8-Oxoguanine DNA Glycosylases: One Lesion, Three Subfamilies


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
1.1. Guanine Oxidation
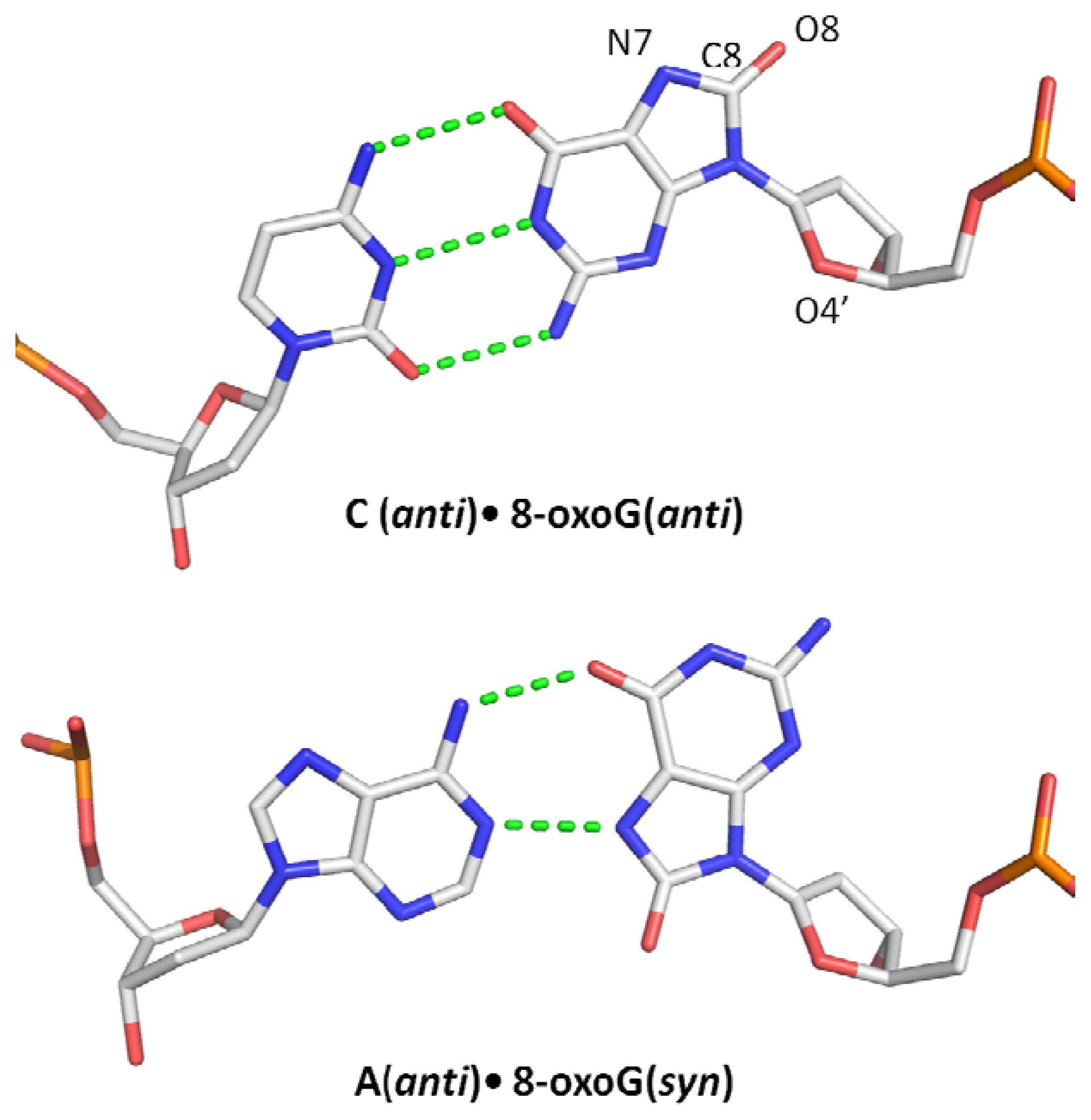
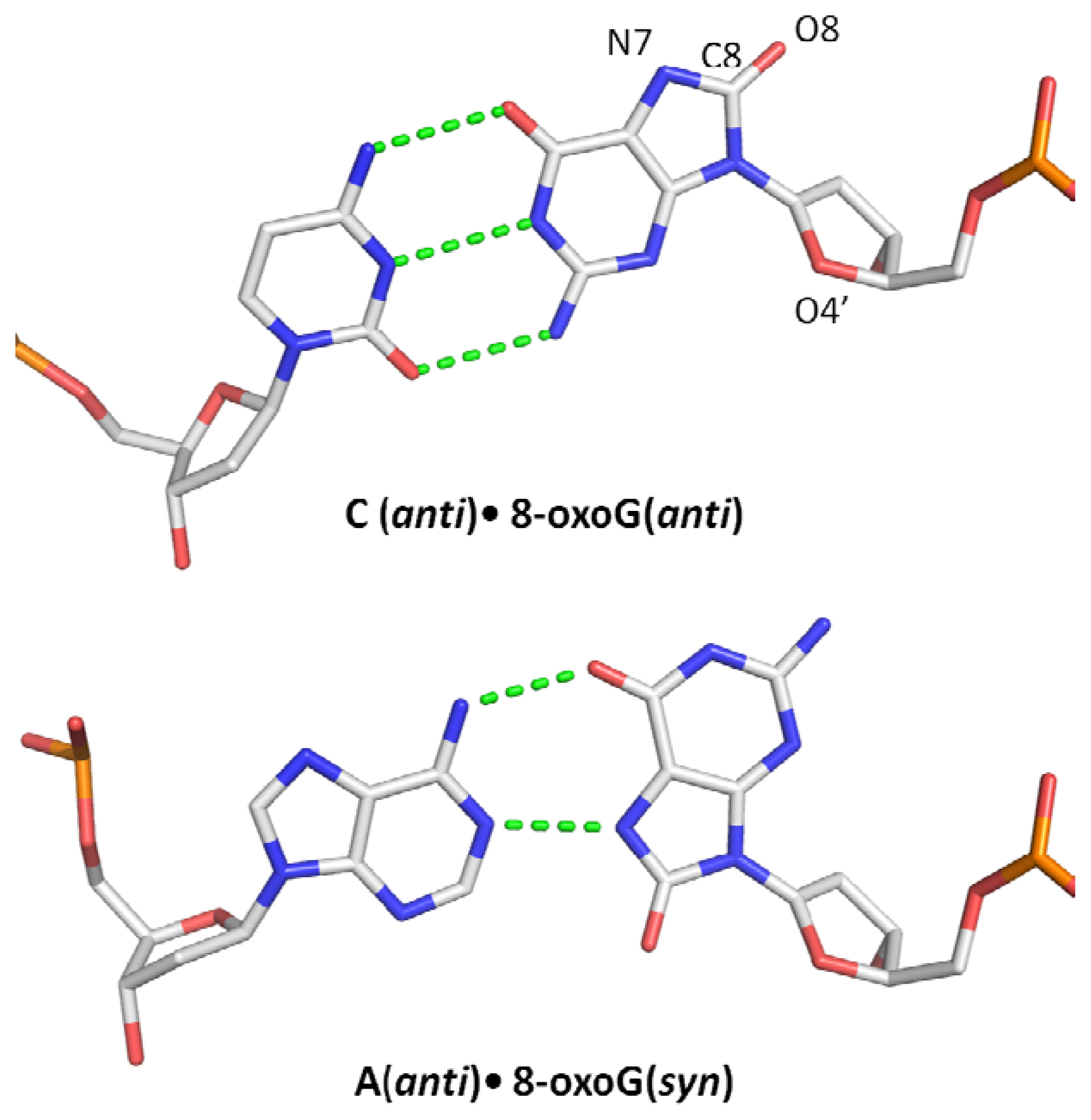
1.2. Miscoding Properties of 8-oxoG
2. 8-oxoguanine DNA Glycosylase
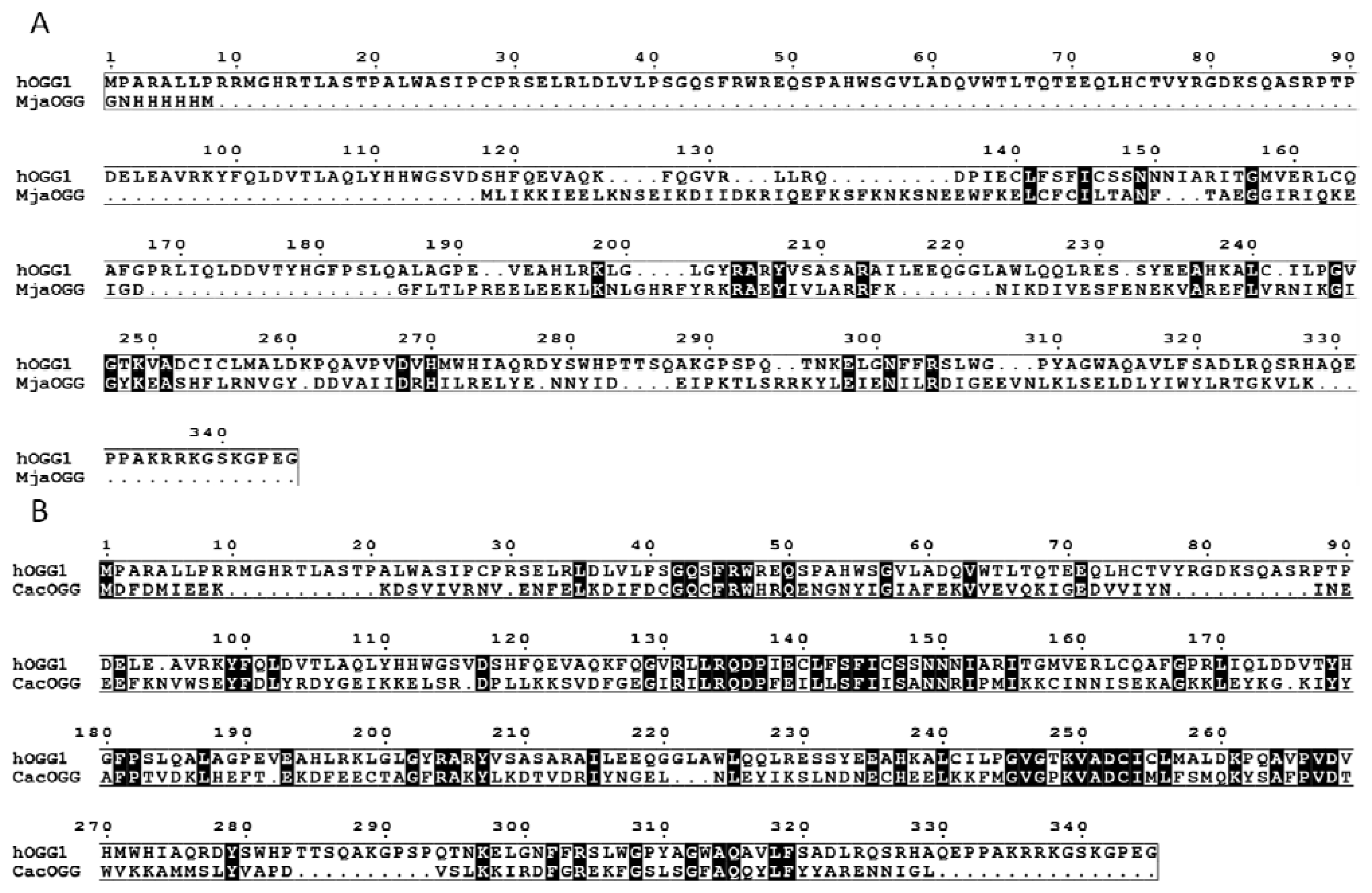
2.1. OGG, Three Families, One Lesion
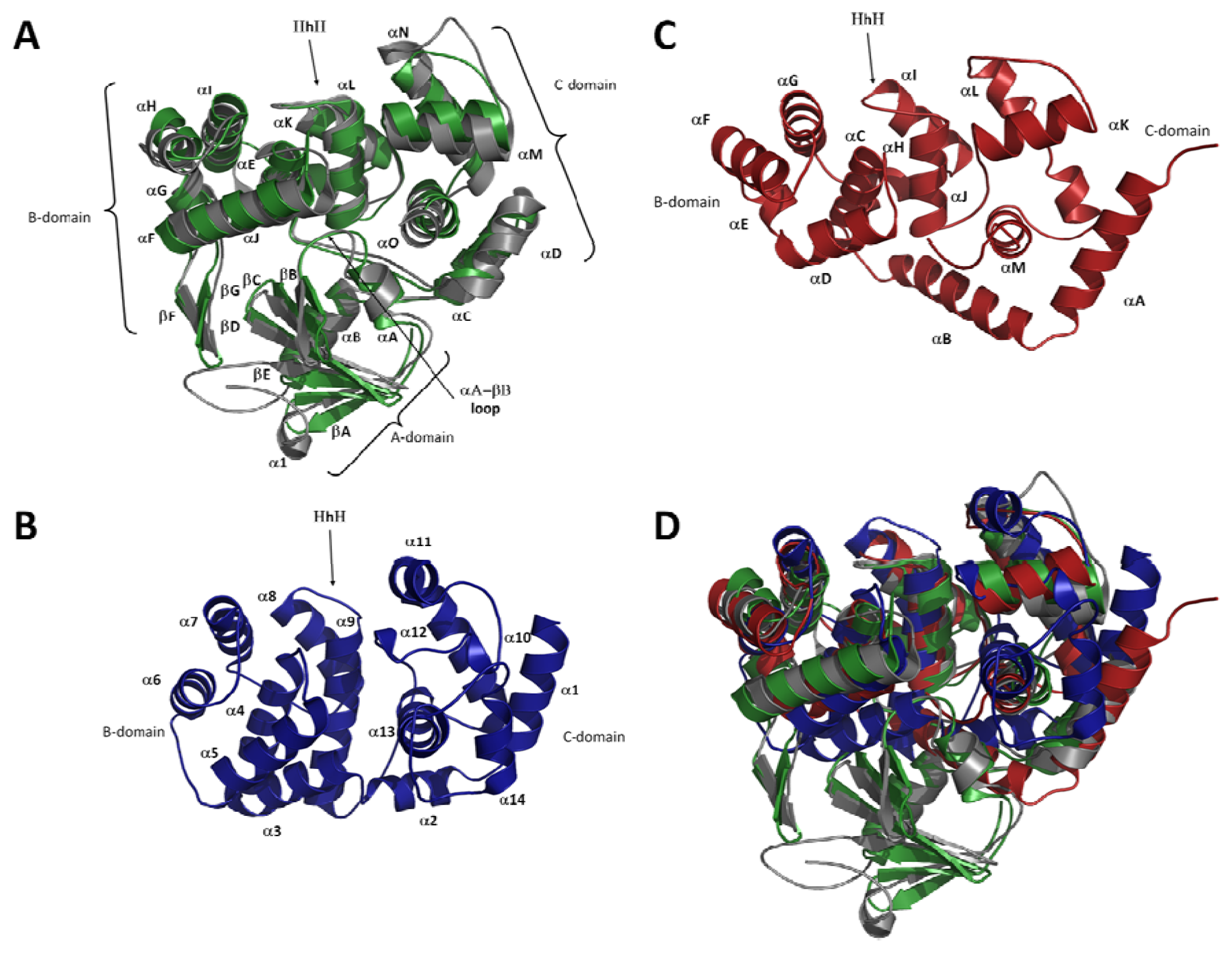
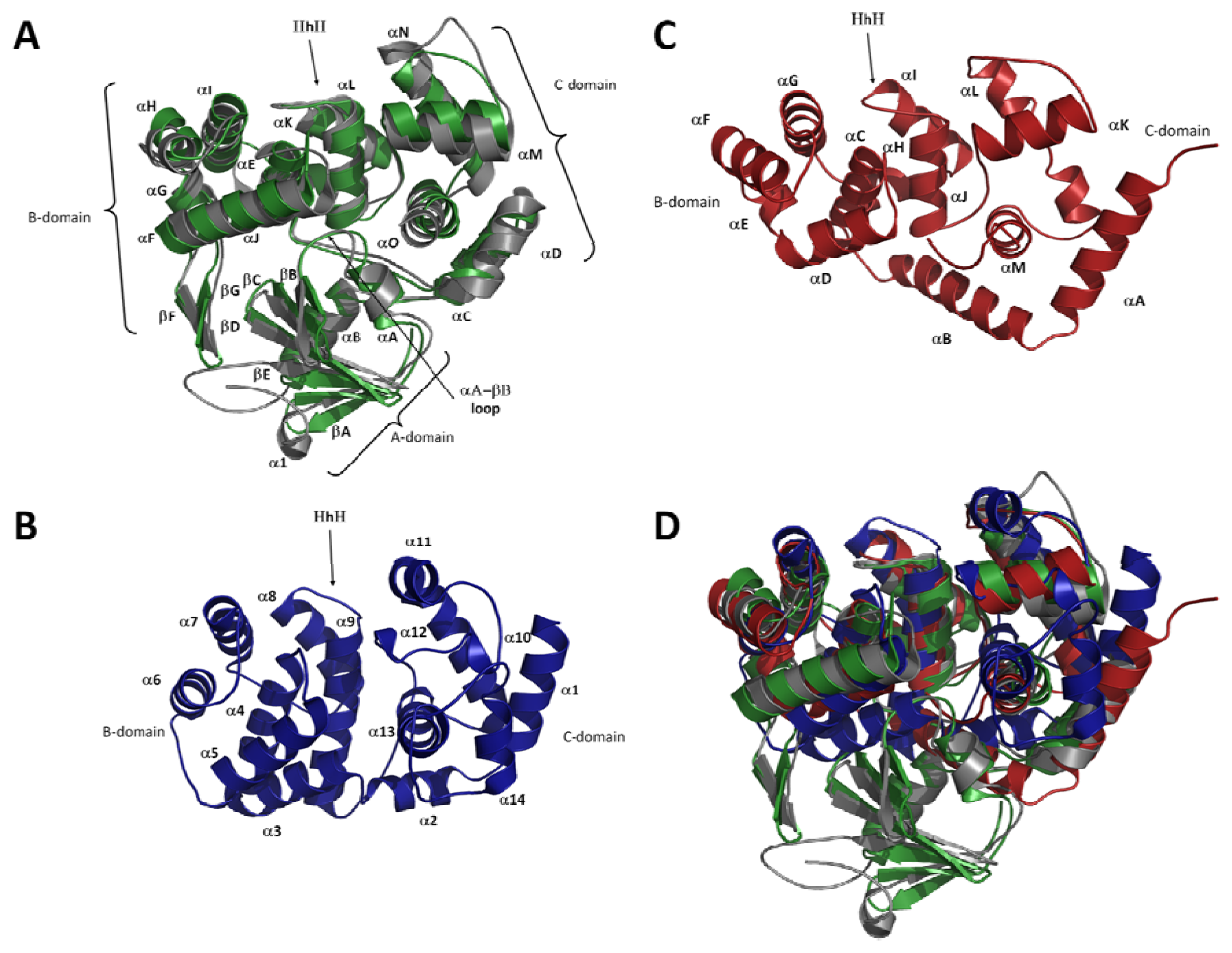
2.2. Overall Fold of OGG
2.3. The Helix-Hairpin Helix (HhH) Motif
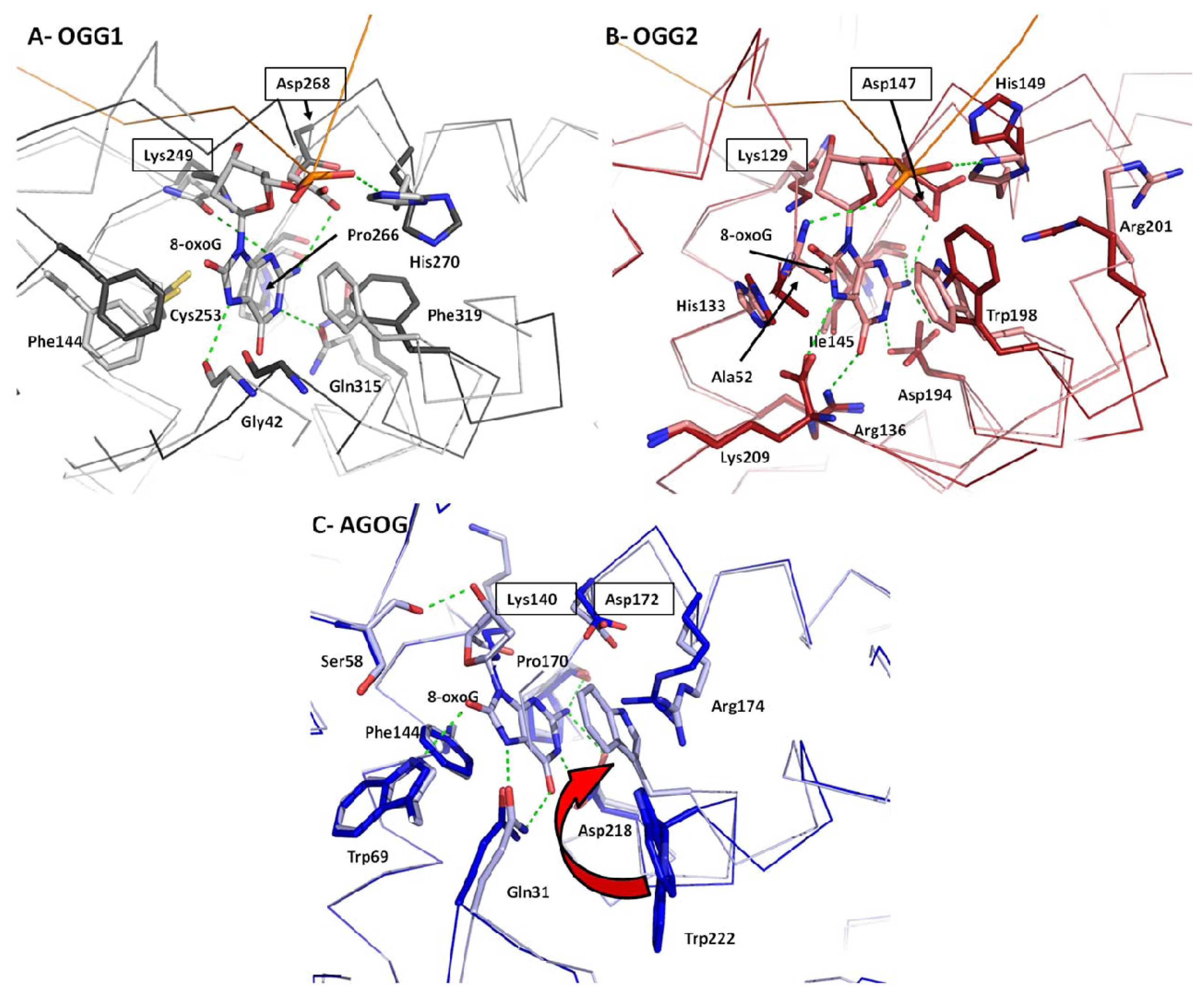
2.4. 8-oxoG Recognition
2.5. Conformational Change upon 8-oxoG Binding
2.6. Catalytic Mechanism
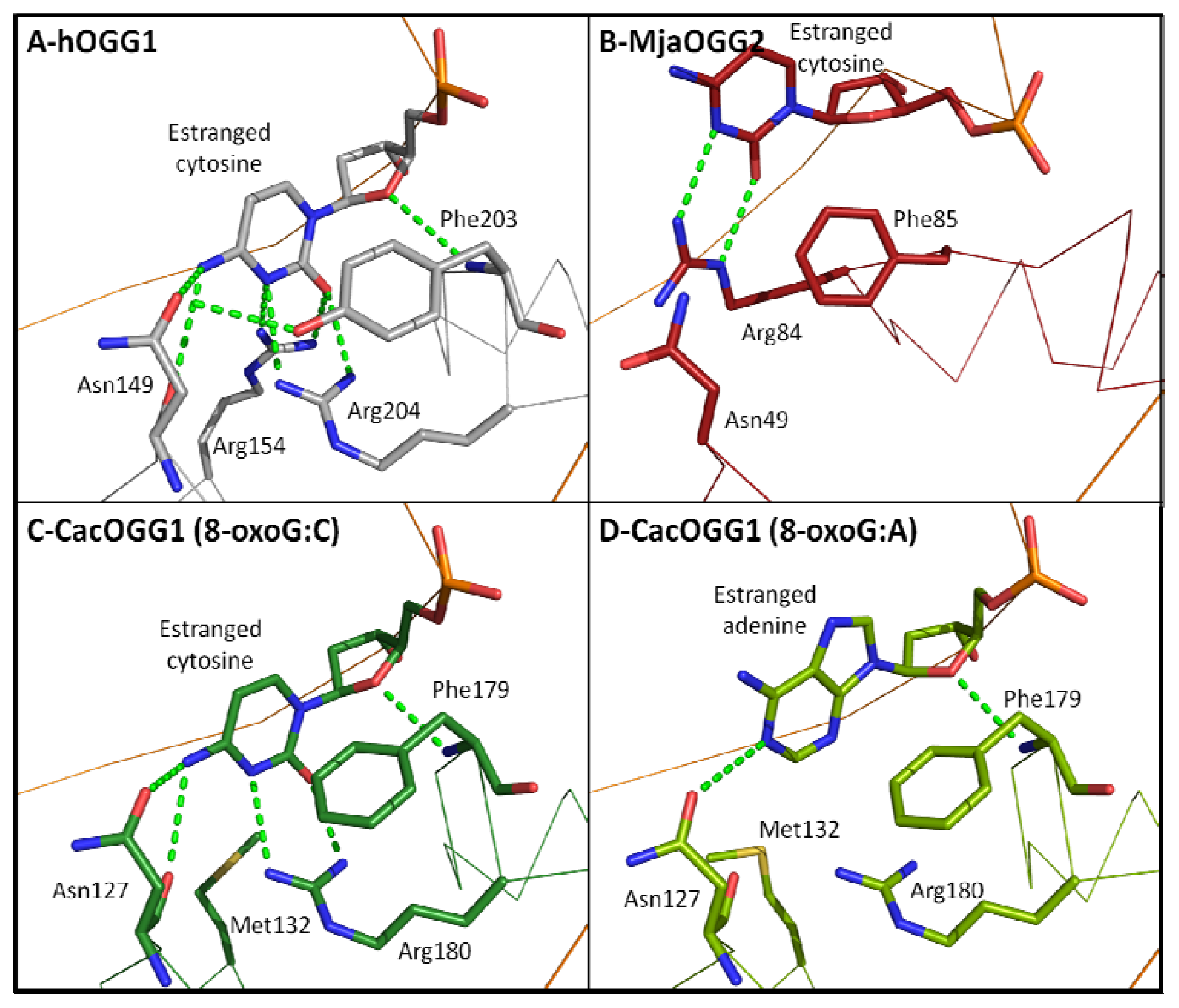
2.7. Estranged Base Binding Site
3. Conclusions
Acknowledgments
References
- Fraga, C.G.; Shigenaga, M.K.; Park, J.W.; Degan, P.; Ames, B.N. Oxidative damage to DNA during aging: 8-hydroxy-2′-deoxyguanosine in rat organ DNA and urine. Proc. Natl. Acad. Sci. USA 1990, 87, 4533–4537. [Google Scholar]
- Steenken, S.; Jovanovic, S.V. How easily oxidizable is DNA? One-electron reduction potentials of adenosine and guanosine radicals in aqueous solution. J. Am. Chem. Soc 1997, 119, 617–618. [Google Scholar]
- Neeley, W.L.; Essigmann, J.M. Mechanisms of formation, genotoxicity, and mutation of guanine oxidation products. Chem. Res. Toxicol 2006, 19, 491–505. [Google Scholar]
- Kasai, H.; Crain, P.F.; Kuchino, Y.; Nishimura, S.; Ootsuyama, A.; Tanooka, H. Formation of 8-hydroxyguanine moiety in cellular DNA by agents producing oxygen radicals and evidence for its repair. Carcinogenesis 1986, 7, 1849–1851. [Google Scholar]
- Gedik, C.M.; Collins, A. Establishing the background level of base oxidation in human lymphocyte DNA: Results of an interlaboratory validation study. FASEB J 2005, 19, 82–84. [Google Scholar]
- Klaunig, J.E.; Kamendulis, L.M. The role of oxidative stress in carcinogenesis. Annu. Rev. Pharmacol. Toxicol 2004, 44, 239–267. [Google Scholar]
- Saito, I.; Takayama, M.; Nakamura, T.; Sugiyama, H.; Komeda, Y.; Iwasaki, M. The most electron-donating sites in duplex DNA: Guanine-guanine stacking rule. Nucleic Acids Symp. Ser 1995, 191–192. [Google Scholar]
- Sugiyama, H.; Saito, I. Theoretical studies of GC-specific photocleavage of DNA via electron transfer: Significant lowering of ionization potential and 5′-localization of HOMO of stacked GG bases in B-form DNA. J. Am. Chem. Soc 1996, 118, 7063–7068. [Google Scholar]
- Giese, B. Long-distance electron transfer through DNA. Annu. Rev. Biochem 2002, 71, 51–70. [Google Scholar]
- Culp, S.J.; Cho, B.P.; Kadlubar, F.F.; Evans, F.E. Structural and conformational analyses of 8-hydroxy-2′-deoxyguanosine. Chem. Res. Toxicol 1989, 2, 416–422. [Google Scholar]
- Uesugi, S.; Ikehara, M. Carbon-13 magnetic resonance spectra of 8-substituted purine nucleosides: Characteristic shifts for the syn conformation. J. Am. Chem. Soc 1977, 99, 3250–3253. [Google Scholar]
- Grollman, A.P.; Moriya, M. Mutagenesis by 8-oxoguanine: An enemy within. Trends Genet 1993, 9, 246–249. [Google Scholar]
- Kuchino, Y.; Mori, F.; Kasai, H.; Inoue, H.; Iwai, S.; Miura, K.; Ohtsuka, E.; Nishimura, S. Misreading of DNA templates containing 8-hydroxydeoxyguanosine at the modified base and at adjacent residues. Nature 1987, 327, 77–79. [Google Scholar]
- Shibutani, S.; Takeshita, M.; Grollman, A.P. Insertion of specific bases during DNA synthesis past the oxidation-damaged base 8-oxodG. Nature 1991, 349, 431–434. [Google Scholar]
- Wood, M.L.; Dizdaroglu, M.; Gajewski, E.; Essigmann, J.M. Mechanistic studies of ionizing radiation and oxidative mutagenesis: Genetic effects of a single 8-hydroxyguanine (7-hydro-8-oxoguanine) residue inserted at a unique site in a viral genome. Biochemistry 1990, 29, 7024–7032. [Google Scholar]
- Sarma, R.H.; Lee, C.H.; Evans, F.E.; Yathindra, N.; Sundaralingam, M. Probing the interrelation between the glycosyl torsion, sugar pucker, and the backbone conformation in C(8) substituted adenine nucleotides by 1H and 1H-(31P) fast Fourier transform nuclear magnetic resonance methods and conformational energy calculations. J. Am. Chem. Soc 1974, 96, 7337–7348. [Google Scholar]
- Tavale, S.S.; Sobell, H.M. Crystal and molecular structure of 8-bromoguanosine and 8-bromoadenosine, two purine nucleosides in the syn conformation. J. Mol. Biol 1970, 48, 109–123. [Google Scholar]
- Brieba, L.G.; Eichman, B.F.; Kokoska, R.J.; Doublié, S.; Kunkel, T.A.; Ellenberger, T. Structural basis for the dual coding potential of 8-oxoguanosine by a high-fidelity DNA polymerase. EMBO J 2004, 23, 3452–3461. [Google Scholar]
- Hsu, G.W.; Ober, M.; Carell, T.; Beese, L.S. Error-prone replication of oxidatively damaged DNA by a high-fidelity DNA polymerase. Nature 2004, 431, 217–221. [Google Scholar]
- Denver, D.R.; Swenson, S.L.; Lynch, M. An evolutionary analysis of the helix-hairpin-helix superfamily of DNA repair glycosylases. Mol. Biol. Evol 2003, 20, 1603–1611. [Google Scholar]
- Castaing, B.; Geiger, A.; Seliger, H.; Nehls, P.; Laval, J.; Zelwer, C.; Boiteux, S. Cleavage and binding of a DNA fragment containing a single 8-oxoguanine by wild type and mutant FPG proteins. Nucleic Acids Res 1993, 21, 2899–2905. [Google Scholar]
- Michaels, M.L.; Pham, L.; Cruz, C.; Miller, J.H. MutM, a protein that prevents G.C→T.A transversions, is formamidopyrimidine-DNA glycosylase. Nucleic Acids Res 1991, 19, 3629–3632. [Google Scholar]
- Matsumoto, Y.; Zhang, Q.M.; Takao, M.; Yasui, A.; Yonei, S. Escherichia coli Nth and human hNTH1 DNA glycosylases are involved in removal of 8-oxoguanine from 8-oxoguanine/guanine mispairs in DNA. Nucleic Acids Res 2001, 29, 1975–1981. [Google Scholar]
- Robey-Bond, S.M.; Barrantes-Reynolds, R.; Bond, J.P.; Wallace, S.S.; Bandaru, V. Clostridium acetobutylicum 8-oxoguanine DNA glycosylase (Ogg) differs from eukaryotic Oggs with respect to opposite base discrimination. Biochemistry 2008, 47, 7626–7636. [Google Scholar]
- Faucher, F.; Robey-Bond, S.M.; Wallace, S.S.; Doublié, S. Structural characterization of Clostridium acetobutylicum 8-oxoguanine DNA glycosylase in its apo form and in complex with 8-oxodeoxyguanosine. J. Mol. Biol 2009, 387, 669–679. [Google Scholar]
- Bjørås, M.; Seeberg, E.; Luna, L.; Pearl, L.H.; Barrett, T.E. Reciprocal “flipping” underlies substrate recognition and catalytic activation by the human 8-oxo-guanine DNA glycosylase. J. Mol. Biol 2002, 317, 171–177. [Google Scholar]
- Faucher, F.; Duclos, S.; Bandaru, V.; Wallace, S.S.; Doublié, S. Crystal structures of two archaeal 8-oxoguanine DNA glycosylases provide structural insight into guanine/8-oxoguanine distinction. Structure 2009, 17, 703–712. [Google Scholar]
- Faucher, F.; Wallace, S.S.; Doublié, S. The C-terminal lysine of Ogg2 DNA glycosylases is a major molecular determinant for guanine/8-oxoguanine distinction. J. Mol. Biol 2010, 397, 46–56. [Google Scholar]
- Bruner, S.D.; Norman, D.P.; Verdine, G.L. Structural basis for recognition and repair of the endogenous mutagen 8-oxoguanine in DNA. Nature 2000, 403, 859–866. [Google Scholar]
- Lingaraju, G.M.; Sartori, A.A.; Kostrewa, D.; Prota, A.E.; Jiricny, J.; Winkler, F.K. A DNA glycosylase from Pyrobaculum aerophilum with an 8-oxoguanine binding mode and a noncanonical helix-hairpin-helix structure. Structure 2005, 13, 87–98. [Google Scholar]
- Labahn, J.; Schärer, O.D.; Long, A.; Ezaz-Nikpay, K.; Verdine, G.L.; Ellenberger, T.E. Structural basis for the excision repair of alkylation-damaged DNA. Cell 1996, 86, 321–329. [Google Scholar]
- Dantzer, F.; Luna, L.; Bjørås, M.; Seeberg, E. Human OGG1 undergoes serine phosphorylation and associates with the nuclear matrix and mitotic chromatin in vivo. Nucleic Acids Res 2002, 30, 2349–2357. [Google Scholar]
- Nishioka, K.; Ohtsubo, T.; Oda, H.; Fujiwara, T.; Kang, D.; Sugimachi, K.; Nakabeppu, Y. Expression and differential intracellular localization of two major forms of human 8-oxoguanine DNA glycosylase encoded by alternatively spliced OGG1 mRNAs. Mol. Biol. Cell 1999, 10, 1637–1652. [Google Scholar]
- Singh, K.K.; Sigala, B.; Sikder, H.A.; Schwimmer, C. Inactivation of Saccharomyces cerevisiae OGG1 DNA repair gene leads to an increased frequency of mitochondrial mutants. Nucleic Acids Res 2001, 29, 1381–1388. [Google Scholar]
- The Human Protein Reference Database. Available online: http://www.hprd.org/index_html accessed on 1 May, 2012.
- Harrison, S.C. A structural taxonomy of DNA-binding domains. Nature 1991, 353, 715–719. [Google Scholar]
- Thayer, M.M.; Ahern, H.; Xing, D.; Cunningham, R.P.; Tainer, J.A. Novel DNA binding motifs in the DNA repair enzyme endonuclease III crystal structure. EMBO J 1995, 14, 4108–4120. [Google Scholar]
- Nash, H.M.; Bruner, S.D.; Schärer, O.D.; Kawate, T.; Addona, T.A.; Spooner, E.; Lane, W.S.; Verdine, G.L. Cloning of a yeast 8-oxoguanine DNA glycosylase reveals the existence of a base-excision DNA-repair protein superfamily. Curr. Biol 1996, 6, 968–980. [Google Scholar]
- Faucher, F.; Wallace, S.S.; Doublié, S. Structural basis for the lack of opposite base specificity of Clostridium acetobutylicum 8-oxoguanine DNA glycosylase. DNA Repair (Amst) 2009, 8, 1283–1289. [Google Scholar]
- Banerjee, A.; Yang, W.; Karplus, M.; Verdine, G.L. Structure of a repair enzyme interrogating undamaged DNA elucidates recognition of damaged DNA. Nature 2005, 434, 612–618. [Google Scholar]
- Radom, C.T.; Banerjee, A.; Verdine, G.L. Structural characterization of human 8-oxoguanine DNA glycosylase variants bearing active site mutations. J. Biol. Chem 2007, 282, 9182–9194. [Google Scholar]
- Asagoshi, K.; Yamada, T.; Terato, H.; Ohyama, Y.; Ide, H. Enzymatic properties of Escherichia coli and human 7,8-dihydro-8-oxoguanine DNA glycosylases. Nucleic Acids Symp. Ser 2000, 11–12. [Google Scholar]
- Asagoshi, K.; Yamada, T.; Terato, H.; Ohyama, Y.; Monden, Y.; Arai, T.; Nishimura, S.; Aburatani, H.; Lindahl, T.; Ide, H. Distinct repair activities of human 7,8-dihydro-8-oxoguanine DNA glycosylase and formamidopyrimidine DNA glycosylase for formamidopyrimidine and 7, 8-dihydro-8-oxoguanine. J. Biol. Chem 2000, 275, 4956–4964. [Google Scholar]
- David, S.S.; O’Shea, V.L.; Kundu, S. Base-excision repair of oxidative DNA damage. Nature 2007, 447, 941–950. [Google Scholar]
- Hamm, M.L.; Gill, T.J.; Nicolson, S.C.; Summers, M.R. Substrate specificity of Fpg (MutM) and hOGG1, two repair glycosylases. J. Am. Chem. Soc 2007, 129, 7724–7725. [Google Scholar]
- Lingaraju, G.M.; Prota, A.E.; Winkler, F.K. Mutational studies of Pa-AGOG DNA glycosylase from the hyperthermophilic crenarchaeon Pyrobaculum aerophilum. DNA Repair (Amst.) 2009, 8, 857–864. [Google Scholar]
- Dalhus, B.; Forsbring, M.; Helle, I.H.; Vik, E.S.; Forstrom, R.J.; Backe, P.H.; Alseth, I.; Bjørås, M. Separation-of-function mutants unravel the dual-reaction mode of human 8-oxoguanine DNA glycosylase. Structure 2011, 19, 117–127. [Google Scholar]
- Crenshaw, C.M.; Nam, K.; Oo, K.; Kutchukian, P.S.; Bowman, B.R.; Karplus, M.; Verdine, G.L. Enforced presentation of an extrahelical guanine to the lesion-recognition pocket of the human 8-oxoguanine DNA glycosylase, hOGG1. J. Biol. Chem 2012, (in press). [Google Scholar]
- Gogos, A.; Clarke, N.D. Characterization of an 8-oxoguanine DNA glycosylase from Methanococcus jannaschii. J. Biol. Chem 1999, 274, 30447–30450. [Google Scholar]
- McCann, J.A.; Berti, P.J. Transition-state analysis of the DNA repair enzyme MutY. J. Am. Chem. Soc 2008, 130, 5789–5797. [Google Scholar]
- Werner, R.M.; Stivers, J.T. Kinetic isotope effect studies of the reaction catalyzed by uracil DNA glycosylase: Evidence for an oxocarbenium ion-uracil anion intermediate. Biochemistry 2000, 39, 14054–14064. [Google Scholar]
- Lau, A.Y.; Schärer, O.D.; Samson, L.; Verdine, G.L.; Ellenberger, T. Crystal structure of a human alkylbase-DNA repair enzyme complexed to DNA: Mechanisms for nucleotide flipping and base excision. Cell 1998, 95, 249–258. [Google Scholar]
- Mol, C.D.; Arvai, A.S.; Slupphaug, G.; Kavli, B.; Alseth, I.; Krokan, H.E.; Tainer, J.A. Crystal structure and mutational analysis of human uracil-DNA glycosylase: Structural basis for specificity and catalysis. Cell 1995, 80, 869–878. [Google Scholar]
- Norman, D.P.; Chung, S.J.; Verdine, G.L. Structural and biochemical exploration of a critical amino acid in human 8-oxoguanine glycosylase. Biochemistry 2003, 42, 1564–1572. [Google Scholar]
- Nash, H.M.; Lu, R.; Lane, W.S.; Verdine, G.L. The critical active-site amine of the human 8-oxoguanine DNA glycosylase, hOgg1: Direct identification, ablation and chemical reconstitution. Chem. Biol 1997, 4, 693–702. [Google Scholar]
- Norman, D.P.; Bruner, S.D.; Verdine, G.L. Coupling of substrate recognition and catalysis by a human base-excision DNA repair protein. J. Am. Chem. Soc 2001, 123, 359–360. [Google Scholar]
- Morland, I.; Luna, L.; Gustad, E.; Seeberg, E.; Bjørås, M. Product inhibition and magnesium modulate the dual reaction mode of hOgg1. DNA Repair (Amst.) 2005, 4, 381–387. [Google Scholar]
- Allinson, S.L.; Dianova, I.I.; Dianov, G.L. DNA polymerase beta is the major dRP lyase involved in repair of oxidative base lesions in DNA by mammalian cell extracts. EMBO J 2001, 20, 6919–6926. [Google Scholar]
- Hill, J.W.; Hazra, T.K.; Izumi, T.; Mitra, S. Stimulation of human 8-oxoguanine-DNA glycosylase by AP-endonuclease: Potential coordination of the initial steps in base excision repair. Nucleic Acids Res 2001, 29, 430–438. [Google Scholar]
- Vidal, A.E.; Hickson, I.D.; Boiteux, S.; Radicella, J.P. Mechanism of stimulation of the DNA glycosylase activity of hOGG1 by the major human AP endonuclease: Bypass of the AP lyase activity step. Nucleic Acids Res 2001, 29, 1285–1292. [Google Scholar]
- Audebert, M.; Radicella, J.P.; Dizdaroglu, M. Effect of single mutations in the OGG1 gene found in human tumors on the substrate specificity of the Ogg1 protein. Nucleic Acids Res 2000, 28, 2672–2678. [Google Scholar]



© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Faucher, F.; Doublié, S.; Jia, Z. 8-Oxoguanine DNA Glycosylases: One Lesion, Three Subfamilies. Int. J. Mol. Sci. 2012, 13, 6711-6729. https://doi.org/10.3390/ijms13066711
Faucher F, Doublié S, Jia Z. 8-Oxoguanine DNA Glycosylases: One Lesion, Three Subfamilies. International Journal of Molecular Sciences. 2012; 13(6):6711-6729. https://doi.org/10.3390/ijms13066711
Chicago/Turabian StyleFaucher, Frédérick, Sylvie Doublié, and Zongchao Jia. 2012. "8-Oxoguanine DNA Glycosylases: One Lesion, Three Subfamilies" International Journal of Molecular Sciences 13, no. 6: 6711-6729. https://doi.org/10.3390/ijms13066711