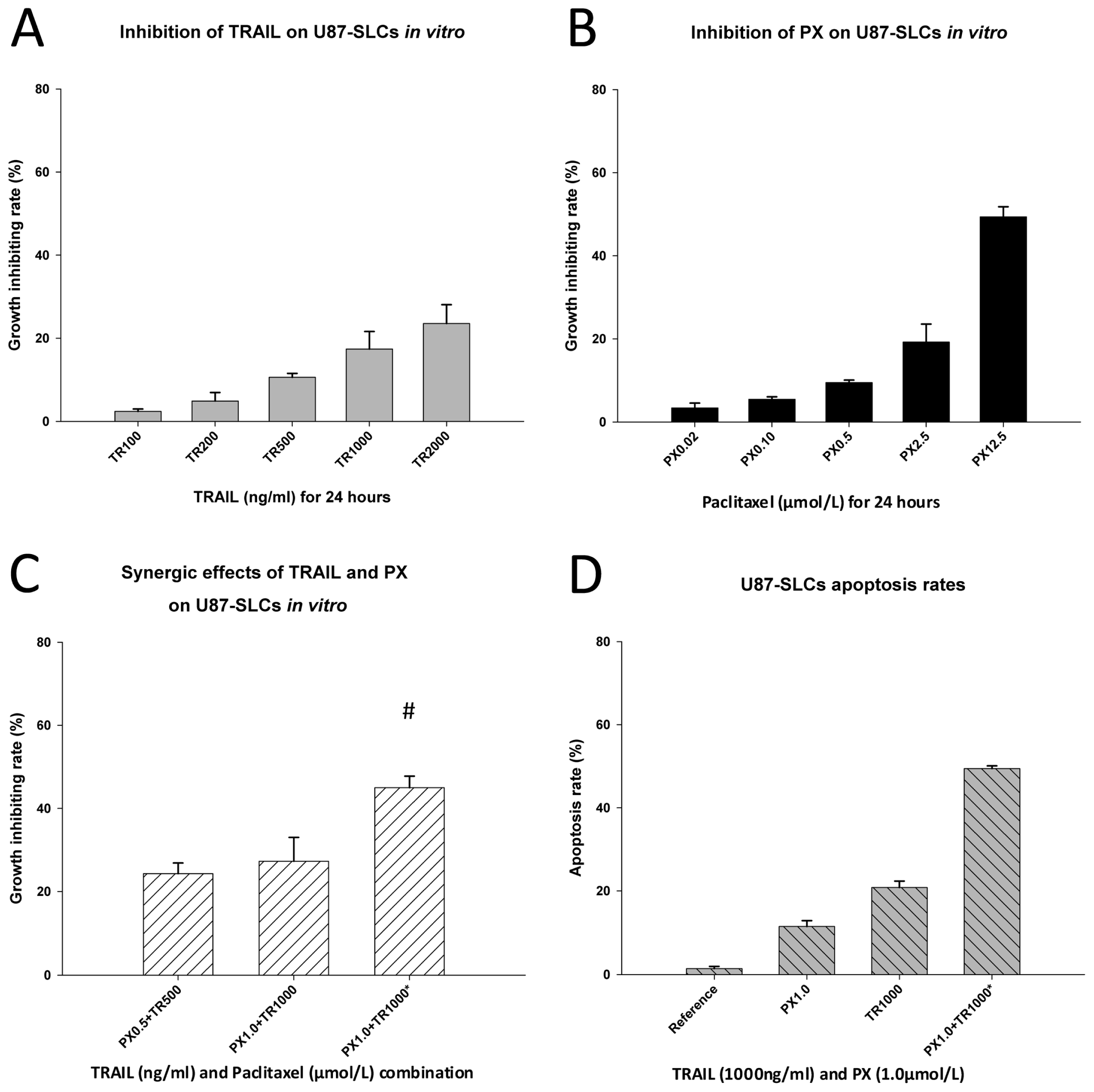
2.4. Inhibitory and Apoptosis-Inducing Effects of TRAIL and PX on U87-SLCs
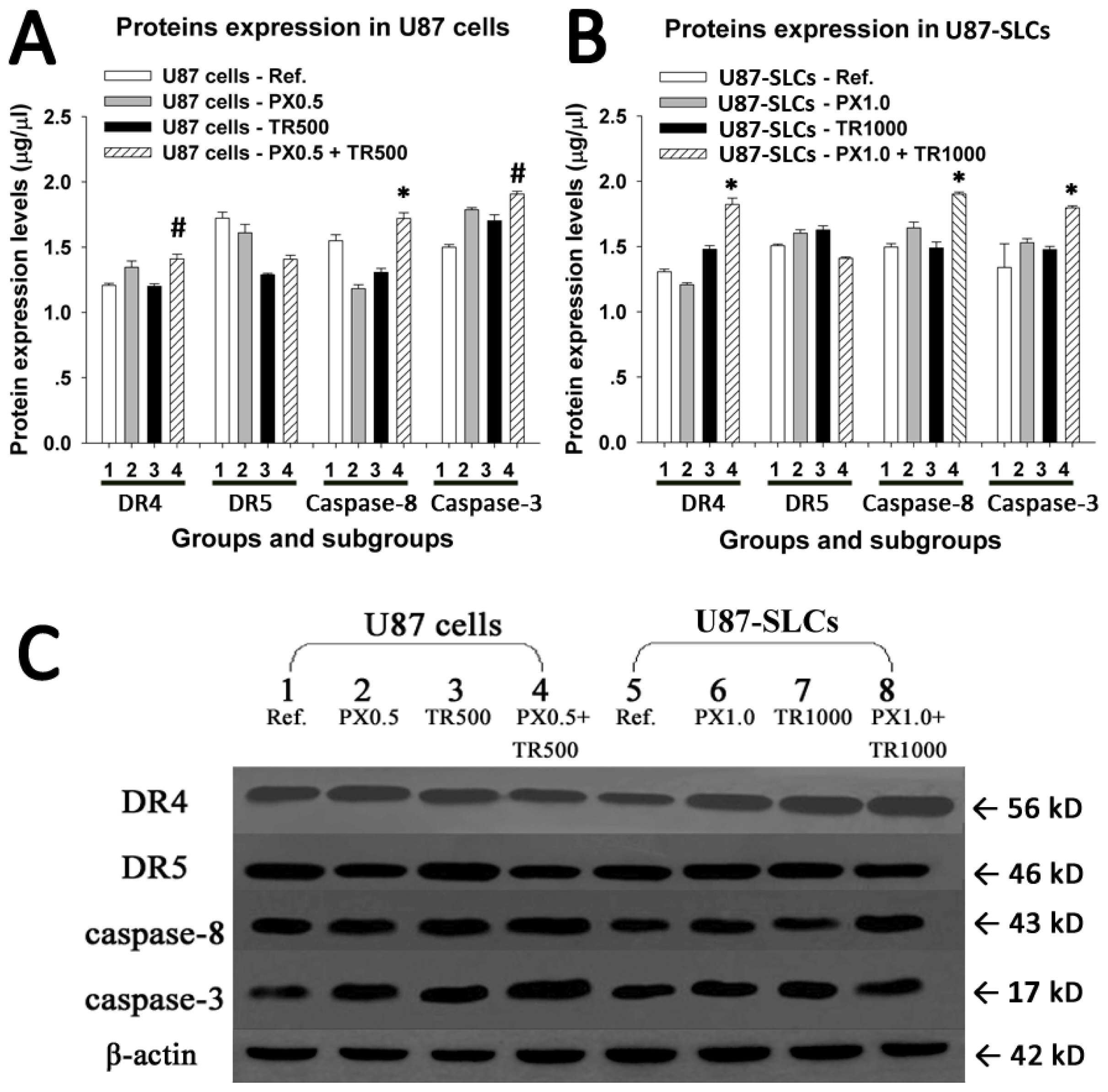
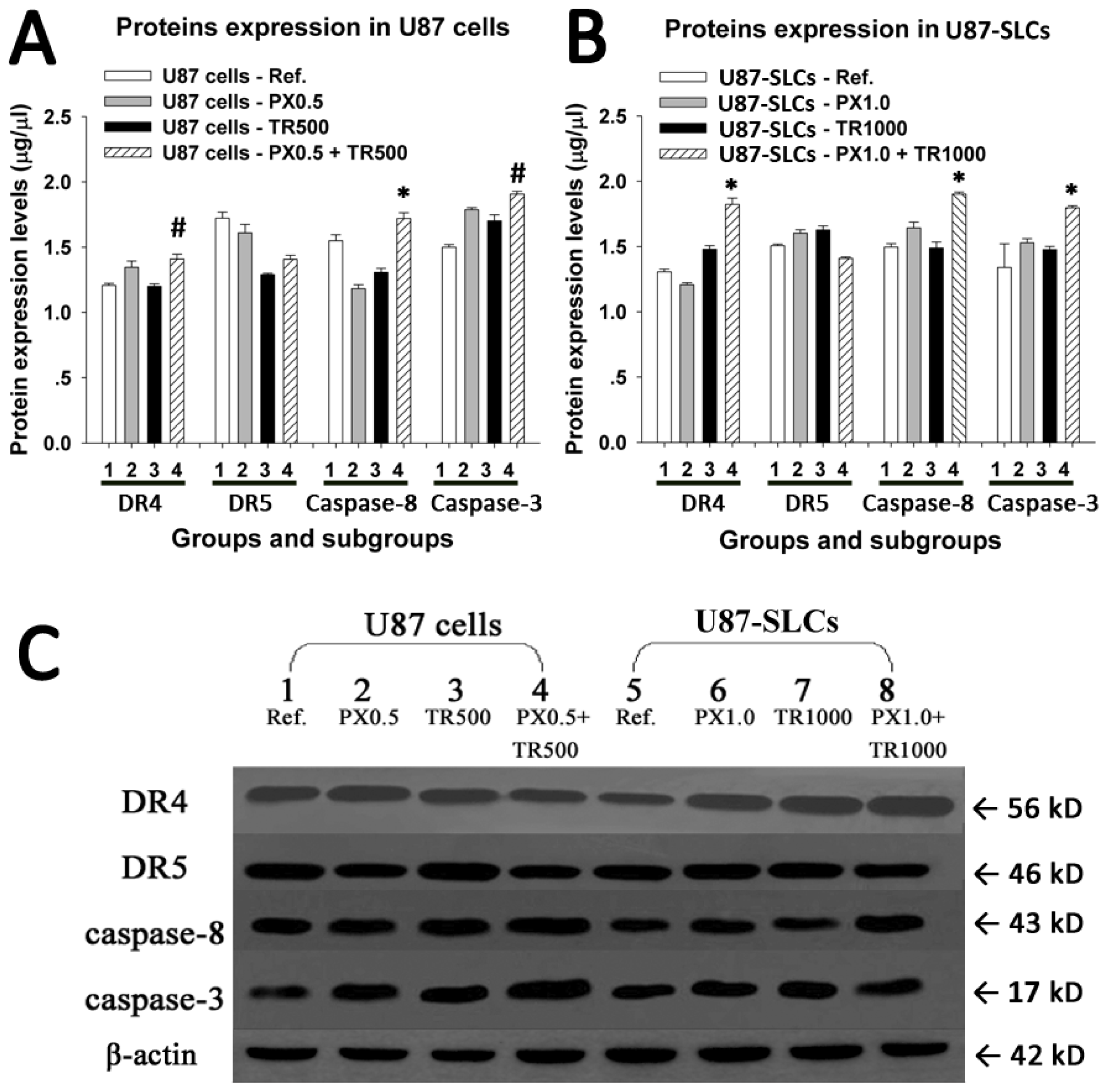
2.5. TRAIL/PX Synergy Occurs through Upregulation of DR4, Caspase-8 and Caspase-3
3. Experimental Section
3.1. Cell Culture
3.2. Induced Differentiation of Tumor Sphere Cells
3.3. Immunocytochemistry of Cultured Cells
3.4. Flow Cytometric Analysis of the Cell Cycle
3.5. Respective and Synergic Effects of TRAIL and PX on U87 Cells
3.6. Respective and Synergic Effects of TRAIL and PX on U87-SLCs Cells
3.7. Assessment of Proteins Related to TRAIL-Induced Apoptosis
3.8. Statistical Analysis
4. Conclusions
Acknowledgments
References
- Bondy, M.L.; Scheurer, M.E.; Malmer, B.; Barnholtz-Sloan, J.S.; Davis, F.G.; Il’yasova, D.; Kruchko, C.; McCarthy, B.J.; Rajaraman, P.; Schwartzbaum, J.A.; et al. Brain tumor epidemiology: Consensus from the Brain Tumor Epidemiology Consortium. Cancer 2008, 113, 1953–1968. [Google Scholar]
- Hess, K.R.; Broglio, K.R.; Bondy, M.L. Adult glioma incidence trends in the United States, 1977–2000. Cancer 2004, 101, 2293–2299. [Google Scholar]
- Park, D.M.; Rich, J.N. Biology of glioma cancer stem cells. Mol. Cells 2009, 28, 7–12. [Google Scholar]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med 2005, 352, 987–996. [Google Scholar]
- Stummer, W.; Kamp, M.A. The importance of surgical resection in malignant glioma. Curr. Opin. Neurol 2009, 22, 645–649. [Google Scholar]
- Stummer, W.; van den Bent, M.J.; Westphal, M. Cytoreductive surgery of glioblastoma as the key to successful adjuvant therapies: New arguments in an old discussion. Acta. Neurochir. (Wien) 2011, 153, 1211–1218. [Google Scholar]
- Holdhoff, M.; Grossman, S.A. Controversies in the adjuvant therapy of high-grade gliomas. Oncologist 2011, 16, 351–358. [Google Scholar]
- Stupp, R.; Hegi, M.E.; Gilbert, M.R.; Chakravarti, A. Chemoradiotherapy in malignant glioma: Standard of care and future directions. J. Clin. Oncol 2007, 25, 4127–4136. [Google Scholar]
- Duesberg, P.; Li, R.; Sachs, R.; Fabarius, A.; Upender, M.B.; Hehlmann, R. Cancer drug resistance: The central role of the karyotype. Drug Resist. Updates 2007, 10, 51–58. [Google Scholar]
- Dean, M.; Fojo, T.; Bates, S. Tumour stem cells and drug resistance. Nat. Rev. Cancer 2005, 5, 275–284. [Google Scholar]
- Frosina, G. Frontiers in targeting glioma stem cells. Eur. J. Cancer 2011, 47, 496–507. [Google Scholar]
- Lamszus, K.; Gunther, H.S. Glioma stem cells as a target for treatment. Target Oncol 2010, 5, 211–215. [Google Scholar]
- Liu, G.; Yuan, X.; Zeng, Z.; Tunici, P.; Ng, H.; Abdulkadir, I.R.; Lu, L.; Irvin, D.; Black, K.L.; Yu, J.S. Analysis of gene expression and chemoresistance of CD133+ cancer stem cells in glioblastoma. Mol. Cancer 2006, 5, 67. [Google Scholar]
- Galli, R.; Binda, E.; Orfanelli, U.; Cipelletti, B.; Gritti, A.; de Vitis, S.; Fiocco, R.; Foroni, C.; Dimeco, F.; Vescovi, A. Isolation and characterization of tumorigenic, stem-like neural precursors from human glioblastoma. Cancer Res 2004, 64, 7011–7021. [Google Scholar]
- Singh, S.K.; Clarke, I.D.; Terasaki, M.; Bonn, V.E.; Hawkins, C.; Squire, J.; Dirks, P.B. Identification of a cancer stem cell in human brain tumors. Cancer Res 2003, 63, 5821–5828. [Google Scholar]
- Yuan, X.; Curtin, J.; Xiong, Y.; Liu, G.; Waschsmann-Hogiu, S.; Farkas, D.L.; Black, K.L.; Yu, J.S. Isolation of cancer stem cells from adult glioblastoma multiforme. Oncogene 2004, 23, 9392–9400. [Google Scholar]
- Dalerba, P.; Cho, R.W.; Clarke, M.F. Cancer stem cells: Models and concepts. Annu. Rev. Med 2007, 58, 267–284. [Google Scholar]
- Eyler, C.E.; Rich, J.N. Survival of the fittest: Cancer stem cells in therapeutic resistance and angiogenesis. J. Clin. Oncol 2008, 26, 2839–2845. [Google Scholar]
- Hide, T.; Takezaki, T.; Nakamura, H.; Kuratsu, J.; Kondo, T. Brain tumor stem cells as research and treatment targets. Brain Tumor Pathol 2008, 25, 67–72. [Google Scholar]
- Venere, M.; Fine, H.A.; Dirks, P.B.; Rich, J.N. Cancer stem cells in gliomas: Identifying and understanding the apex cell in cancer’s hierarchy. Glia 2011, 59, 1148–1154. [Google Scholar]
- Suliman, A.; Lam, A.; Datta, R.; Srivastava, R.K. Intracellular mechanisms of TRAIL: Apoptosis through mitochondrial-dependent and -independent pathways. Oncogene 2001, 20, 2122–2133. [Google Scholar]
- Smyth, M.J.; Takeda, K.; Hayakawa, Y.; Peschon, J.J.; van den Brink, M.R.; Yagita, H. Nature’s TRAIL—on a path to cancer immunotherapy. Immunity 2003, 18, 1–6. [Google Scholar]
- Kuijlen, J.M.; Bremer, E.; Mooij, J.J.; den Dunnen, W.F.; Helfrich, W. Review: On TRAIL for malignant glioma therapy? Neuropathol. Appl. Neurobiol 2010, 36, 168–182. [Google Scholar]
- Mora, R.; Abschuetz, A.; Kees, T.; Dokic, I.; Joschko, N.; Kleber, S.; Geibig, R.; Mosconi, E.; Zentgraf, H.; Martin-Villalba, A.; et al. TNF-α- and TRAIL-resistant glioma cells undergo autophagy-dependent cell death induced by activated microglia. Glia 2009, 57, 561–581. [Google Scholar]
- Shankar, S.; Srivastava, R.K. Enhancement of therapeutic potential of TRAIL by cancer chemotherapy and irradiation: Mechanisms and clinical implications. Drug Resist. Updates 2004, 7, 139–156. [Google Scholar]
- Zhang, L.; Fang, B. Mechanisms of resistance to TRAIL-induced apoptosis in cancer. Cancer Gene Ther 2005, 12, 228–237. [Google Scholar]
- Baritaki, S.; Huerta-Yepez, S.; Sakai, T.; Spandidos, D.A.; Bonavida, B. Chemotherapeutic drugs sensitize cancer cells to TRAIL-mediated apoptosis: Up-regulation of DR5 and inhibition of Yin Yang 1. Mol. Cancer Ther 2007, 6, 1387–1399. [Google Scholar]
- Kim, M.; Liao, J.; Dowling, M.L.; Voong, K.R.; Parker, S.E.; Wang, S.; El-Deiry, W.S.; Kao, G.D. TRAIL inactivates the mitotic checkpoint and potentiates death induced by microtubule-targeting agents in human cancer cells. Cancer Res 2008, 68, 3440–3449. [Google Scholar]
- Chang, S.M.; Kuhn, J.G.; Robins, H.I.; Schold, S.C., Jr; Spence, A.M.; Berger, M.S.; Mehta, M.; Pollack, I.F.; Rankin, C.; Prados, M.D. A Phase II study of paclitaxel in patients with recurrent malignant glioma using different doses depending upon the concomitant use of anticonvulsants: A North American Brain Tumor Consortium report. Cancer 2001, 91, 417–422. [Google Scholar]
- Postma, T.J.; Heimans, J.J.; Luykx, S.A.; van Groeningen, C.J.; Beenen, L.F.; Hoekstra, O.S.; Taphoorn, M.J.; Zonnenberg, B.A.; Klein, M.; Vermorken, J.B. A phase II study of paclitaxel in chemonaive patients with recurrent high-grade glioma. Ann. Oncol 2000, 11, 409–413. [Google Scholar]
- Dorsey, J.F.; Mintz, A.; Tian, X.; Dowling, M.L.; Plastaras, J.P.; Dicker, D.T.; Kao, G.D.; El-Deiry, W.S. Tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) and paclitaxel have cooperative in vivo effects against glioblastoma multiforme cells. Mol. Cancer Ther 2009, 8, 3285–3295. [Google Scholar]
- Sun, X.; Pang, Z.; Ye, H.; Qiu, B.; Guo, L.; Li, J.; Ren, J.; Qian, Y.; Zhang, Q.; Chen, J.; et al. Co-delivery of pEGFP-hTRAIL and paclitaxel to brain glioma mediated by an angiopep-conjugated liposome. Biomaterials 2012, 33, 916–924. [Google Scholar]
- Yu, C.C.; Chiou, G.Y.; Lee, Y.Y.; Chang, Y.L.; Huang, P.I.; Cheng, Y.W.; Tai, L.K.; Ku, H.H.; Chiou, S.H.; Wong, T.T. Medulloblastoma-derived tumor stem-like cells acquired resistance to TRAIL-induced apoptosis and radiosensitivity. Childs Nerv. Syst 2010, 26, 897–904. [Google Scholar]
- Jordan, C.T.; Guzman, M.L.; Noble, M. Cancer stem cells. N. Engl. J. Med 2006, 355, 1253–1261. [Google Scholar]
- Wang, J.; Wang, X.; Jiang, S.; Lin, P.; Zhang, J.; Wu, Y.; Xiong, Z.; Ren, J.J.; Yang, H. Partial biological characterization of cancer stem-like cell line (WJ(2)) of human glioblastoma multiforme. Cell Mol. Neurobiol 2008, 28, 991–1003. [Google Scholar]
- Capper, D.; Gaiser, T.; Hartmann, C.; Habel, A.; Mueller, W.; Herold-Mende, C.; von Deimling, A.; Siegelin, M.D. Stem-cell-like glioma cells are resistant to TRAIL/Apo2L and exhibit down-regulation of caspase-8 by promoter methylation. Acta Neuropathol 2009, 117, 445–456. [Google Scholar]
- Lu, C.; Shervington, A. Chemoresistance in gliomas. Mol. Cell Biochem 2008, 312, 71–80. [Google Scholar]
- Cai, S.; Xu, Y.; Cooper, R.J.; Ferkowicz, M.J.; Hartwell, J.R.; Pollok, K.E.; Kelley, M.R. Mitochondrial targeting of human O6-methylguanine DNA methyltransferase protects against cell killing by chemotherapeutic alkylating agents. Cancer Res 2005, 65, 3319–3327. [Google Scholar]
- Hirschmann-Jax, C.; Foster, A.E.; Wulf, G.G.; Nuchtern, J.G.; Jax, T.W.; Gobel, U.; Goodell, M.A.; Brenner, M.K. A distinct “side population” of cells with high drug efflux capacity in human tumor cells. Proc. Natl. Acad. Sci. USA 2004, 101, 14228–14233. [Google Scholar]
- Mellor, H.R.; Ferguson, D.J.; Callaghan, R. A model of quiescent tumour microregions for evaluating multicellular resistance to chemotherapeutic drugs. Br. J. Cancer 2005, 93, 302–309. [Google Scholar]
- Asakuma, J.; Sumitomo, M.; Asano, T.; Hayakawa, M. Selective Akt inactivation and tumor necrosis actor-related apoptosis-inducing ligand sensitization of renal cancer cells by low concentrations of paclitaxel. Cancer Res 2003, 63, 1365–1370. [Google Scholar]
- Hunter, T.B.; Manimala, N.J.; Luddy, K.A.; Catlin, T.; Antonia, S.J. Paclitaxel and TRAIL synergize to kill paclitaxel-resistant small cell lung cancer cells through a caspase-independent mechanism mediated through AIF. Anticancer Res 2011, 31, 3193–3204. [Google Scholar]
- Park, S.J.; Wu, C.H.; Gordon, J.D.; Zhong, X.; Emami, A.; Safa, A.R. Taxol induces caspase-10-dependent apoptosis. J. Biol. Chem 2004, 279, 51057–51067. [Google Scholar]
- Singh, T.R.; Shankar, S.; Chen, X.; Asim, M.; Srivastava, R.K. Synergistic interactions of chemotherapeutic drugs and tumor necrosis factor-related apoptosis-inducing ligand/Apo-2 ligand on apoptosis and on regression of breast carcinoma in vivo. Cancer Res 2003, 63, 5390–5400. [Google Scholar]
- Saqr, H.E.; Omran, O.M.; Oblinger, J.L.; Yates, A.J. TRAIL-induced apoptosis in U-1242 MG glioma cells. J. Neuropathol. Exp. Neurol 2006, 65, 152–161. [Google Scholar]
- Walczak, H.; Bouchon, A.; Stahl, H.; Krammer, P.H. Tumor necrosis factor-related apoptosis-inducing ligand retains its apoptosis-inducing capacity on Bcl-2- or Bcl-xL-overexpressing chemotherapy-resistant tumor cells. Cancer Res 2000, 60, 3051–3057. [Google Scholar]
- Qiu, B.; Zhang, D.; Tao, J.; Tie, X.; Wu, A.; Wang, Y. Human brain glioma stem cells are more invasive than their differentiated progeny cells in vitro. J. Clin. Neurosci 2012, 19, 130–134. [Google Scholar]
- Cao, S.S.; Zhen, Y.S. Potentiation of antimetabolite antitumor activity in vivo by dipyridamole and amphotericin B. Cancer Chemother. Pharmacol 1989, 24, 181–186. [Google Scholar]
- Kogel, D.; Schomburg, R.; Copanaki, E.; Prehn, J.H. Regulation of gene expression by the amyloid precursor protein: Inhibition of the JNK/c-Jun pathway. Cell Death Differ 2005, 12, 1–9. [Google Scholar]
© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Qiu, B.; Sun, X.; Zhang, D.; Wang, Y.; Tao, J.; Ou, S. TRAIL and Paclitaxel Synergize to Kill U87 Cells and U87-Derived Stem-Like Cells in Vitro. Int. J. Mol. Sci. 2012, 13, 9142-9156. https://doi.org/10.3390/ijms13079142
Qiu B, Sun X, Zhang D, Wang Y, Tao J, Ou S. TRAIL and Paclitaxel Synergize to Kill U87 Cells and U87-Derived Stem-Like Cells in Vitro. International Journal of Molecular Sciences. 2012; 13(7):9142-9156. https://doi.org/10.3390/ijms13079142
Chicago/Turabian StyleQiu, Bo, Xiyang Sun, Dongyong Zhang, Yong Wang, Jun Tao, and Shaowu Ou. 2012. "TRAIL and Paclitaxel Synergize to Kill U87 Cells and U87-Derived Stem-Like Cells in Vitro" International Journal of Molecular Sciences 13, no. 7: 9142-9156. https://doi.org/10.3390/ijms13079142