Leukocyte Mitochondrial DNA Alteration in Systemic Lupus Erythematosus and Its Relevance to the Susceptibility to Lupus Nephritis

 and
and
Abstract
:1. Introduction
2. Results and Discussion
2.1. Demographic Data of the SLE Patients and Controls
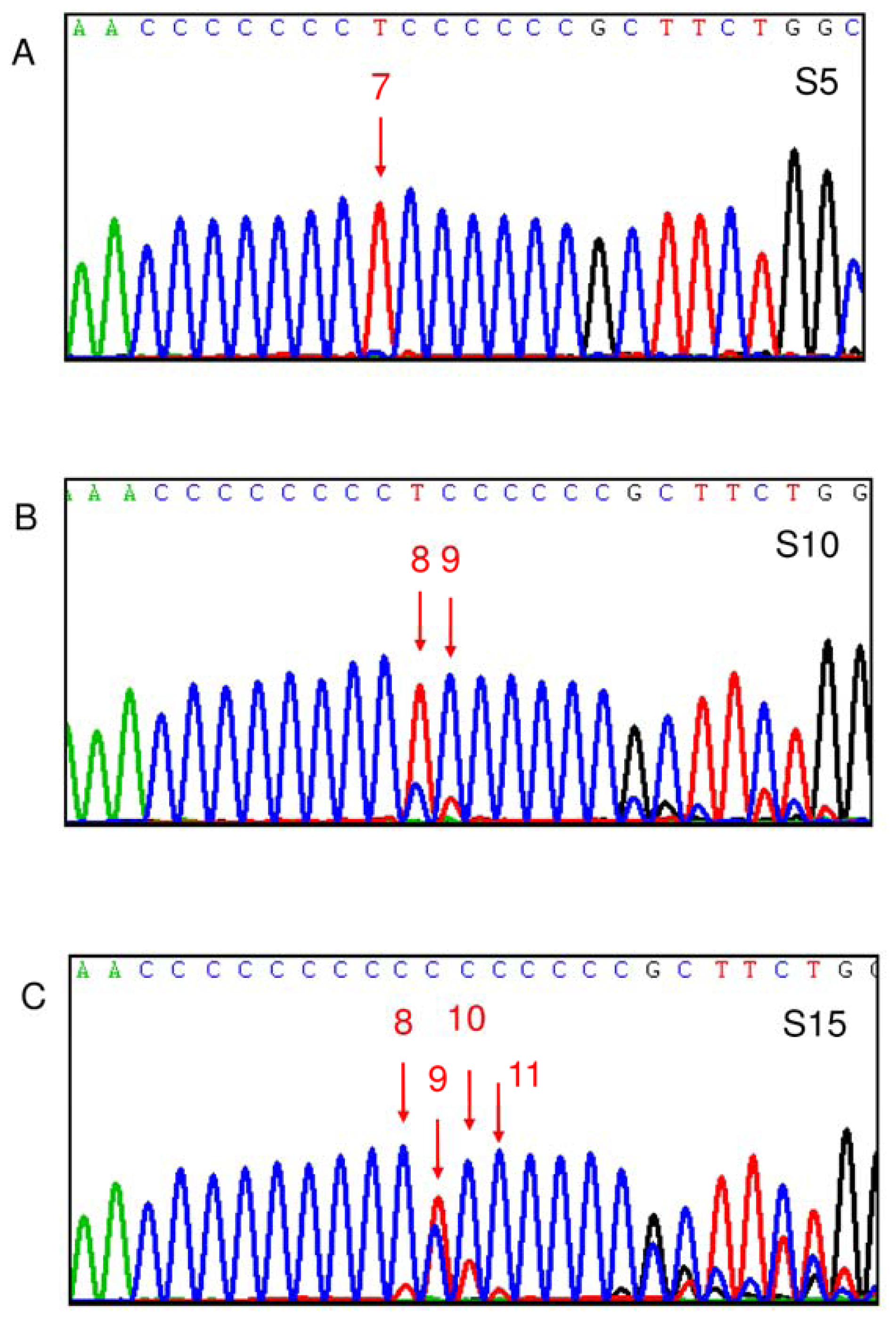
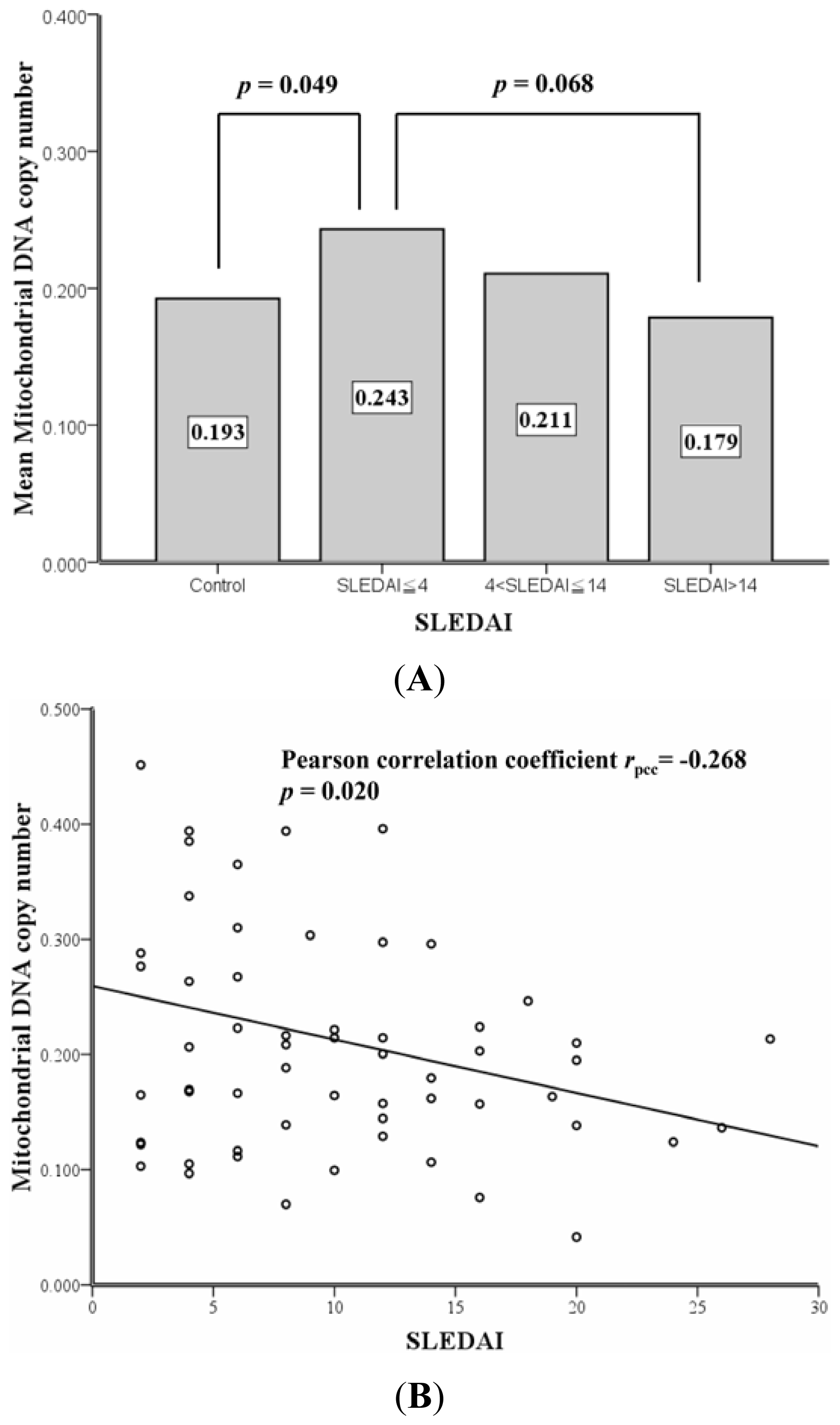
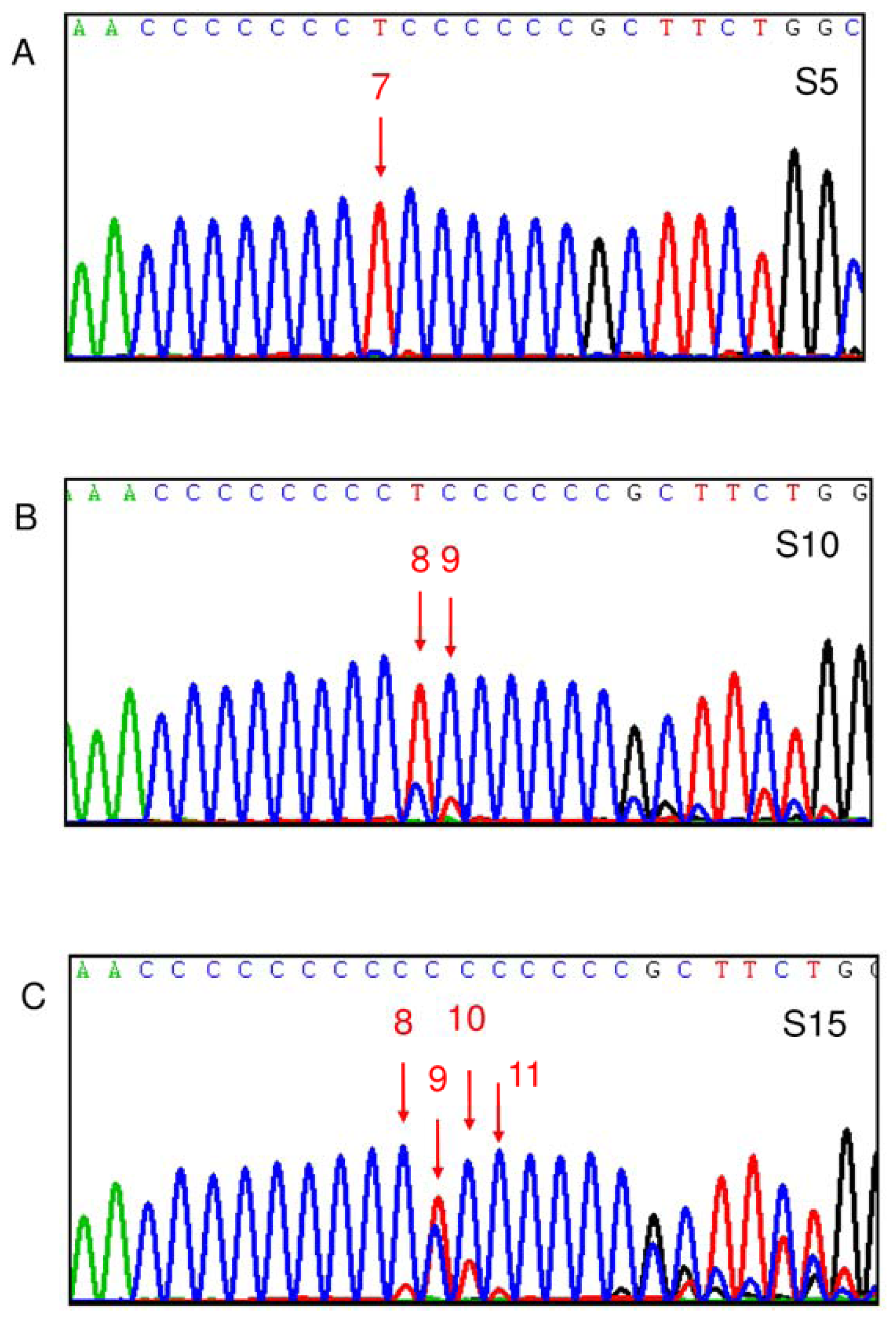
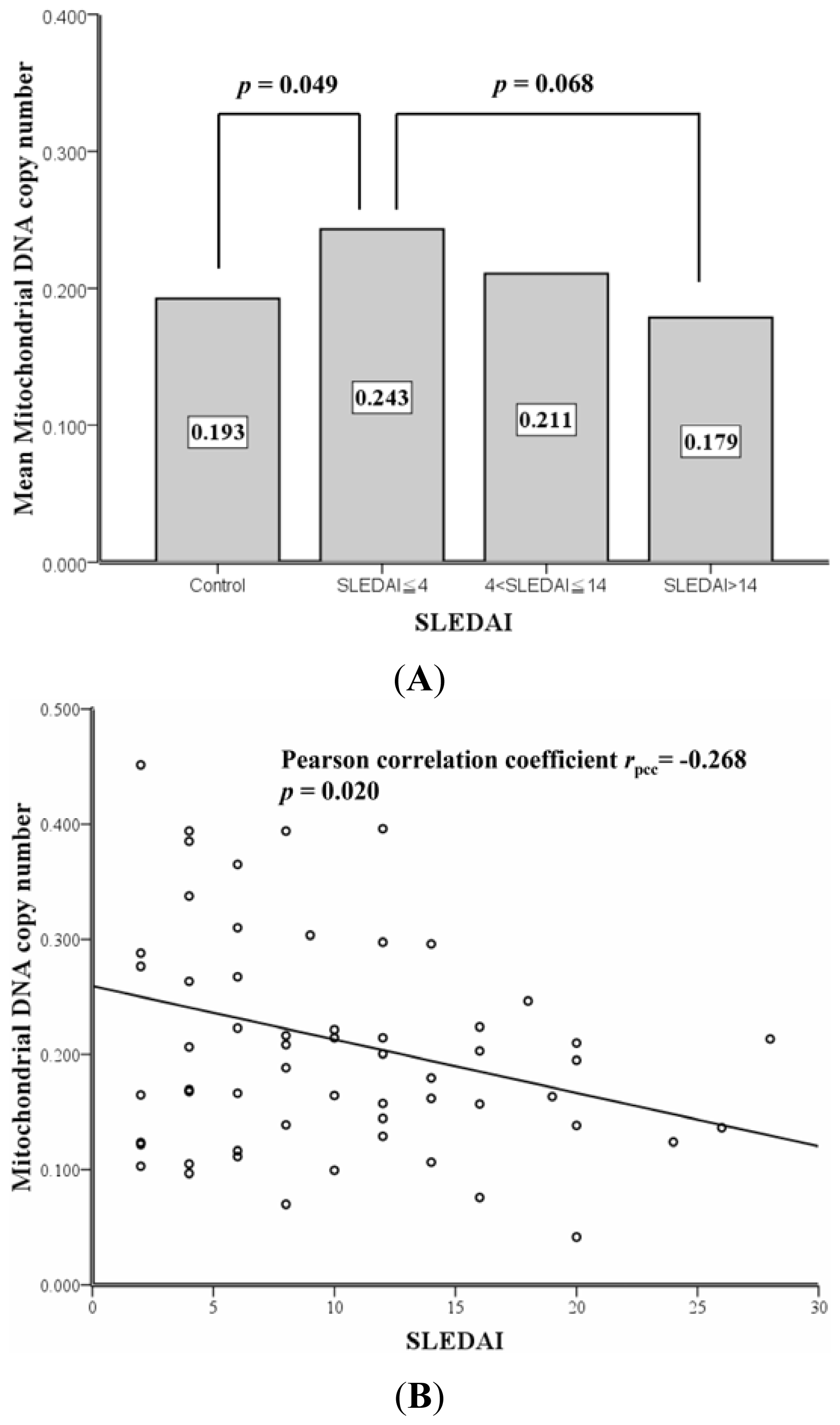
2.2. Correlation between SLEDAI and Leukocyte mtDNA in SLE Patients
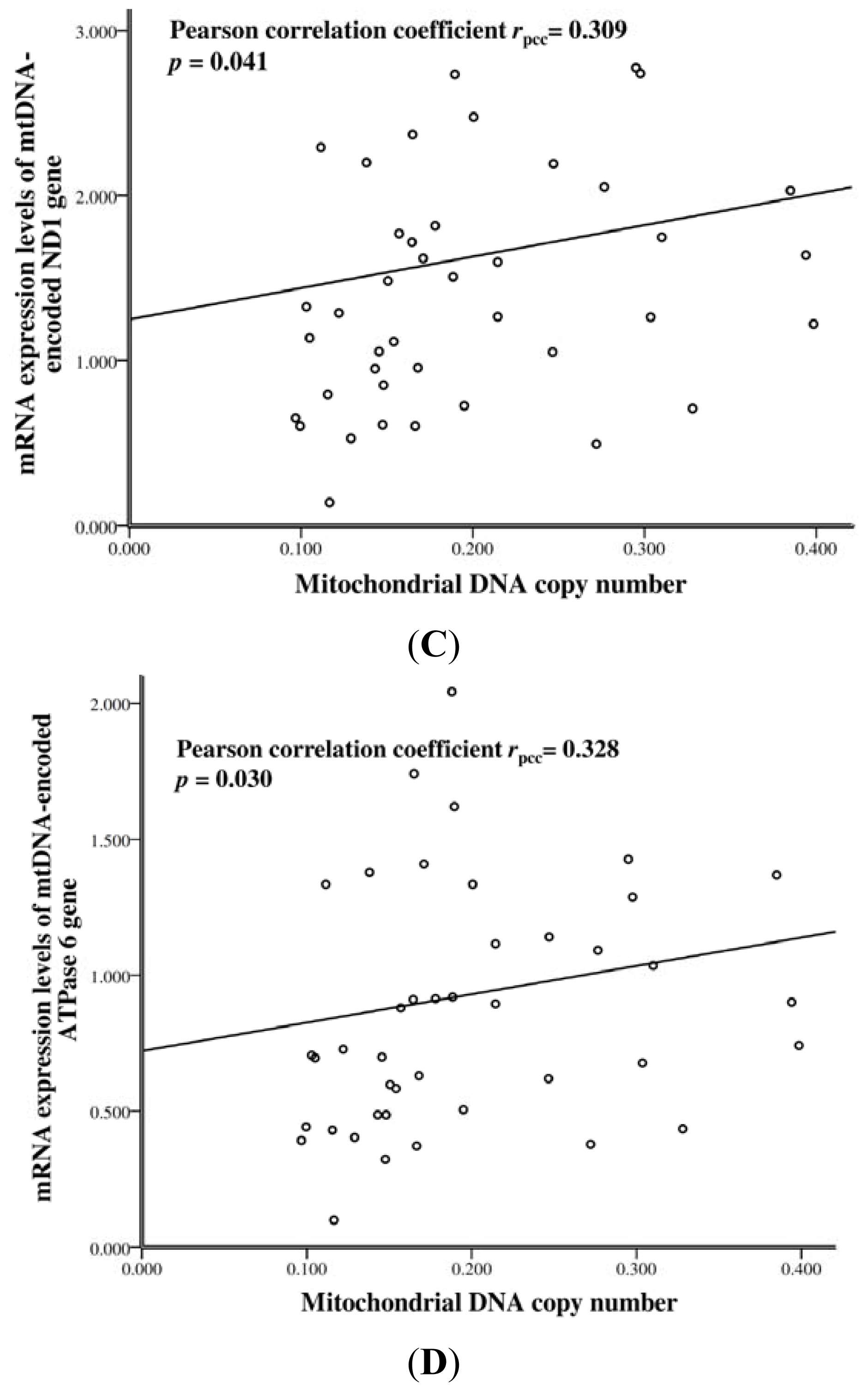
2.3. The mtDNA Copy Number and mRNA Levels of mtDNA-Encoded Genes in SLE Patients
2.4. Correlation between D310 Heteroplasmy and Clinical Manifestations of SLE
2.5. D310 Sequence Variation and Susceptibility to Clinical Manifestations of SLE
3. Experimental Section
3.1. Patient Recruitment, Blood Sample Collection, and Leukocyte DNA and RNA Extraction
3.2. Sequencing of the D310 Region of mtDNA
3.3. Standard Curves for DNA and RNA Quantification
3.4. Determination of mtDNA Copy Number and the Expression of mtDNA-Encoded Genes
3.5. Statistical Analysis
4. Conclusions
Acknowledgments
References
- Perl, A. Pathogenic mechanisms in systemic lupus erythematosus. Autoimmunity 2010, 43, 1–6. [Google Scholar]
- Blank, M.; Shoenfeld, Y.; Perl, A. Cross-talk of the environment with the host genome and the immune system through endogenous retroviruses in systemic lupus erythematosus. Lupus 2009, 18, 1136–1143. [Google Scholar]
- Fernandez, D.; Perl, A. Metabolic control of T cell activation and death in SLE. Autoimmun. Rev 2009, 8, 184–189. [Google Scholar]
- Perl, A.; Gergely, P., Jr; Nagy, G.; Koncz, A.; Banki, K. Mitochondrial hyperpolarization: A checkpoint of T-cell life, death and autoimmunity. Trends Immunol 2004, 25, 360–367. [Google Scholar]
- Perl, A.; Gergely, P., Jr; Banki, K. Mitochondrial dysfunction in T cells of patients with systemic lupus erythematosus. Int. Rev. Immunol 2004, 23, 293–313. [Google Scholar]
- Lee, H.C.; Wei, Y.H. Mitochondrial role in life and death of the cell. J. Biomed. Sci 2000, 7, 2–15. [Google Scholar]
- Jonsen, A.; Yu, X.; Truedsson, L.; Nived, O.; Sturfelt, G.; Ibrahim, S.; Bengtsson, A. Mitochondrial DNA polymorphisms are associated with susceptibility and phenotype of systemic lupus erythematosus. Lupus 2009, 18, 309–312. [Google Scholar]
- Vyshkina, T.; Sylvester, A.; Sadiq, S.; Bonilla, E.; Canter, J.A.; Perl, A.; Kalman, B. Association of common mitochondrial DNA variants with multiple sclerosis and systemic lupus erythematosus. Clin. Immunol 2008, 129, 31–35. [Google Scholar]
- Venhoff, N.; Thiel, J.; Lebrecht, D.; Foocharoen, C.; Effelsberg, N.M.; Trendelenburg, M.; Hasler, P. Circulating Mitochondrial DNA Copy Numbers As a Highly Sensitive Diagnostic Marker of Systemic Lupus Erythematosus and An Independent Predictor of SLE Activity. Proceedings of the Annual Scientific Meeting, Chicago, IL, USA, 4–9 November 2011.
- Andrews, R.M.; Kubacka, I.; Chinnery, P.F.; Lightowlers, R.N.; Turnbull, D.M.; Howell, N. Reanalysis and revision of the Cambridge reference sequence for human mitochondrial DNA. Nat. Genet 1999, 23, 147. [Google Scholar]
- Mitomap: A Human Mitochondrial Genome Database (r61). Available online: http://www.mitomap.org accessed on 14 June 2012.
- Moraes, C.T. What regulates mitochondrial DNA copy number in animal cells? Trends Genet 2001, 17, 199–205. [Google Scholar]
- Lee, H.C.; Wei, Y.H. Mitochondrial biogenesis and mitochondrial DNA maintenance of mammalian cells under oxidative stress. Int. J. Biochem. Cell Biol 2005, 37, 822–834. [Google Scholar]
- Marchington, D.R.; Hartshorne, G.M.; Barlow, D.; Poulton, J. Homopolymeric tract heteroplasmy in mtDNA from tissues and single oocytes: Support for a genetic bottleneck. Am. J. Hum. Genet 1997, 60, 408–416. [Google Scholar]
- Lightowlers, R.N.; Chinnery, P.F.; Turnbull, D.M.; Howell, N. Mammalian mitochondrial genetics: Heredity, heteroplasmy and disease. Trends Genet 1997, 13, 450–455. [Google Scholar]
- Wallace, D.C. The mitochondrial genome in human adaptive radiation and disease: On the road to therapeutics and performance enhancement. Gene 2005, 354, 169–180. [Google Scholar]
- Croteau, D.L.; Stierum, R.H.; Bohr, V.A. Mitochondrial DNA repair pathways. Mutat. Res 1999, 434, 137–148. [Google Scholar]
- Yakes, F.M.; van Houten, B. Mitochondrial DNA damage is more extensive and persists longer than nuclear DNA damage in human cells following oxidative stress. Proc. Natl. Acad. Sci. USA 1997, 94, 514–519. [Google Scholar]
- Mambo, E.; Gao, X.; Cohen, Y.; Guo, Z.; Talalay, P.; Sidransky, D. Electrophile and oxidant damage of mitochondrial DNA leading to rapid evolution of homoplasmic mutations. Proc. Natl. Acad. Sci. USA 2003, 100, 1838–1843. [Google Scholar]
- Wallace, D.C. Mitochondrial DNA mutations in disease and aging. Environ. Mol. Mutagen 2010, 51, 440–450. [Google Scholar]
- Lin, C.S.; Chang, S.C.; Wang, L.S.; Chou, T.Y.; Hsu, W.H.; Wu, Y.C.; Wei, Y.H. The role of mitochondrial DNA alterations in esophageal squamous cell carcinomas. J. Thorac. Cardiovasc. Surg 2010, 139, 189–197. [Google Scholar]
- Wang, P.N.; Lee, H.C.; Wang, C.H.; Ping, Y.H.; Liu, T.Y.; Chi, C.W.; Lin, K.N.; Liu, H.C. Heteroplasmy of mitochondrial D310 mononucleotide repeat region in the blood of patients with Alzheimer’s disease. J. Alzheimers Dis 2009, 18, 345–353. [Google Scholar]
- Tseng, L.M.; Yin, P.H.; Chi, C.W.; Hsu, C.Y.; Wu, C.W.; Lee, L.M.; Wei, Y.H.; Lee, H.C. Mitochondrial DNA mutations and mitochondrial DNA depletion in breast cancer. Genes Chromosomes Cancer 2006, 45, 629–638. [Google Scholar]
- Wu, C.W.; Yin, P.H.; Hung, W.Y.; Li, A.F.; Li, S.H.; Chi, C.W.; Wei, Y.H.; Lee, H.C. Mitochondrial DNA mutations and mitochondrial DNA depletion in gastric cancer. Genes Chromosomes Cancer 2005, 44, 19–28. [Google Scholar]
- Shin, M.G.; Levin, B.C.; Kim, H.J.; Kim, H.R.; Lee, I.K.; Cho, D.; Kee, S.J.; Shin, J.H.; Suh, S.P.; Ryang, D.W. Profiling of length heteroplasmies in the human mitochondrial DNA control regions from blood cells in the Korean population. Electrophoresis 2006, 27, 1331–1340. [Google Scholar]
- Kim, M.M.; Clinger, J.D.; Masayesva, B.G.; Ha, P.K.; Zahurak, M.L.; Westra, W.H.; Califano, J.A. Mitochondrial DNA quantity increases with histopathologic grade in premalignant and malignant head and neck lesions. Clin. Cancer Res 2004, 10, 8512–8515. [Google Scholar]
- Lee, H.C.; Lim, M.L.; Lu, C.Y.; Liu, V.W.; Fahn, H.J.; Zhang, C.; Nagley, P.; Wei, Y.H. Concurrent increase of oxidative DNA damage and lipid peroxidation together with mitochondrial DNA mutation in human lung tissues during aging—Smoking enhances oxidative stress on the aged tissues. Arch. Biochem. Biophys 1999, 362, 309–316. [Google Scholar]
- Lee, H.C.; Lu, C.Y.; Fahn, H.J.; Wei, Y.H. Aging- and smoking-associated alteration in the relative content of mitochondrial DNA in human lung. FEBS Lett 1998, 441, 292–296. [Google Scholar]
- Dolff, S.; Berden, J.H.; Bijl, M. Treatment of lupus nephritis. Expert Rev. Clin. Immunol 2010, 6, 901–911. [Google Scholar] [Green Version]
- Houssiau, F.A.; Ginzler, E.M. Current treatment of lupus nephritis. Lupus 2008, 17, 426–430. [Google Scholar]
- Hochberg, M.C. Updating the American College of Rheumatology revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum 1997, 40, 1725. [Google Scholar]
- Tan, E.M.; Cohen, A.S.; Fries, J.F.; Masi, A.T.; McShane, D.J.; Rothfield, N.F.; Schaller, J.G.; Talal, N.; Winchester, R.J. The 1982 revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum 1982, 25, 1271–1277. [Google Scholar]
- Gladman, D.D.; Ibanez, D.; Urowitz, M.B. Systemic lupus erythematosus disease activity index 2000. J. Rheumatol 2002, 29, 288–291. [Google Scholar]
- Bombardier, C.; Gladman, D.D.; Urowitz, M.B.; Caron, D.; Chang, C.H. Derivation of the SLEDAI. A disease activity index for lupus patients. The Committee on Prognosis Studies in SLE. Arthritis Rheum 1992, 35, 630–640. [Google Scholar]
- Liu, C.S.; Kuo, C.L.; Cheng, W.L.; Huang, C.S.; Lee, C.F.; Wei, Y.H. Alteration of the copy number of mitochondrial DNA in leukocytes of patients with hyperlipidemia. Ann. N. Y. Acad. Sci 2005, 1042, 70–75. [Google Scholar]
- Chomczynski, P.; Mackey, K. Short technical reports. Modification of the TRI reagent procedure for isolation of RNA from polysaccharide- and proteoglycan-rich sources. Biotechniques 1995, 19, 942–945. [Google Scholar]
- Bai, R.K.; Perng, C.L.; Hsu, C.H.; Wong, L.J. Quantitative PCR analysis of mitochondrial DNA content in patients with mitochondrial disease. Ann. N. Y. Acad. Sci 2004, 1011, 304–309. [Google Scholar]
- Chen, C.T.; Shih, Y.R.; Kuo, T.K.; Lee, O.K.; Wei, Y.H. Coordinated changes of mitochondrial biogenesis and antioxidant enzymes during osteogenic differentiation of human mesenchymal stem cells. Stem Cells 2008, 26, 960–968. [Google Scholar]
- Ma, Y.S.; Wu, S.B.; Lee, W.Y.; Cheng, J.S.; Wei, Y.H. Response to the increase of oxidative stress and mutation of mitochondrial DNA in aging. Biochim. Biophys. Acta 2009, 1790, 1021–1029. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
| Demographic data/mtDNA characteristics | Controls | SLE patients | p-value |
|---|---|---|---|
| Case number (%) | 45 (100.0) | 85 (100.0) | - |
| Gender | - | - | 0.825 * |
| Male (%) | 7 (15.6) | 12 (14.1) | - |
| Female (%) | 38 (84.4) | 73 (85.9) | - |
| Age (mean ± SD) years | 42.6 ± 9.0 | 44.6 ± 12.0 | 0.335 § |
| SLEDAI | - | - | - |
| Min~Max, Median | - | 2~28, 8 | - |
| Mitochondrial DNA | |||
| D310 sequence variation, predominant variant | - | - | 0.079 * |
| C5TC6 (%) | 1 (2.2) | 0 (0.0) | - |
| C7TC6 (%) | 19 (42.2) | 27 (31.8) | - |
| C8TC6 (%) | 18 (40.0) | 36 (42.4) | - |
| C9TC6 (%) | 5 (11.1) | 21 (24.7) | - |
| C10TC6 (%) | 2 (4.4) | 0 (0.0) | - |
| C11TC6 (%) | 0 (0.0) | 1 (1.2) | - |
| D310 patterns | - | - | 0.022 * |
| Heteroplasmy (%) | 22 (48.9) | 59 (69.4) | - |
| Homoplasmy (%) | 23 (51.1) | 26 (30.6) | - |
| Number of D310 variants (mean ± SD) | 1.7 ± 0.8 | 2.2 ± 1.1 | 0.014 § |
| mtDNA copy number (mean ± SD) | 0.193 ± 0.065 | 0.214 ± 0.113 | 0.169 § |
| Case number (%) † | 26 (100.0) | 44 (100.0) | - |
| mtDNA encoded mRNA expression | |||
| ND1 (mean ± SD) | 2.845 ± 2.000 | 1.625 ± 1.029 | 0.001 § |
| ATPase 6 (mean ± SD) | 1.538 ± 1.108 | 0.928 ± 0.526 | 0.003 § |
| Control | SLE | p-value | |||
|---|---|---|---|---|---|
| SLEDAI ≤ 4.0@ | 4.0@ < SLEDAI ≤ 14.0@ | SLEDAI > 14.0@ | |||
| Case number (%) | 45 (100.0) | 26 (100.0) | 42 (100.0) | 17 (100.0) | - |
| Mitochondrial DNA | |||||
| D310 pattern | - | - | - | - | 0.021 * |
| Heteroplasmy (%) | 22 (48.9) | 17 (65.4) | 29 (69.0) | 13 (76.5) | - |
| Homoplasmy (%) | 23 (51.1) | 9 (34.6) | 13 (31.0) | 4 (23.5) | - |
| mtDNA copy number (mean ± SD) | 0.193 ± 0.065 | 0.243 ± 0.147 | 0.211 ± 0.098 | 0.179 ± 0.077 | 0.119 ** |
| 0.193 ± 0.065 | 0.243 ± 0.147 | - | - | 0.049 a | |
| - | 0.243 ± 0.147 | - | 0.179 ± 0.077 | 0.068 b | |
| D310 pattern | Number of D310 variants | ||||
|---|---|---|---|---|---|
| Case number | Heteroplasmic No. (%) | Homoplasmic No. (%) | p-value * | (Mean ± SD) | p-value ** |
| CNS involvement | |||||
| No (n = 42) (100%) | 29 (69.0) | 13 (31.0) | 0.943 | 2.1 ± 1.0 | 0.860 |
| Yes (n = 43) (100%) | 30 (69.8) | 13 (30.2) | 2.2 ± 1.2 | ||
| Nephritis | |||||
| No (n = 52) (100%) | 35 (67.3) | 17 (32.7) | 0.597 | 2.0 ± 0.9 | 0.035 |
| Yes (n = 33) (100%) | 24 (72.7) | 9 (27.3) | 2.5 ± 1.3 | ||
| Skin rash | |||||
| No (n = 65) (100%) | 45 (69.2) | 20 (30.8) | 0.948 | 2.2 ± 1.1 | 0.604 |
| Yes (n = 20) (100%) | 14 (70.0) | 6 (30.0) | 2.1 ± 1.1 | ||
| Alopecia | |||||
| No (n = 59) (100%) | 39 (66.1) | 20 (33.9) | 0.318 | 2.1 ± 1.1 | 0.325 |
| Yes (n = 26) (100%) | 20 (76.9) | 6 (23.1) | 2.3 ± 1.2 | ||
| Oral ulcer | |||||
| No (n = 72) (100%) | 52 (72.2) | 20 (27.8) | 0.186 | 2.2 ± 1.1 | 0.268 |
| Yes (n = 13) (100%) | 7 (53.8) | 6 (46.2) | 1.8 ± 1.1 | ||
| Decreased complement | |||||
| No (n = 54) (100%) | 36 (66.7) | 18 (33.3) | 0.469 | 2.2 ± 1.2 | 0.413 |
| Yes *** (n = 31) (100%) | 23 (74.2) | 8 (25.8) | 2.0 ± 0.9 | ||
| D310 sequence variation, predominant variant (Case number, %) | |||||
|---|---|---|---|---|---|
| C7TC6 | C8TC6 | C9TC6 | C12TC6 | ||
| n = 27 (100) | n = 36 (100) | n = 21 (100) | n = 1 (100) | p-value * | |
| CNS involvement | |||||
| No (n = 42) | 14 (51.9) | 16 (44.4) | 12 (57.1) | 0 (0.0) | 0.594 |
| Yes (n = 43) | 13 (48.1) | 20 (55.6) | 9 (42.9) | 1 (100.0) | |
| Nephritis | |||||
| No (n = 52) | 18 (66.7) | 24 (66.7) | 10 (47.6) | 0 (0.0) | 0.084 |
| Yes (n = 33) | 9 (33.3) | 12 (33.3) | 11 (52.4) | 1 (100.0) | |
| Skin rash | |||||
| No (n = 65) | 21 (77.8) | 24 (66.7) | 19 (90.5) | 1 (100.0) | 0.208 |
| Yes (n = 20) | 6 (22.2) | 12 (33.3) | 2 (9.5) | 0 (0.0) | |
| Alopecia | |||||
| No (n = 59) | 22 (81.5) | 22 (61.1) | 14 (66.7) | 1 (100.0) | 0.316 |
| Yes (n = 26) | 5 (18.5) | 14 (38.9) | 7 (33.3) | 0 (0.0) | |
| Oral ulcer | |||||
| No (n = 72) | 21 (77.8) | 31 (86.1) | 19 (90.5) | 1 (100.0) | 0.620 |
| Yes (n = 13) | 6 (22.2) | 5 (13.9) | 2 (9.5) | 0 (0.0) | |
| Decreased complement | |||||
| No (n = 54) | 19 (70.4) | 19 (52.8) | 15 (71.4) | 1 (100.0) | 0.323 |
| Yes (n = 31) | 8 (29.6) | 17 (47.2) | 6 (28.6) | 0 (0.0) | |
© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Lee, H.-T.; Lin, C.-S.; Chen, W.-S.; Liao, H.-T.; Tsai, C.-Y.; Wei, Y.-H. Leukocyte Mitochondrial DNA Alteration in Systemic Lupus Erythematosus and Its Relevance to the Susceptibility to Lupus Nephritis. Int. J. Mol. Sci. 2012, 13, 8853-8868. https://doi.org/10.3390/ijms13078853
Lee H-T, Lin C-S, Chen W-S, Liao H-T, Tsai C-Y, Wei Y-H. Leukocyte Mitochondrial DNA Alteration in Systemic Lupus Erythematosus and Its Relevance to the Susceptibility to Lupus Nephritis. International Journal of Molecular Sciences. 2012; 13(7):8853-8868. https://doi.org/10.3390/ijms13078853
Chicago/Turabian StyleLee, Hui-Ting, Chen-Sung Lin, Wei-Sheng Chen, Hsien-Tzung Liao, Chang-Youh Tsai, and Yau-Huei Wei. 2012. "Leukocyte Mitochondrial DNA Alteration in Systemic Lupus Erythematosus and Its Relevance to the Susceptibility to Lupus Nephritis" International Journal of Molecular Sciences 13, no. 7: 8853-8868. https://doi.org/10.3390/ijms13078853