Homology Modeling and Analysis of Structure Predictions of the Bovine Rhinitis B Virus RNA Dependent RNA Polymerase (RdRp)


Abstract
:1. Introduction
2. Results and Discussion
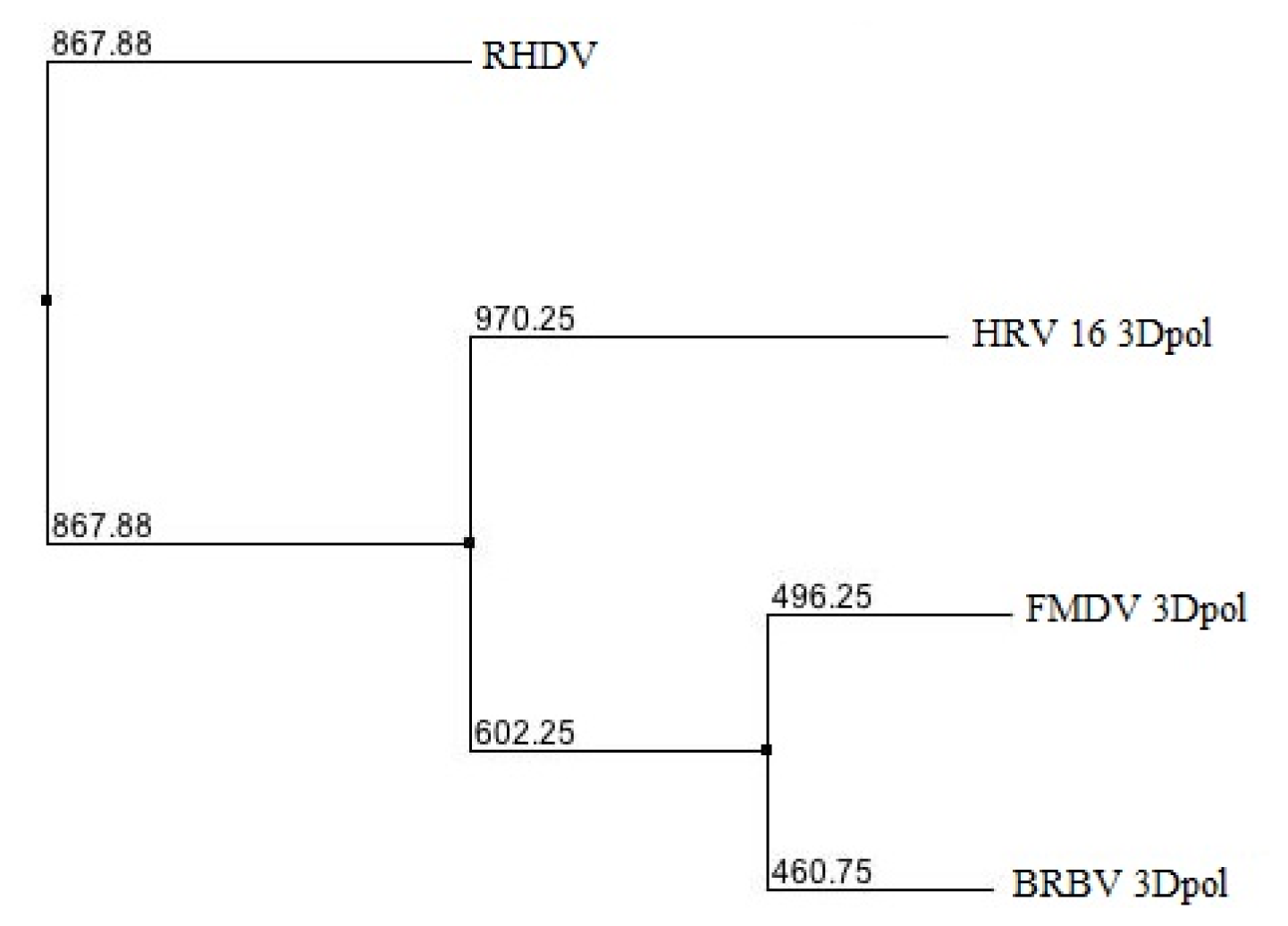
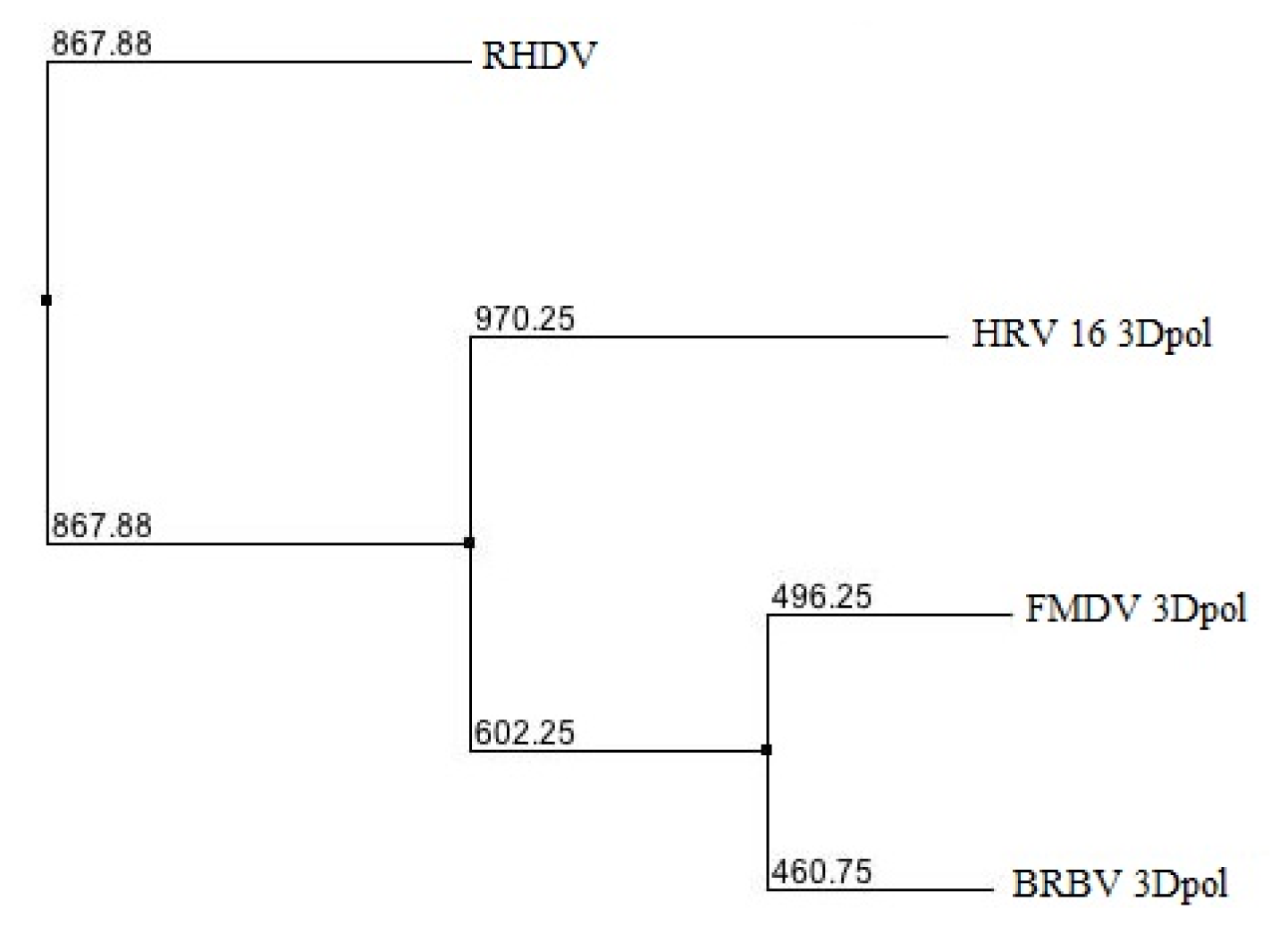
2.1. Phylogenetic Analysis of BRBV RdRp
2.2. Structure Based Sequence Alignment
2.3. Search for Structural Homologues to BRBV 3Dpol via deconSTRUCT Web Server
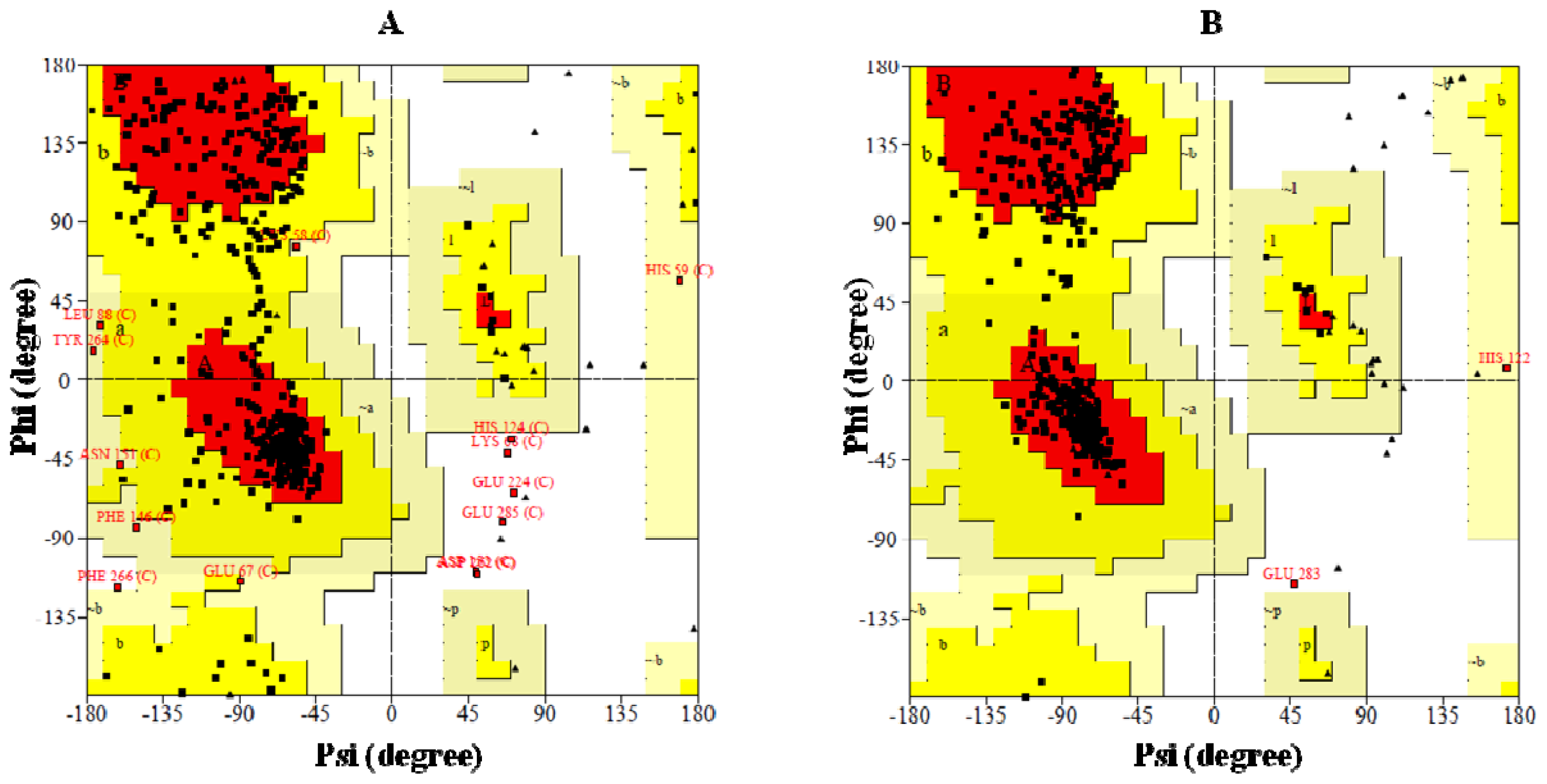
2.4. Preparation of Homology Model of BRBV 3Dpol and the Assessment of Model Accuracy
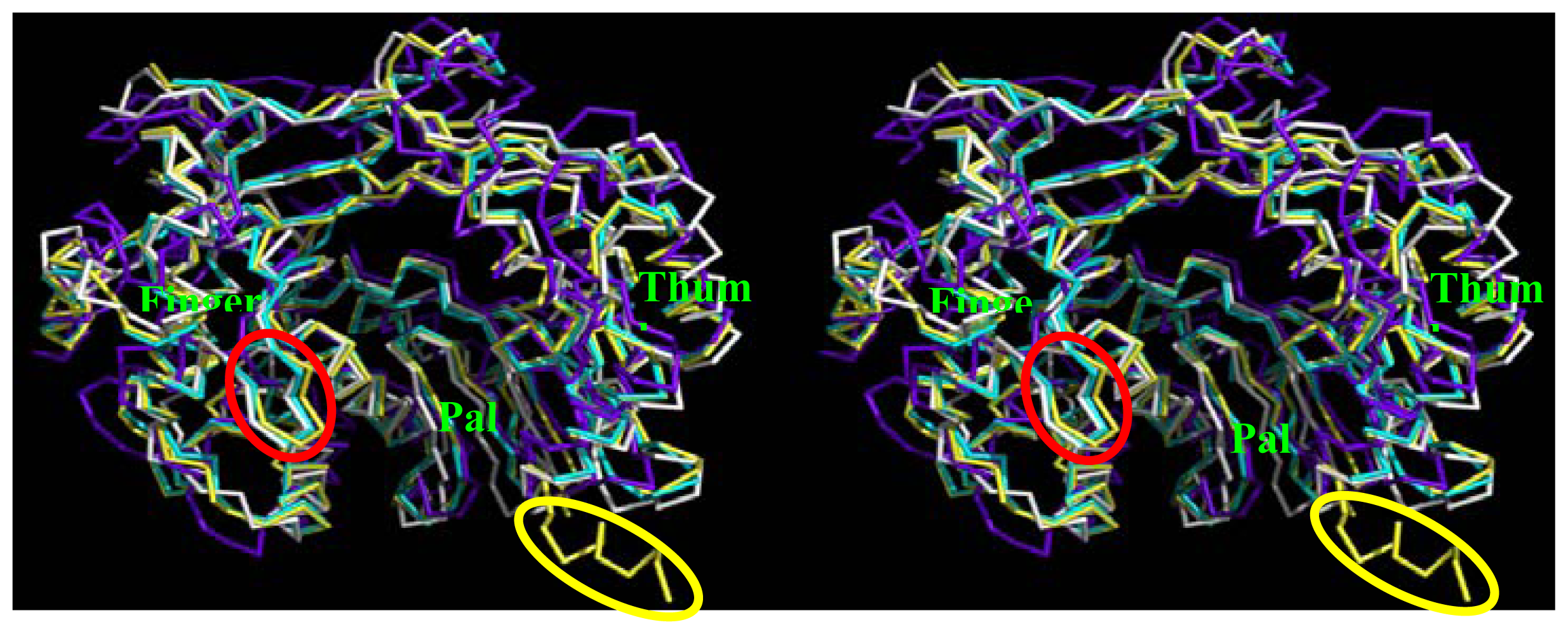
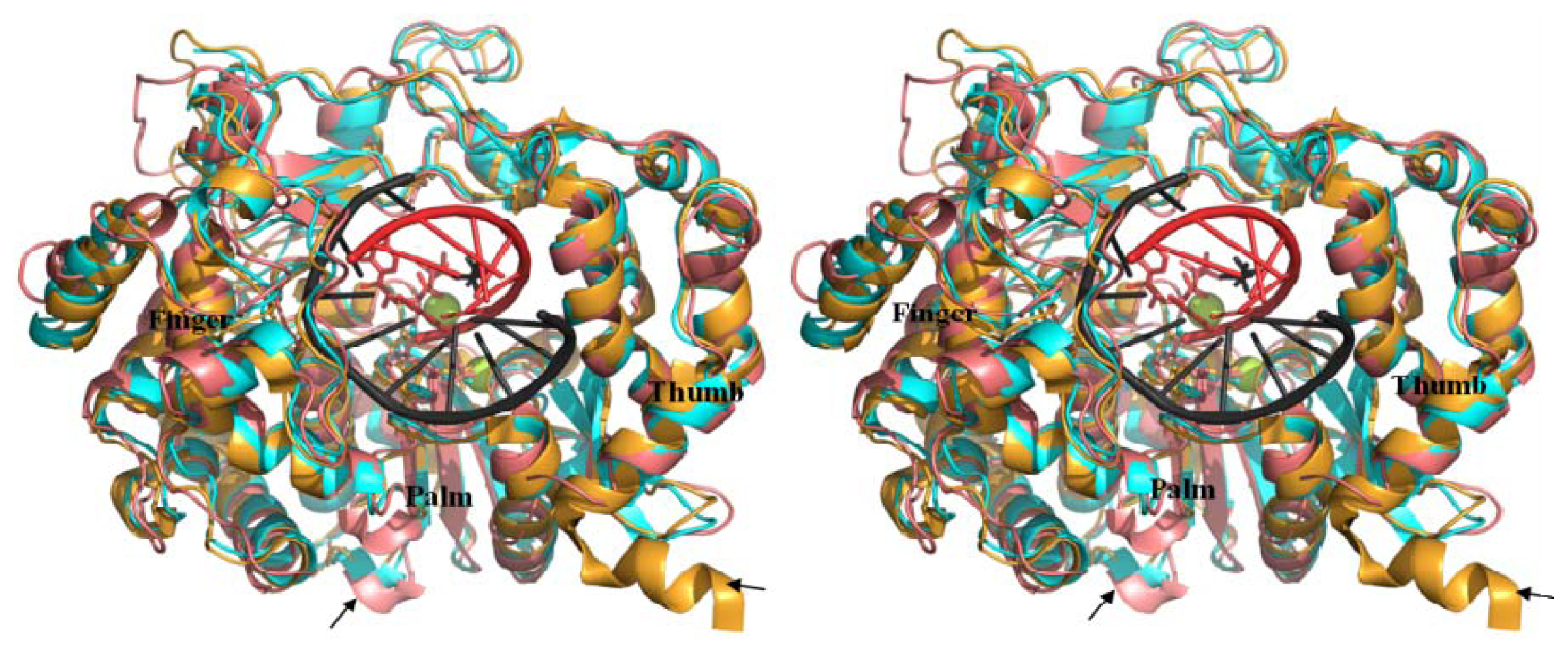
2.5. Comparison of BRBV 3Dpol and Other 3Dpol Crystal Structures
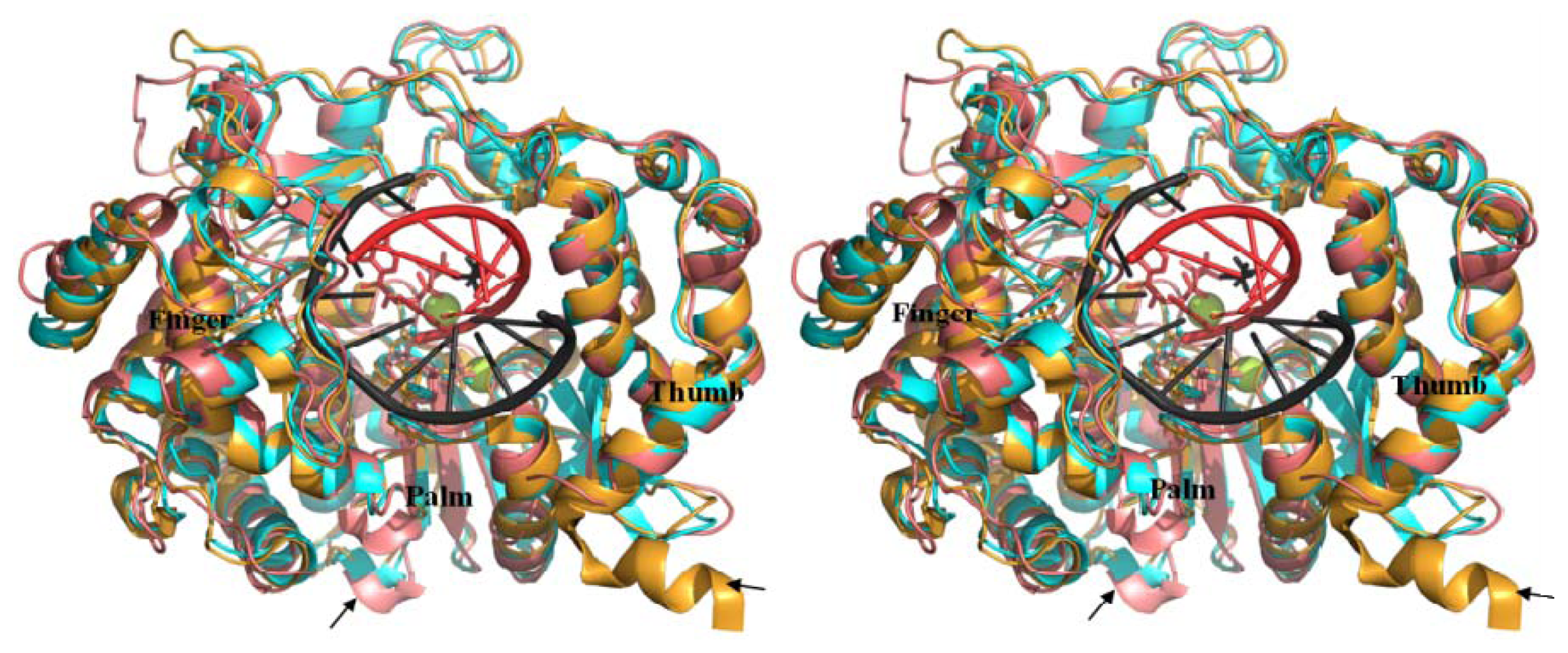
2.6. Analysis of Key Structural Features of FMDV 3Dpol and BRBV Homology Model
2.7. Analysis on Non-Covalent Interactions in FMDV and BRBV 3Dpol
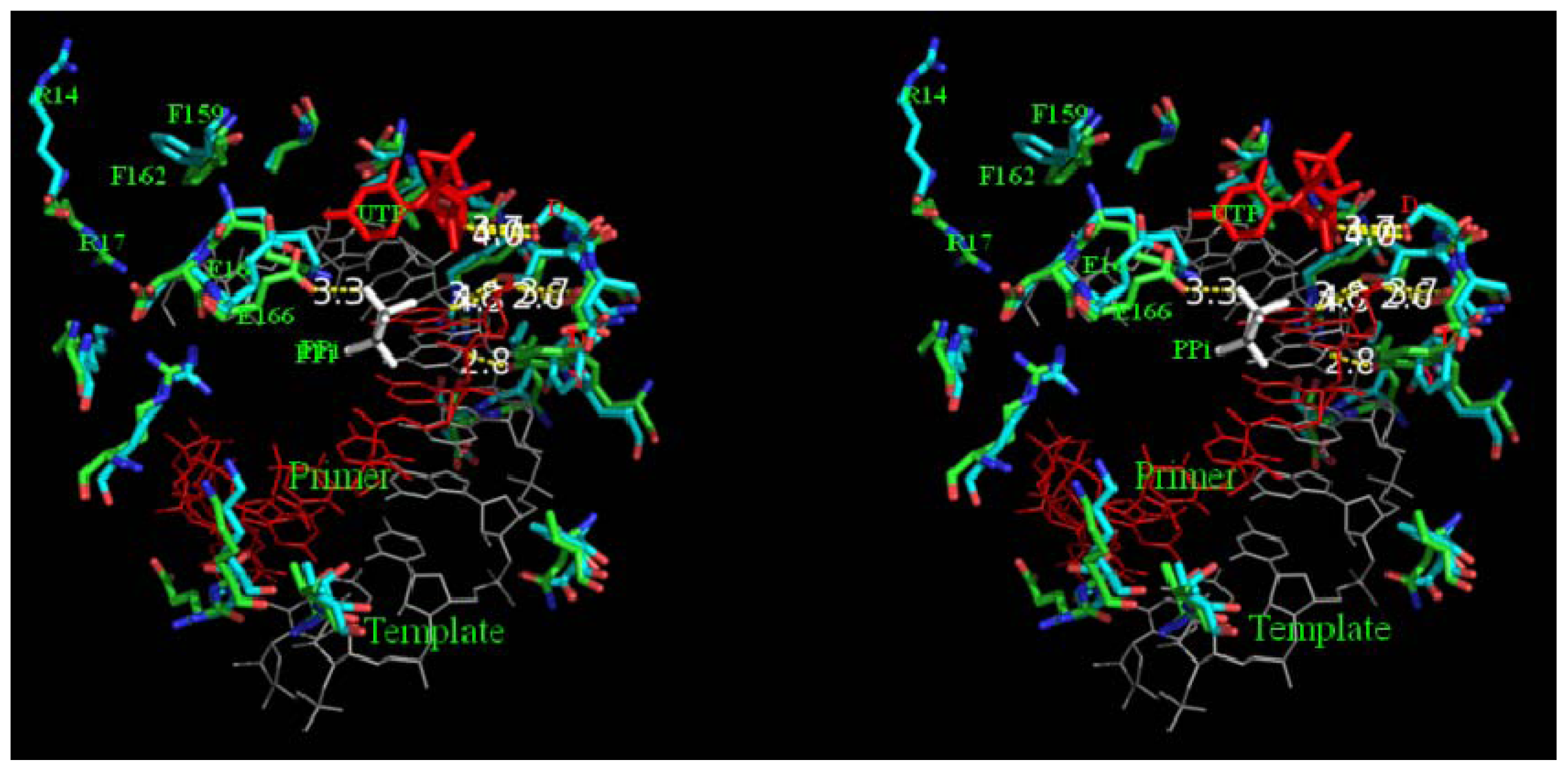
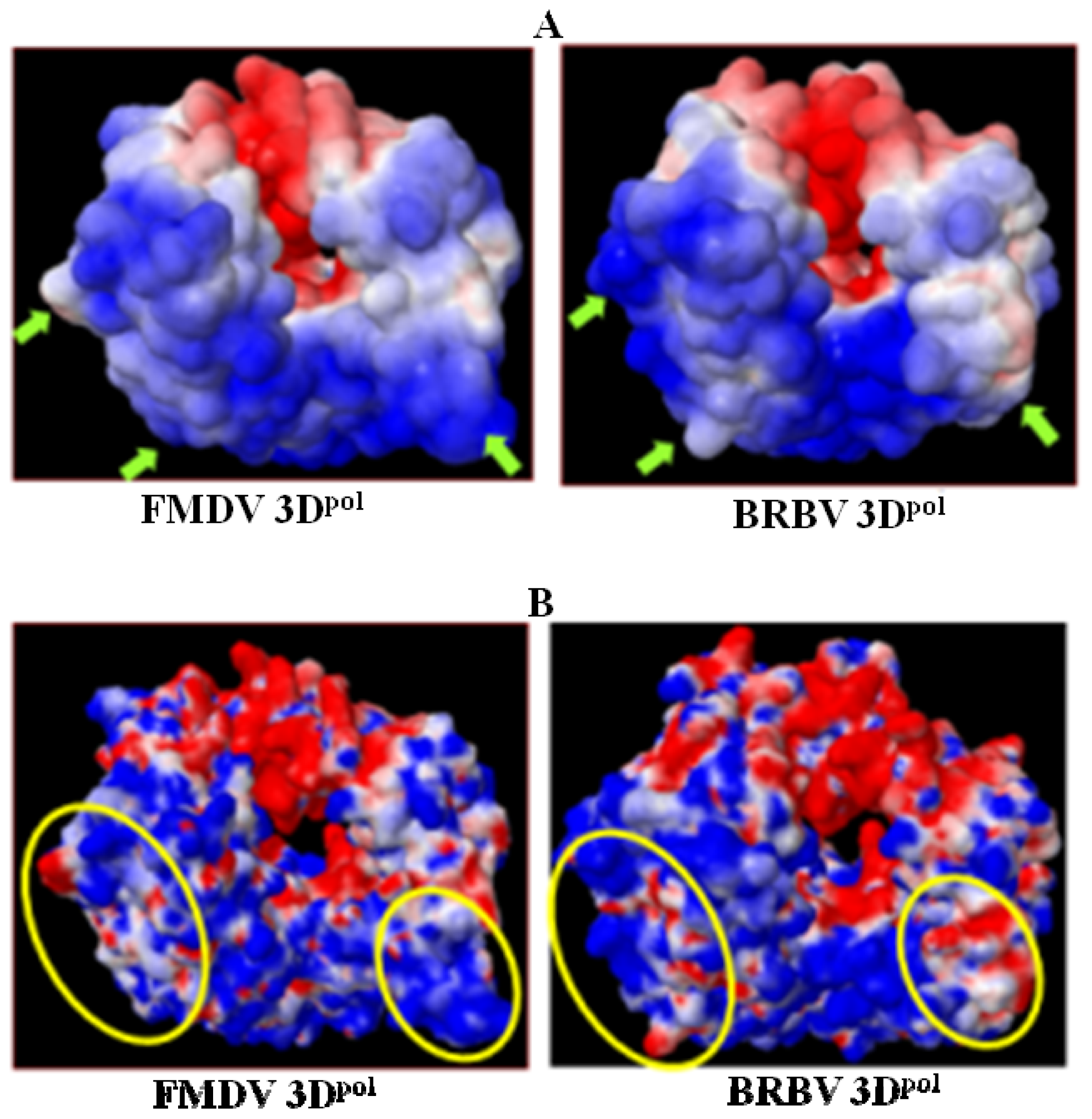
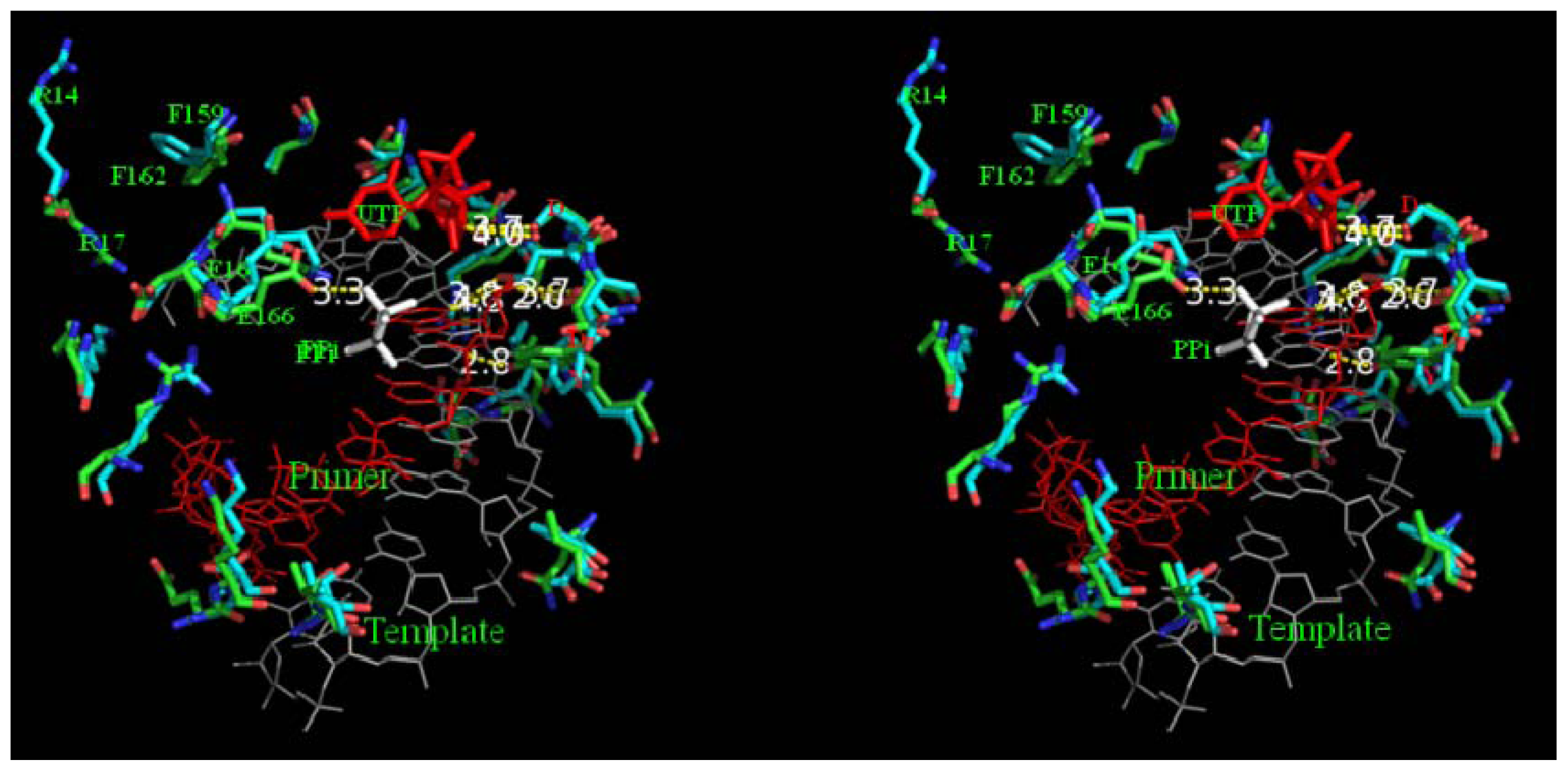
2.8. Analysis of Template/Primer (T/P) Binding Interface and Active Site of BRBV 3Dpol
3. Experimental Section
3.1. Sequence
3.2. Phylogenetic Analysis
3.3. Structure based Alignment of the Polymerase Sequences
3.4. Preparation of Homology Model of BRBV 3Dpol
- Geno3D web serverCrystal structures of FMDV (PDB, 1U09) and HRV-16 3Dpol (PDB, 1XR7) were utilized as templates for model generation. After the completion of run 10 homology models were prepared and their quality was assessed through PROCHECK [20,28]. Geno3D ( http://geno3d-pbil.ibcp.fr) is an automated web server for protein molecular modeling [20]. Starting with a query protein sequence, the server performs the homology modeling in six successive steps: first, it identifies homologous proteins with known structures by using PSI-BLAST [21]. This provides the user all potential templates for target selection. After the user defines the templates and submits the job, the server performs the alignment of both query and subject sequences and extracts geometrical restraints (dihedral angles and distances) for corresponding atoms between the query and the template. Lastly, it performs the 3D construction of the protein by using a distance geometry approach.
- SWISS-MODELThe crystal structure of FMDV 3Dpol (PDB, 1U09) was identified as the closest match to the BRV counterpart and later utilized as template by SWISS-MODEL workspace, which is an integrated web-based modeling environment. For a given target protein it searches a library of experimental protein structures to identify suitable templates. On the basis of a sequence alignment between the target protein and the template structure, a three-dimensional model for the target protein is generated. The alignment produced by SWISS-MODEL was verified using another alignment algorithm (T-coffee). In homology modeling the most crucial steps is the evaluation and refinement of the raw model and SWISS-MODEL utilizes a set of unique analysis tools in addition to commonly tools such as PROCHECK and WHAT CHECK [22–26,28,34] to achieve this. These tools include atomic empirical mean force potential (ANOLEA) which performs energy calculations on a protein chain, evaluating the “Non- Local Environment” (NLE) of each heavy atom in the molecule and is used to assess packing quality of the models; QMEAN which is a composite scoring function for both the estimation of the global quality of the entire model as well as for the local per-residue analysis of different regions within a model; DFIRE, which is an all-atom statistical potential based on a distance-scaled finite ideal-gas reference state and reflects the quality of the model to indicate that a that a model is lower energy closer to the native conformation [35–37]. Finally, it minimizes the structure using GROMOS9639 performing 200 cycles by the steepest descent method and 300 cycles by the conjugate gradient method to minimize the steric clashes [38]. With stringent quality controls SWISS-MODEL workspace usually generates models with a reasonably reliable quality.
3.5. DeconSTRUCT Analysis of the Homology Models of BRBV 3Dpol
3.6. Calculation of Electrostatic Surface Potential
3.7. Calculation of Non-Covalent Interactions
3.8. Preparation of Structures
4. Conclusions
Acknowledgements
- Conflict of InterestThe authors declare that they have no conflict of interest.
References
- Knowles, N.J.; Hovi, T; Hyypiä, T.; King, A.M.Q.; Lindberg, M.; Pallansch, M.A.; Palmenberg, A.C.; Simmonds, P.; Skern, T.; Stanway, G.; et al. Picornaviridae. In Virus Taxonomy: Classification and Nomenclature of Viruses: Ninth Report of the International Committee on Taxonomy of Viruses; King, A.M.Q., Adams, M.J., Carstens, E.B., Lefkowitz, E.J., Eds.; Elsevier: San Diego, CA, USA, 2011; pp. 855–880. [Google Scholar]
- Hollister, J.R.; Vagnozzi, A.; Knowles, N.J.; Rieder, E. Molecular and phylogenetic analyses of bovine rhinovirus type 2 shows it is closely related to foot-and-mouth disease virus. Virology 2008, 373, 411–425. [Google Scholar]
- Betts, A.O.; Edington, N.; Jennings, A.R.; Reed, S.E. Studies on a rhinovirus (EC11) derived from a calf. II. Disease in calves. J. Comp. Pathol 1971, 81, 41–48. [Google Scholar]
- Ng, K.K.; Cherney, M.M.; Vazquez, A.L.; Machin, A.; Alonso, J.M.; Parra, F.; James, M.N. Crystal structures of active and inactive conformations of a caliciviral RNA-dependent RNA polymerase. J. Biol. Chem 2002, 277, 1381–1387. [Google Scholar]
- Love, R.A.; Maegley, K.A.; Yu, X.; Ferre, R.A.; Lingardo, L.K.; Diehl, W.; Parge, H.E.; Dragovich, P.S.; Fuhrman, S.A. The crystal structure of the RNA-dependent RNA polymerase from human rhinovirus: A dual function target for common cold antiviral therapy. Structure 2004, 12, 1533–1544. [Google Scholar]
- Thompson, A.A.; Peersen, O.B. Structural basis for proteolysis-dependent activation of the poliovirus RNA-dependent RNA polymerase. EMBO J 2004, 23, 3462–3471. [Google Scholar]
- Ferrer-Orta, C.; Arias, A.; Escarmís, C.; Verdaguer, N. A comparison of viral RNA-dependent RNA polymerases. Curr. Opin. Struct. Biol 2006, 16, 27–34. [Google Scholar]
- Ferrer-Orta, C.; Arias, A.; Perez-Luque, R.; Escarmis, C.; Domingo, E.; Verdaguer, N. Structure of foot-and-mouth disease virus RNA-dependent RNA polymerase and its complex with a template-primer RNA. J. Biol. Chem 2005, 279, 47212–47221. [Google Scholar]
- Gruez, A.; Selisko, B.; Roberts, M.; Bricogne, G.; Bussetta, C.; Jabafi, I.; Coutard, B.; de Palma, A.M.; Neyts, J.; Canard, B. The crystal structure of coxsackievirus B3 RNA-dependent RNA polymerase in complex with its protein primer VPg confirms the existence of a second VPg binding site on Picornaviridae polymerases. J. Virol 2008, 82, 9577–9790. [Google Scholar]
- Steitz, T.A. DNA and RNA polymerases: Structural diversity and common mechanisms. Harvey Lect. 1997–1998, 93, 75–93. [Google Scholar]
- Gong, P.; Peersen, O.B. Structural basis for active site closure by the poliovirus RNA-dependent RNA polymerase. Proc. Natl. Acad. Sci. USA 2010, 107, 22505–22510. [Google Scholar]
- Bruenn, J.A. A structural and primary sequence comparison of the viral RNA-dependent RNA polymerases. Nucleic Acids Res 2003, 31, 1821–1829. [Google Scholar]
- Cameron, C.E.; Gohara, D.W.; Arnold, J.J. Poliovirus RNA-Dependent RNA Polymerase (3Dpol): Structure, Function and Mechanisms. In Molecular Biology of Picornaviruses; Semler, B.L., Wimmer, E., Eds.; ASM Press: Washington DC, USA, 2002; pp. 255–267. [Google Scholar]
- Van Dijk, A.A.; Makeyev, E.V.; Bamford, D.H. Structural basis for proteolysis-dependent activation of the poliovirus RNA-dependent RNA polymerase. EMBO J 2004, 23, 3462–3471. [Google Scholar]
- Thompson, A.A.; Albertini, R.A.; Peersen, O.B. Stabilization of poliovirus polymerase by NTP binding and fingers-thumb interactions. J. Mol. Biol 2007, 366, 1459–1474. [Google Scholar]
- Kok, C.; McMinn, P. Picornavirus RNA-dependent RNA polymerase. Int. J. Biochem. Cell Biol 2009, 41, 498–502. [Google Scholar]
- Waterhouse, A.M.; Procter, J.B.; Martin, D.M.A.; Clamp, M.; Barton, G.J. Jalview version 2: A multiple sequence alignment and analysis workbench. Bioinformatics 2009, 25, 1189–1191. [Google Scholar]
- Styczynski, M.P.; Jensen, K.L.; Rigoutsos, I.; Stephanopoulos, G. BLOSUM62 miscalculations improve search performance. Nat. Biotechnol 2008, 26, 274–275. [Google Scholar]
- Guex, N.; Peitsch, M.C. SWISS-MODEL and the Swiss-PDBViewer: An environment for comparative protein modeling. Electrophoresis 1997, 18, 2714–2723. [Google Scholar]
- Combet, C.; Jambon, M.; Deléage, G.; Geourjon, C. Geno3D: Automatic comparative molecular modelling of protein. Bioinformatics 2002, 18, 213–214. [Google Scholar]
- Müller, A.; MacCallum, R.M.; Sternberg, M.J. Benchmarking PSI-BLAST in genome annotation. J. Mol. Biol 1999, 293, 1257–1271. [Google Scholar]
- Arnold, K.; Bordoli, L.; Kopp, J.; Schwede, T. The SWISS-MODEL workspace: A web-based environment for protein structure homology modelling. Bioinformatics 2006, 22, 195–201. [Google Scholar]
- Kiefer, F.; Arnold, K.; Künzli, M.; Bordoli, L.; Schwede, T. The SWISS-MODEL repository and associated resources. Nucleic Acids Res 2009, 37, D387–D392. [Google Scholar]
- Kopp, J.; Schwede, T. The SWISS-MODEL Repository: New features and functionalities. Nucleic Acids Res 2006, 34, D315–D318. [Google Scholar]
- Schwede, T.; Kopp, J.; Guex, N.; Peitsch, M.C. SWISS-MODEL: An automated protein homology-modeling server. Nucleic Acids Res 2003, 31, 3381–3385. [Google Scholar]
- Arnold, K.; Bordoli, L.; Kopp, J.; Schwede, T. The SWISS-MODEL workspace: A web-based environment for protein structure homology modelling. Bioinformatics 2006, 22, 195–201. [Google Scholar]
- Zhang, Z.H.; Bharatham, K.; Sherman, W.A.; Mihalek, I. deconSTRUCT: General purpose protein database search on the substructure level. Nucleic Acids Res 2010, 38, W590–W594. [Google Scholar]
- Laskowski, R.A.; MacArthur, M.W.; Moss, D.S.; Thornton, J.M. PROCHECK-a program to check the stereochemical quality of protein structures. In J. Appl. Cryst; 1996; Volume 26, pp. 283–291. [Google Scholar]
- Dolinsky, T.J.; Nielsen, J.E.; McCammon, J.A.; Baker, N.A. PDB2PQR: An automated pipeline for the setup, execution, and analysis of Poisson-Boltzmann electrostatics calculations. Nucleic Acids Res 2010, 32, W665–W667. [Google Scholar]
- Baker, N.A.; Sept, D.; Joseph, S.; Holst, M.J.; McCammon, J.A. Electrostatics of nanosystems: Application to microtubules and the ribosome. Proc. Natl. Acad. Sci. USA 2001, 98, 10037–10041. [Google Scholar]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph 1996, 14, 33–38. [Google Scholar]
- Gallivan, J.P.; Dougherty, D.A. Cation-pi interactions in structructural biology. Proc. Natl. Acad. Sci. USA 1999, 96, 9459–9464. [Google Scholar]
- Reed, S.E.; Tyrrell, D.A.; Betts, A.O.; Watt, R.G. Studies on a rhinovirus (EC11) derived from a calf. I. Isolation in calf tracheal organ cultures and characterization of the virus. J. Comp. Pathol 1971, 81, 33–40. [Google Scholar]
- Hooft, R.W.; Vriend, G.; Sander, C.; Abola, E. Errors in protein structures. Nature 1996, 381. [Google Scholar] [CrossRef]
- Melo, F.; Feytmans, E. Assessing protein structures with a non-local atomic interaction energy. J. Mol. Biol 1998, 277, 1141–1152. [Google Scholar]
- Benkert, P.; Tosatto, S.C.E.; Schomburg, D. QMEAN: A comprehensive scoring function for model quality assessment. Proteins Struct. Funct. Bioinforma 1998, 71, 261–277. [Google Scholar]
- Zhou, H.; Zhou, Y. Distance-scaled, finite ideal-gas reference state improves structure-derived potentials of mean force for structure selection and stability prediction. Protein Sci 2002, 11, 2714–2726. [Google Scholar]
- Van Gunsteren, W.F.; Billeter, S.R.; Eising, A.A.; Hünenberger, P.H.; Krüger, P.; Mark, A.E.; Scott, W.R.P.; Tironi, I.G. Biomolecular Simulation: The GROMOS96 Manual and User Guide; Verlag der Fachvereine Hochschulverlag AG an der ETH Zurich: Zurich, Switzerland, 1996. [Google Scholar]
- The PyMOL Molecular Graphics System, version 1.3. Available online: http://www.pymol.org/ accessed on 19 July 2012.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PDB id | Aln. Score | Aln. Length | RMSD (Å) | Avg. dL | Geom. Z | Target Molecule |
|---|---|---|---|---|---|---|
| 1U09A | 330.50 | 370 | 1.42 | 1.88 | −7.20 | FMDV RdRp |
| 2B43A | 273.36 | 337 | 2.65 | 2.11 | −4.04 | Hepatitis C virus (HCV) RdRp |
| 1KHWA | 259.87 | 316 | 2.43 | 2.75 | −3.44 | RHDV RdRp |
| 1S48A | 182.59 | 231 | 2.87 | 2.50 | 0.51 | Bovine viral diarrhea virus (BVDV Rna-dependent rna polymerase |
| 2JL9A | 116.57 | 148 | 2.94 | 1.67 | 1.58 | Phi6 RNA polymerase |
| Stereochemical quality | No. of data points | Parameter value | Comparison typical value | Value band width | No. of band widths from mean | Interpretation |
|---|---|---|---|---|---|---|
| % residues in A,B,L | 413 | 73.6 | 83.8 | 10 | −1.0 | WORSE |
| Omega angle St. Dev. | 463 | 0.6 | 6.0 | 3.0 | −1.8 | BETTER |
| Bad Contact/100 Residue | 0 | 0 | 4.2 | 10 | −0.4 | INSIDE |
| Zeta angle St. Dev. | 433 | 1.1 | 3.1 | 1.6 | −1.3 | BETTER |
| H-bond Energy St.Dev. | 283 | 0.7 | 0.8 | 0.2 | −0.5 | INSIDE |
| Overall G-Factor | 465 | 0.2 | −0.4 | 0.3 | 1.9 | BETTER |
| Stereochemical quality | No of data points | Parameter value | Comparison typical value | Value band width | No. of band widths from mean | Interpretation |
|---|---|---|---|---|---|---|
| % residues in A,B,L | 405 | 90.1 | 76.6 | 10 | 1.4 | WORSE |
| Omega angle St. dev. | 452 | 5.2 | 6.0 | 3.0 | −0.3 | BETTER |
| Bad Contact/100 Residue | 0 | 0 | 10.5 | 10 | −1.1 | INSIDE |
| Zeta angle St. Dev. | 425 | 1.0 | 3.1 | 1.6 | −1.3 | BETTER |
| H-bond Energy St. Dev. | 320 | 0.7 | 0.9 | 0.2 | −1.1 | INSIDE |
| Overall G-Factor | 454 | 0.1 | −0.6 | 0.3 | 2.2 | BETTER |
| Stereochemical quality | No. of data points | Parameter value | Comparison typical value | Value band width | No. of band widths from mean | Interpretation |
|---|---|---|---|---|---|---|
| Chi-1 gauche minus St. Dev. | 52 | 10.0 | 22.7 | 6.5 | −1.9 | BETTER |
| Chi-1 trans St. Dev | 118 | 10.1 | 22.7 | 5.3 | −2.4 | BETTER |
| Chi-1 gauche plus St. Dev. | 200 | 10.1 | 21.3 | 4.9 | −2.3 | BETTER |
| Chi-1 pooled St. Dev. | 370 | 10.2 | 22.0 | 4.8 | −2.4 | BETTER |
| Chi-2 transst dev | 108 | 8.6 | 23.1 | 5.0 | −2.9 | BETTER |
| Stereochemical quality | No. of data points | Parameter value | Comparison typical value | Value band width | No. of band widths from mean | Interpretation |
|---|---|---|---|---|---|---|
| Chi-1 gauche minus St. Dev. | 60 | 16.6 | 18.1 | 6.5 | −0.2 | INSIDE |
| Chi-1 trans St. Dev. | 132 | 11.9 | 19 | 5.3 | −1.3 | BETTER |
| Chi-1 gauche plus St. Dev. | 185 | 13.8 | 17.5 | 4.9 | −0.8 | INSIDE |
| Chi-1 pooled St. Dev. | 377 | 14.2 | 18.2 | 4.8 | −0.8 | INSIDE |
| Chi-2 trans St. Dev. | 86 | 14.7 | 20.4 | 5.0 | −1.1 | INSIDE |
© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Rai, D.K.; Rieder, E. Homology Modeling and Analysis of Structure Predictions of the Bovine Rhinitis B Virus RNA Dependent RNA Polymerase (RdRp). Int. J. Mol. Sci. 2012, 13, 8998-9013. https://doi.org/10.3390/ijms13078998
Rai DK, Rieder E. Homology Modeling and Analysis of Structure Predictions of the Bovine Rhinitis B Virus RNA Dependent RNA Polymerase (RdRp). International Journal of Molecular Sciences. 2012; 13(7):8998-9013. https://doi.org/10.3390/ijms13078998
Chicago/Turabian StyleRai, Devendra K., and Elizabeth Rieder. 2012. "Homology Modeling and Analysis of Structure Predictions of the Bovine Rhinitis B Virus RNA Dependent RNA Polymerase (RdRp)" International Journal of Molecular Sciences 13, no. 7: 8998-9013. https://doi.org/10.3390/ijms13078998