Investigation of Antigen-Antibody Interactions of Sulfonamides with a Monoclonal Antibody in a Fluorescence Polarization Immunoassay Using 3D-QSAR Models

Abstract
:1. Introduction
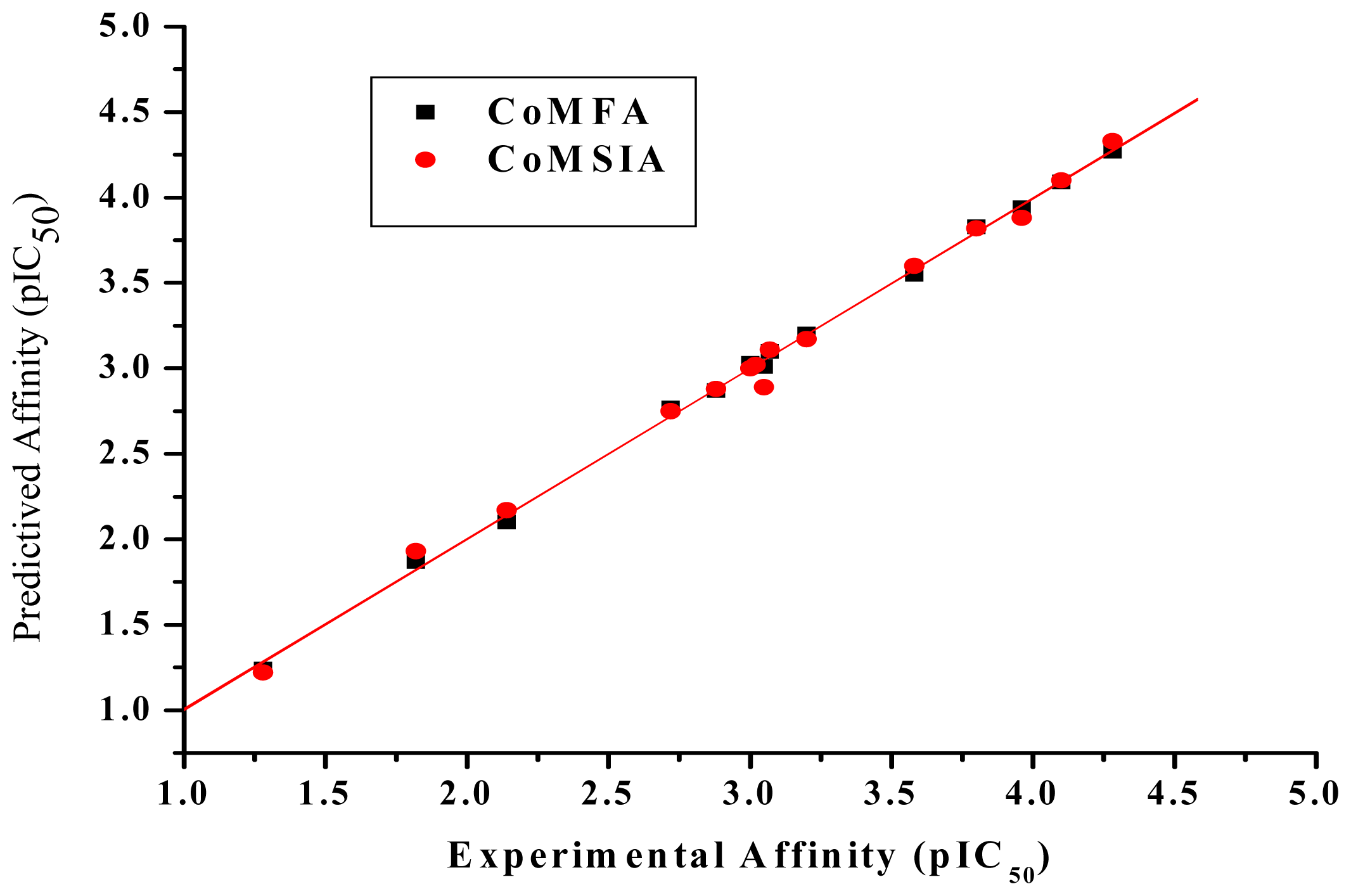
2. Results and Discussion
2.1. Determination of IC50 Values Using FPIA
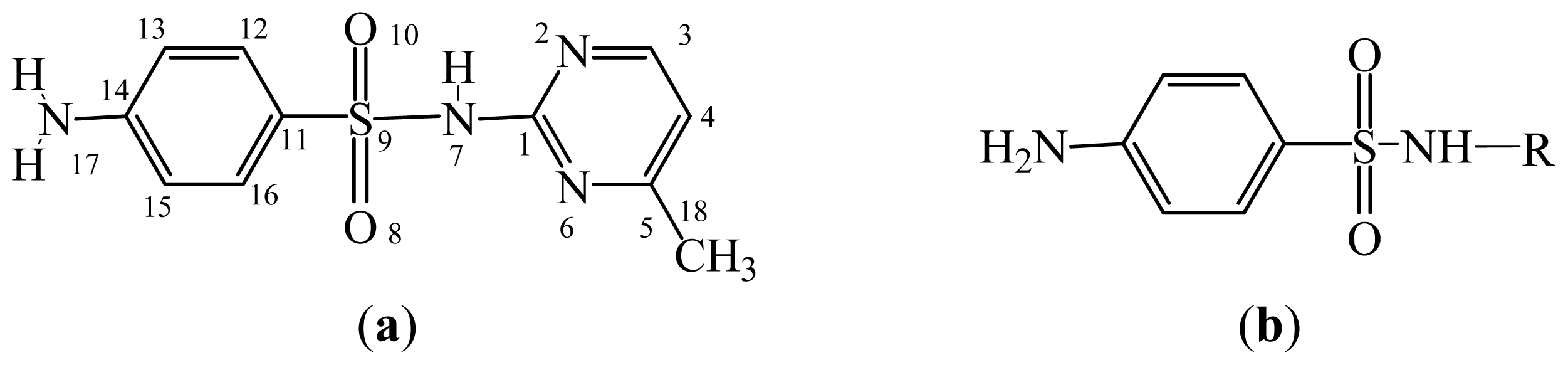
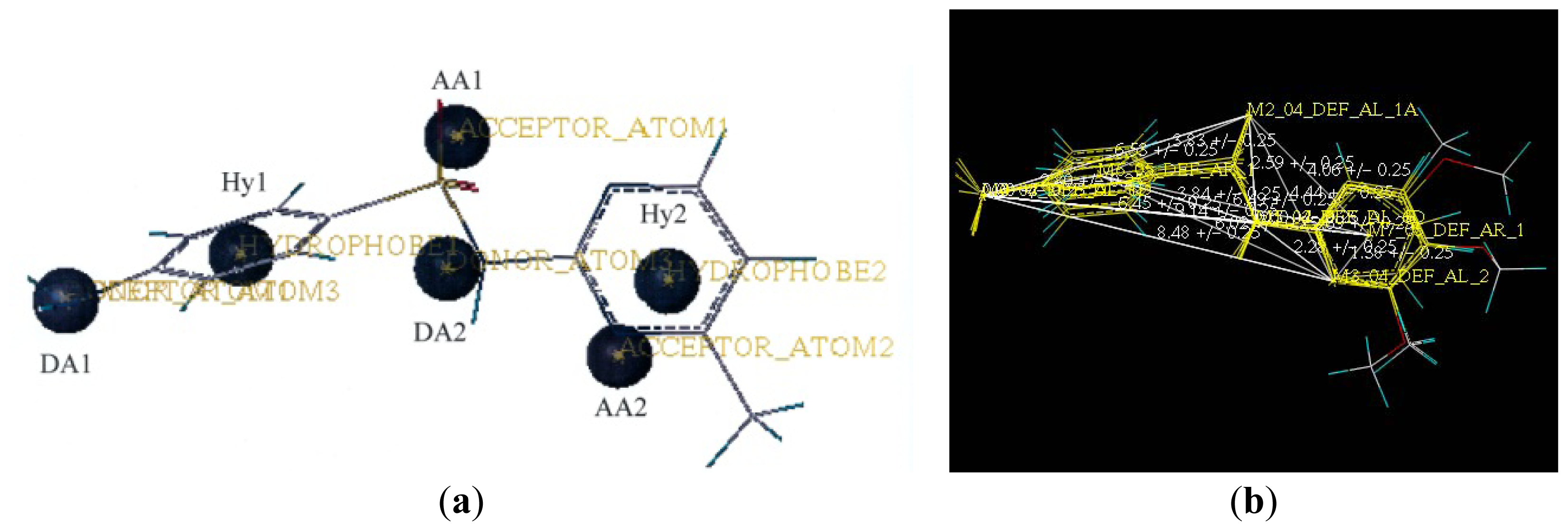
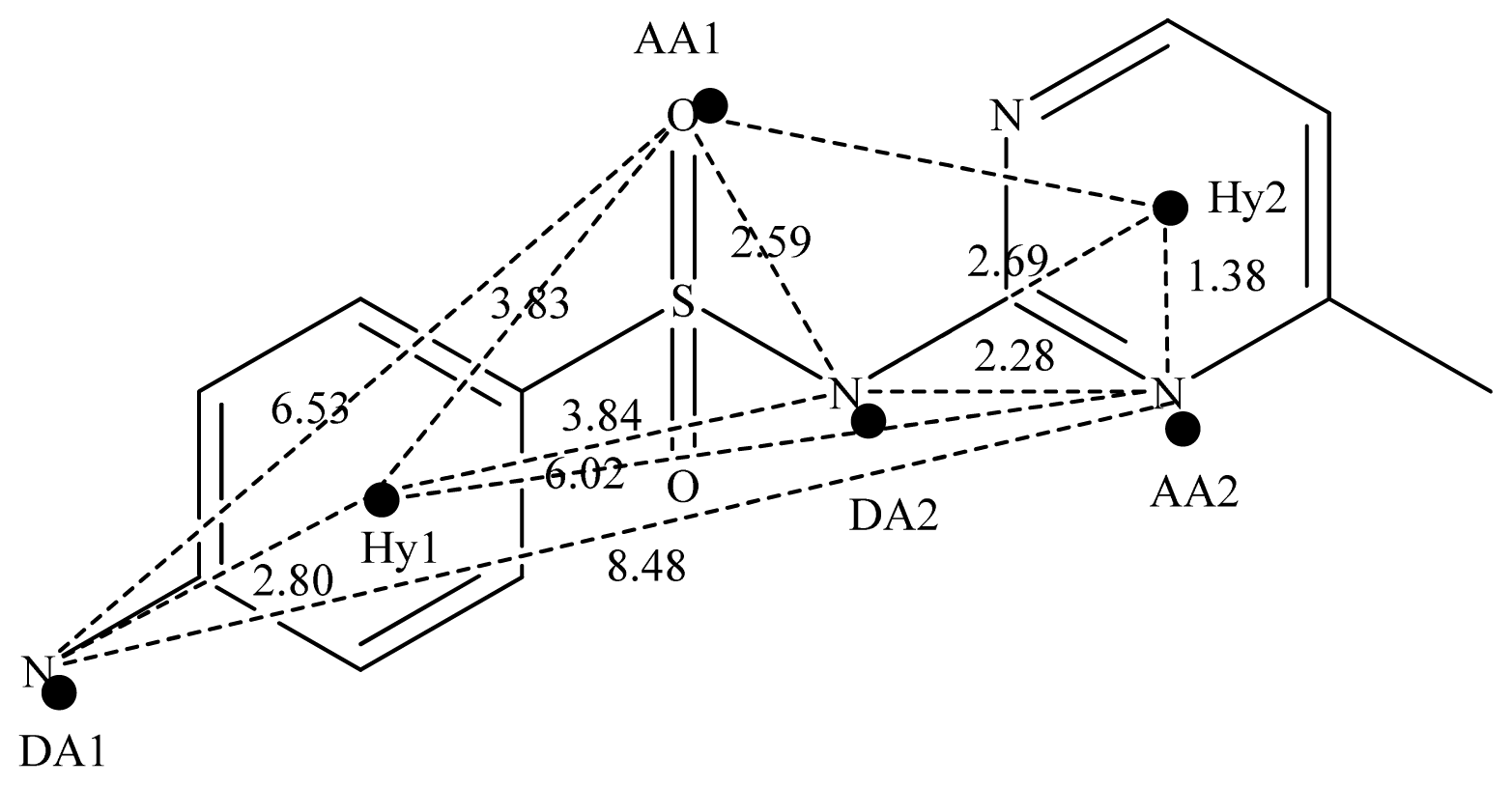
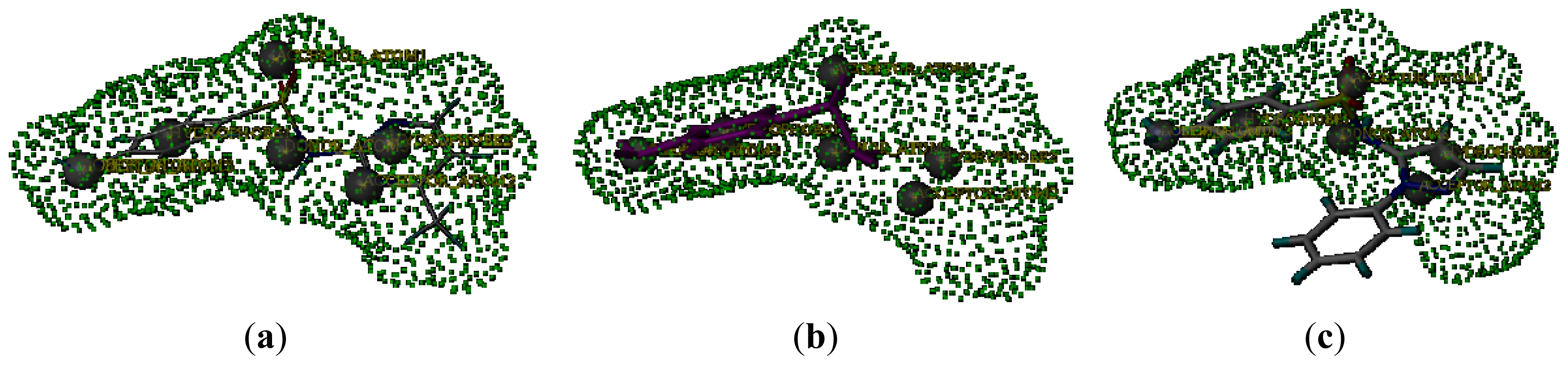
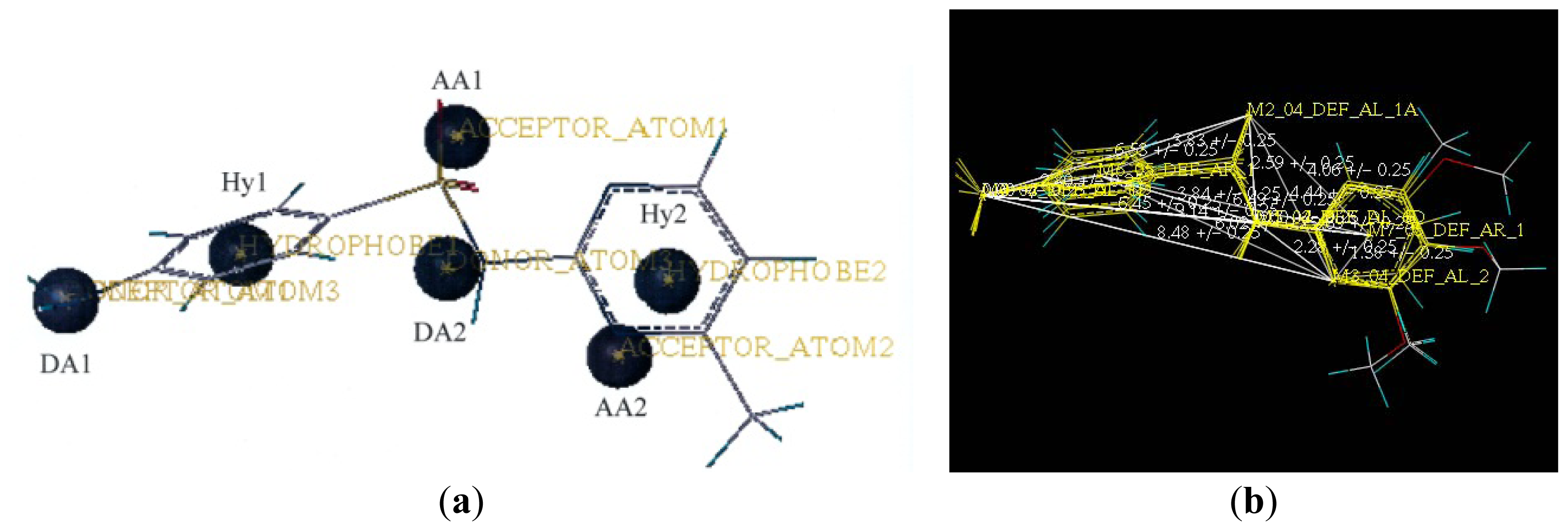
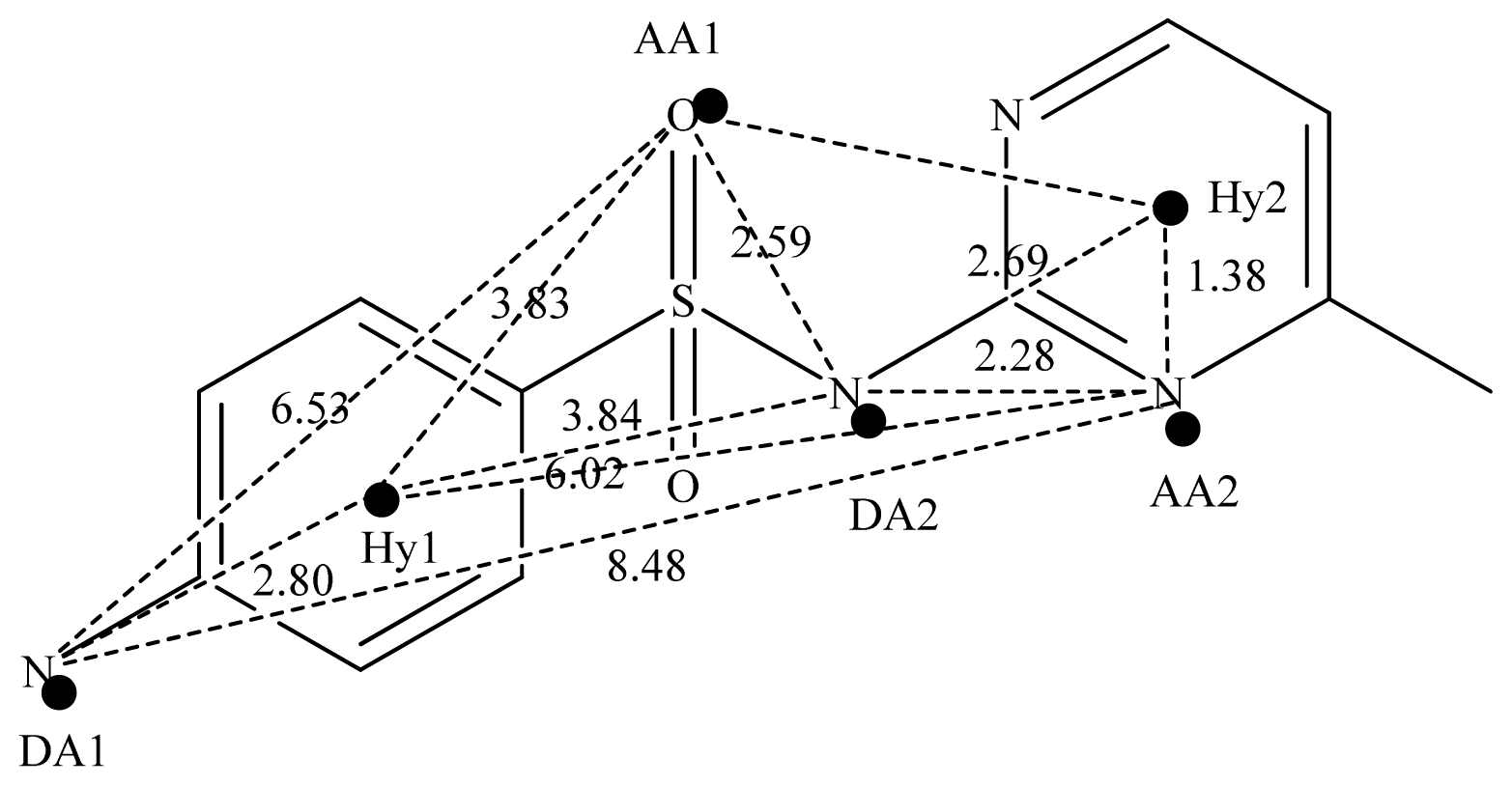
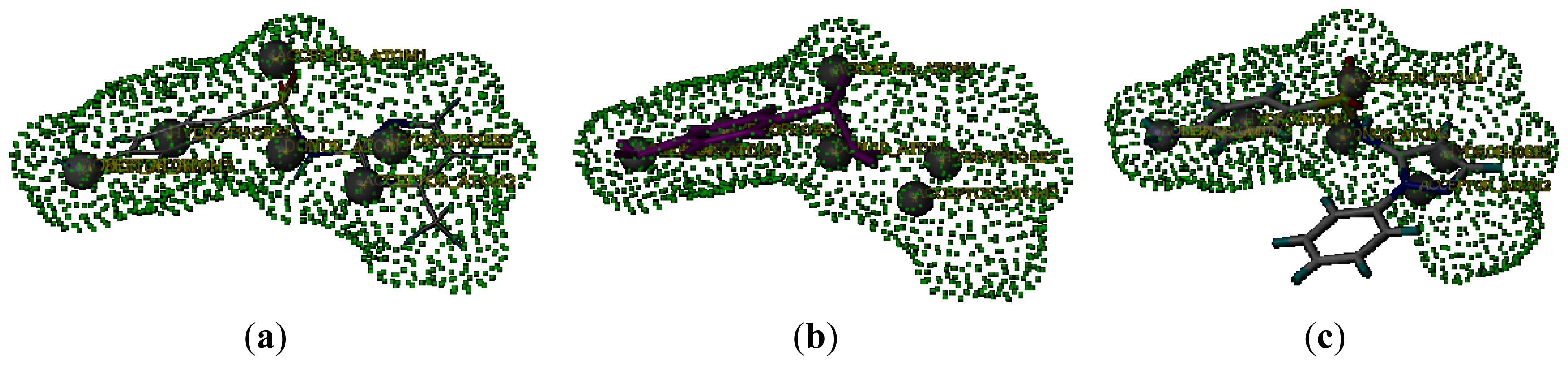
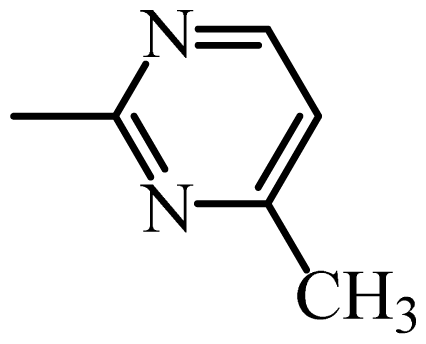
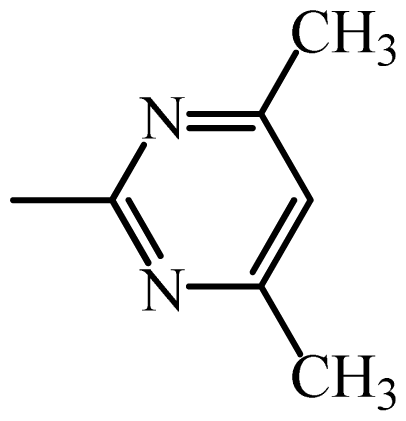
2.2. Pharmacophore and Alignment of the Sulfonamides
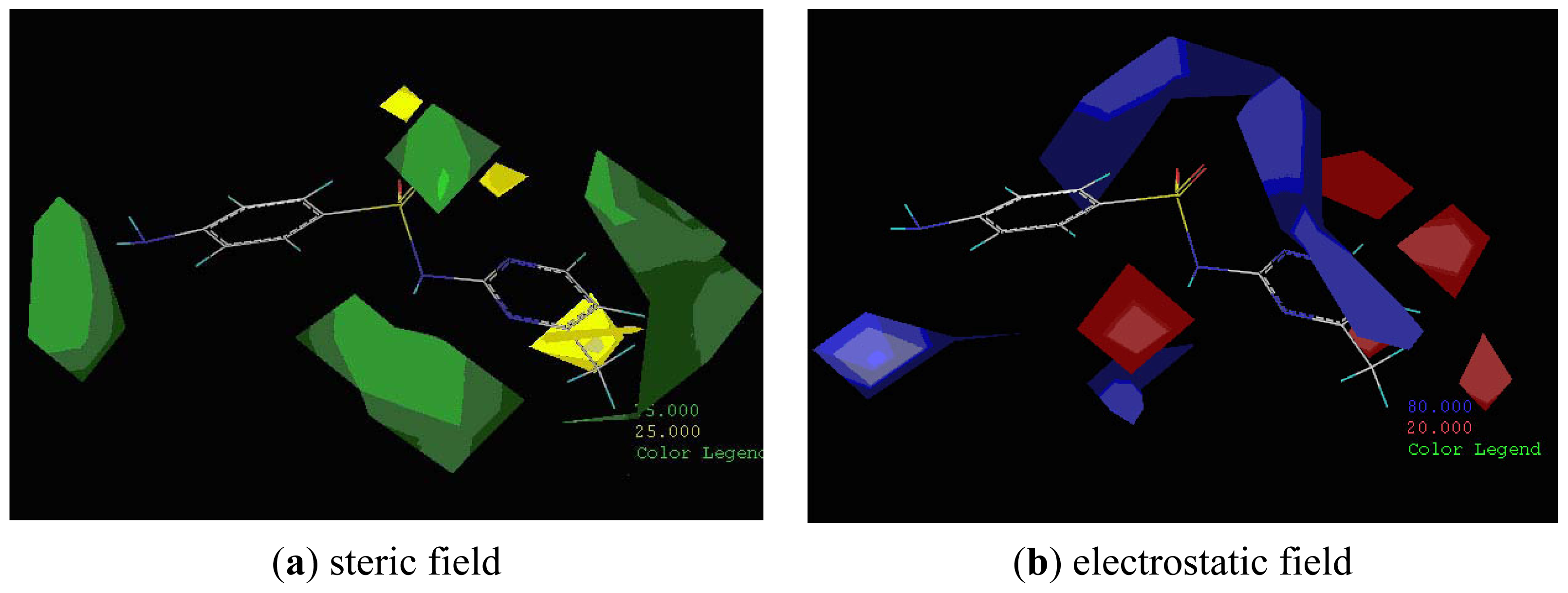
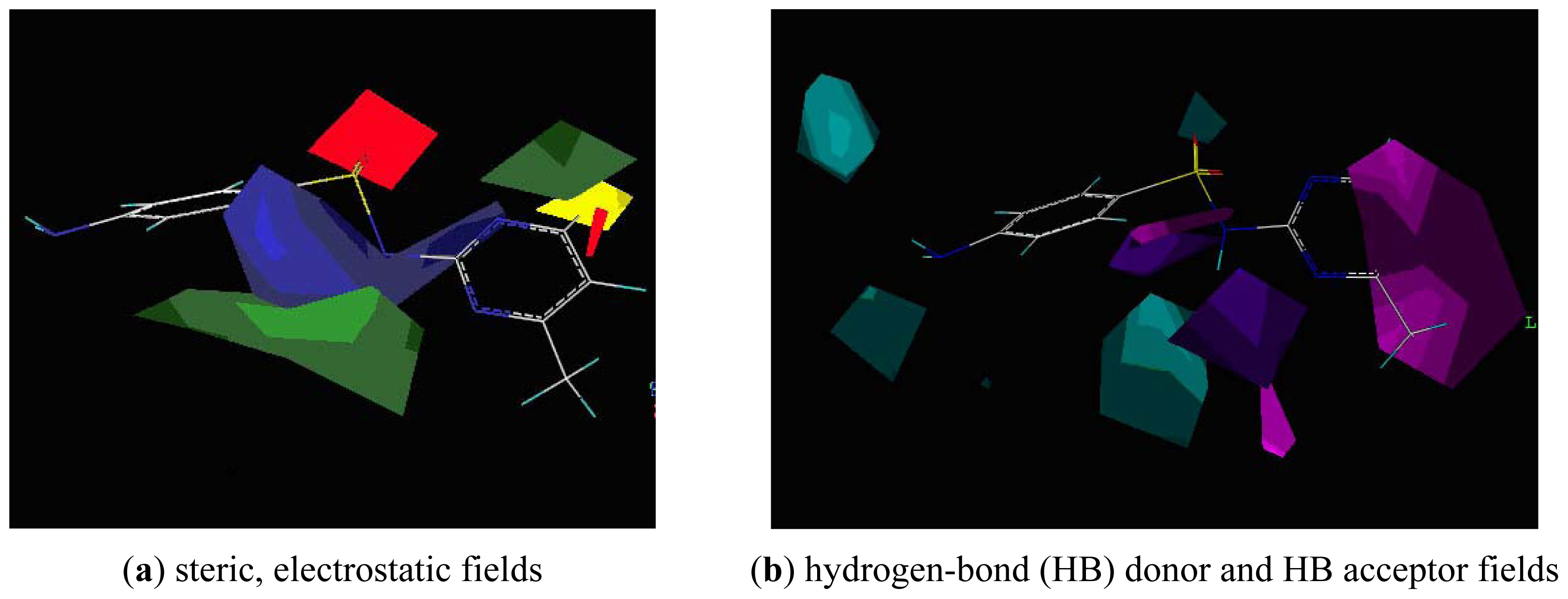
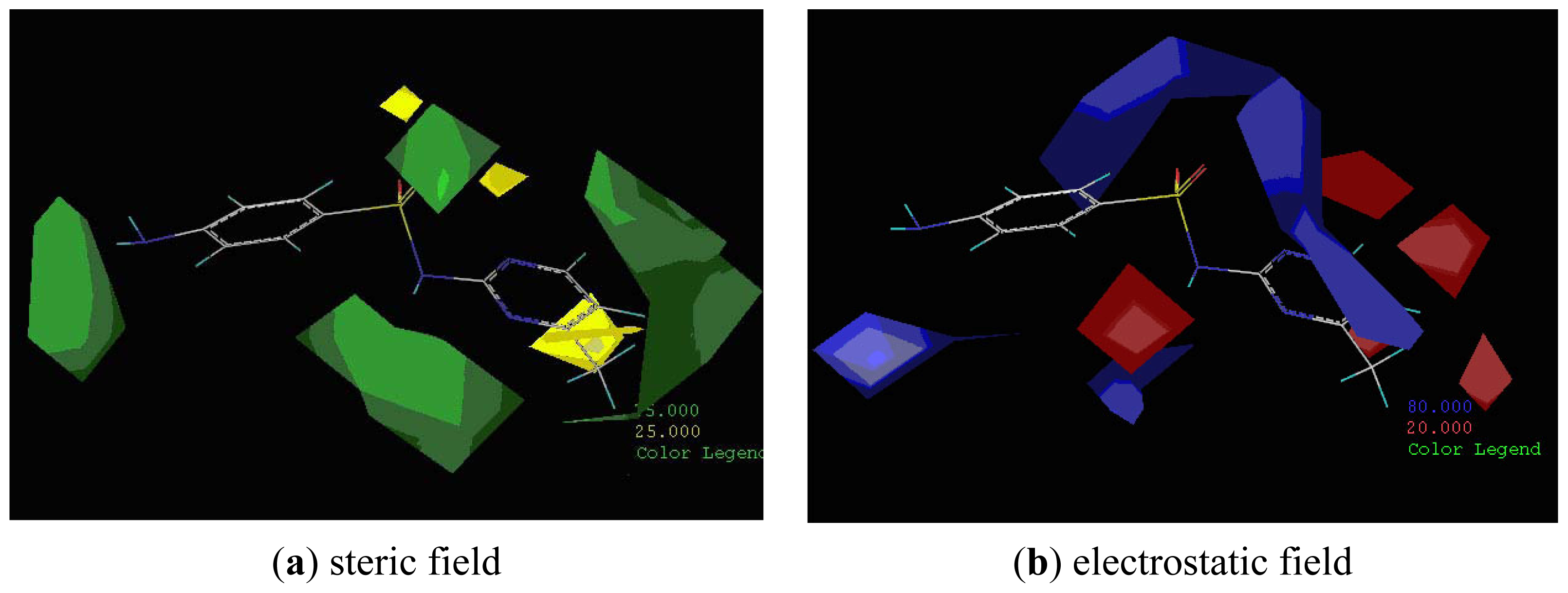
2.3. CoMFA Analysis
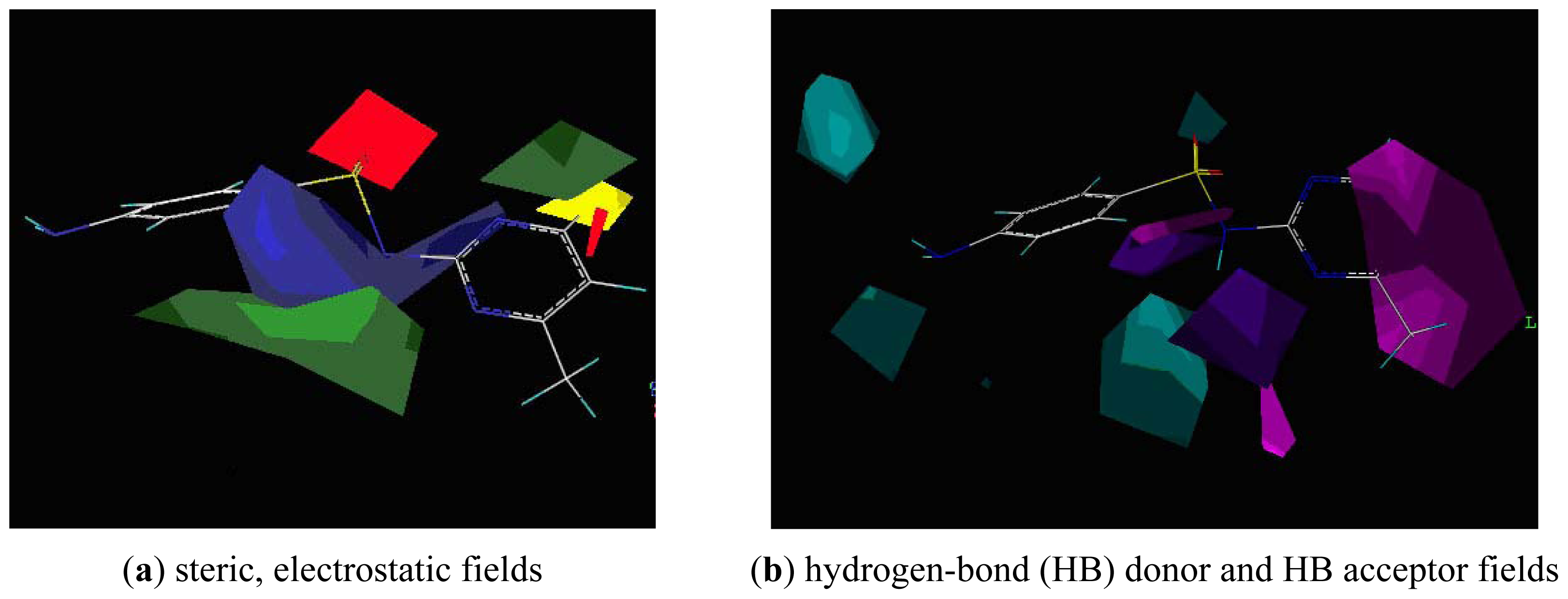
2.4. CoMSIA Analysis
2.5. Connolly Surface Analysis of Sulfonamides
3. Experimental Section
3.1. Tracer and Monoclonal Antibody
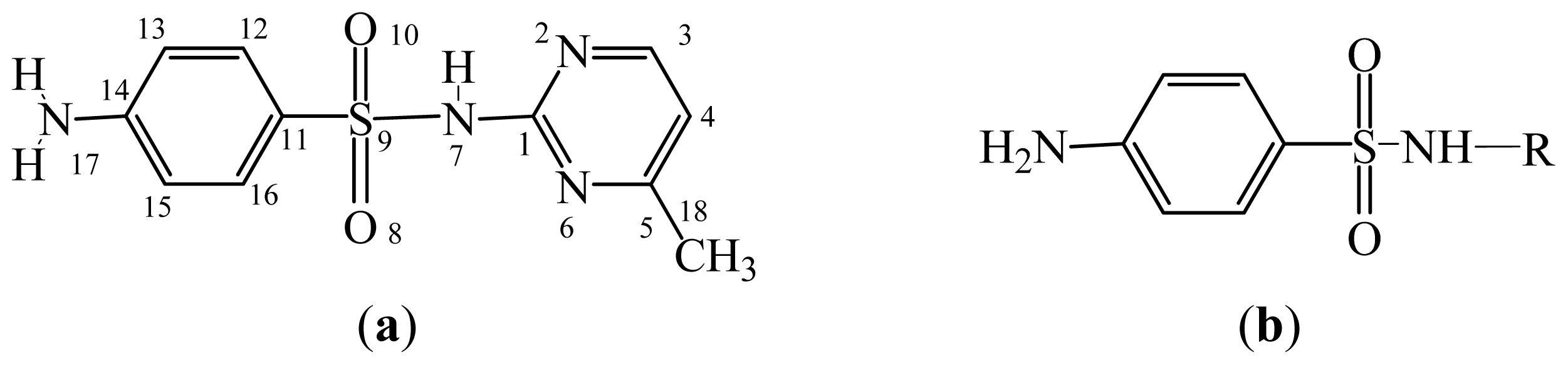
3.2. Sulfonamides
3.3. Fluorescence Polarization Immunoassay
3.4. Energy Minimization
3.5. Data Set Alignment Using DISCOtech
3.6. CoMFA Analysis
3.7. CoMSIA Analysis
3.8. Partial Least Squares (PLS) Regression Analysis
3.9. Connolly Surface
4. Conclusions
Acknowledgments
References
- Raviolo, M.A.; Rambla-Alegre, M.; Clausell-Tormos, J.; Capella-Peiró, M.-E.; Carda-Broch, S.; Esteve-Romero, J. Determination of sulfonamides in milk after precolumn derivatisation by micellar liquid chromatography. Anal. Chim. Acta 2007, 593, 152–156. [Google Scholar]
- Richter, D.; Dünnbier, U.; Massmann, G.; Pekdeger, A. Quantitative determination of three sulfonamides in environmental water samples using liquid chromatography coupled to electrospray tandem mass spectrometry. J. Chromatogr. A 2007, 1157, 115–121. [Google Scholar]
- Granelli, K.; Branzell, C. Rapid multi-residue screening of antibiotics in muscle and kidney by liquid chromatography-electrospray ionization-tandem mass spectrometry. Anal. Chim. Acta 2007, 586, 289–295. [Google Scholar]
- Pang, G.-F.; Cao, Y.-Z.; Zhang, J.-J.; Jia, G.-Q.; Fan, C.-L.; Li, X.-M.; Liu, Y.-M.; Li, Z.-Y.; Shi, Y.-Q. Simultaneous determination of 16 sulfonamides in honey by liquid chromatography/tandemmass spectrometry. J. AOAC Int 2005, 88, 1304–1311. [Google Scholar]
- Maudens, K.E.; Zhang, G.-F.; Lambert, W.E. Quantitative analysis of twelve sulfonamides in honey after acidic hydrolysis by high-performance liquid chromatography with post-column derivatization and fluorescence detection. J. Chromatogr. A 2004, 1047, 85–92. [Google Scholar]
- Crabbe, P.; van Peteghem, C. Rapid and sensitive screening of sulfamethazine in porcine urine with an enzyme-linked immunosorbent assay and a field-portable immunofiltration assay. J. Food Protect 2002, 65, 820–827. [Google Scholar]
- Sheth, H.B.; Sporns, P. Develpoment of a single ELISA for detection of sulfonamides. J. Agric. Food Chem 1991, 39, 1696–1700. [Google Scholar]
- Muldoon, M.T.; Holtzapple, C.K.; Deshpande, S.S.; Beier, R.C.; Stanker, L.H. Development of a monoclonal antibody-based cELISA for the analysis of sulfadimethoxine. 1. Development and characterization of monoclonal antibodies and molecular modeling studies of antibody recognition. J. Agric. Food Chem 2000, 48, 537–544. [Google Scholar]
- Cliquet, P.; Cox, E.; Haasnoot, W.; Schacht, E.; Goddeeris, B.M. Generation of group-specific antibodies against sulfonamides. J. Agric. Food Chem 2003, 51, 5835–5842. [Google Scholar]
- Zhang, H.; Duan, Z.; Wang, L.; Zhang, Y.; Wang, S. Hapten synthesis and development of polyclonal antibody-based multi-sulfonamide immunoassays. J. Agric. Food Chem 2006, 54, 4499–4505. [Google Scholar]
- Spinks, C.A.; Wyatt, G.M.; Lee, H.A.; Morgan, M.R.A. Molecular modeling of hapten structure and relevance to broad specificity immunoassay of sulfonamide antibiotics. Bioconjug. Chem 1999, 10, 583–588. [Google Scholar]
- Avila, C.M.; Romeiro, N.C.; Sperandio da Silva, G.M.; Sant’Anna, C.M.R.; Barreiro, E.J.; Fraga, C.A.M. Development of new CoMFA and CoMSIA 3D-QSAR models for anti-inflammatory phthalimide-containing TNFα modulators. Bioorg. Med. Chem 2006, 14, 6874–6885. [Google Scholar]
- Balogh, B.; Jójárt, B.; Wágner, Z.; Kovács, P.; Máté, G.; Gyires, K.; Zádori, Z.; Falkay, G.; Márki, Á.; Viskolcz, B.; Mátyus, P. 3D QSAR models for α2a-adrenoceptor agonists. Neurochem. Int 2007, 51, 268–276. [Google Scholar]
- Huang, H.; Pan, X.; Tan, N.; Zeng, G.; Ji, C. 3D-QSAR study of sulfonamide inhibitors of human carbonic anhydrase II. Eur. J. Med. Chem 2007, 42, 365–372. [Google Scholar]
- Bhongade, B.A.; Gouripur, V.V.; Gadad, A.K. 3D-QSAR CoMFA studies on trypsin-like serine protease inhibitors: A comparative selectivity analysis. Bioorg. Med. Chem 2005, 13, 2773–2782. [Google Scholar]
- Paula, S.; Tabet, M.R.; Keenan, S.M.; Welsh, W.J.; Ball, W.J., Jr. Three-dimensional structure–activity relationship modeling of cocaine binding to two monoclonal antibodies by comparative molecular field analysis. J. Mol. Biol 2003, 325, 515–530. [Google Scholar]
- Farr, C.D.; Tabet, M.R.; Ball, W.J., Jr; Fishwild, D.M.; Wang, X.; Nair, A.C.; Welsh, W.J. Three-dimensional quantitative structure–activity relationship analysis of ligand binding to human sequence antidigoxin monoclonal antibodies using comparative molecular field analysis. J. Med. Chem 2002, 45, 3257–3270. [Google Scholar]
- Paula, S.; Tabet, M.R.; Farr, C.D.; Norman, A.B.; Ball, W.J., Jr. Three-dimensional quantitative structure-activity relationship modeling of cocaine binding by a novel human monoclonal antibody. J. Med. Chem 2004, 47, 133–142. [Google Scholar]
- Zhang, S.; Wang, Z.; Nesterenko, I.S.; Eremin, S.A.; Shen, J. Fluorescence polarization immunoassay based on a monoclonal antibody for the detection of sulfamethazine in chicken muscle. Int. J. Food Sci. Technol 2007, 42, 36–44. [Google Scholar]
- Eremin, S.A.; Smith, D.S. Fluorescence polarization immunoassays for pesticides. Comb. Chem. High Throughput Screen 2003, 6, 257–266. [Google Scholar]
- Wang, Z.; Zhang, S.; Ding, S.; Eremin, S.A.; Shen, J. Simultaneous determination of sulphamerazine, sulphamethazine and sulphadiazine in honey and chicken muscle by a new monoclonal antibody-based fluorescence polarisation immunoassay. Food Addit. Contam 2008, 25, 574–582. [Google Scholar]
- Sugawara, Y.; Gee, S.J.; Sanborn, J.R.; Gilman, S.D.; Hammock, B.D. Development of a highly sensitive enzyme-linked immunosorbent assay based on polyclonal antibodies for the detection of polychlorinated dibenzo-p-dioxins. Anal. Chem 1998, 70, 1092–1099. [Google Scholar]
- Peoples, M.C.; Karnes, H.T. Microfluidic immunoaffinity separations for bioanalysis. J. Chromatogr. B 2008, 866, 14–25. [Google Scholar]
- Zou, X.-J.; Lai, L.-H.; Jin, G.-Y.; Zhang, Z.-X. Synthesis, fungicidal activity, and 3D-QSAR of pyridazinone-substituted 1,3,4-oxadiazoles and 1,3,4-thiadiazoles. J. Agric. Food Chem 2002, 50, 3757–3760. [Google Scholar]
- Li, Y.; Wang, Y.; Yang, L.; Zhang, S.; Liu, C. Structural determinants of flavones interacting with the C-terminal nucleotide-binding domain as P-glycoprotein Inhibitors. Internet Electron. J. Mol. Des 2006, 5, 1–12. [Google Scholar]
- Inglesby, M.K.; Zeronian, S.H.; Elder, T.J. Aggregation of direct dyes investigated by molecular modeling. Textil. Res. J 2002, 72, 231–239. [Google Scholar]
- Cramer, R.D., III; Patterson, D.E.; Bunce, J.D. Comparative molecular field analysis (CoMFA). 1. Effect of shape on binding of steroids to carrier proteins. J. Am. Chem. Soc 1988, 110, 5959–5967. [Google Scholar]
- Wang, Y.; Han, K.-L.; Yang, S.-L.; Yang, L. Structural determinants of steroids for cytochrome P450 3A4-mediated metabolism. J. Mol. Struct. (Theochem) 2004, 710, 215–221. [Google Scholar]
- Ashek, A.; Lee, C.; Park, H.; Cho, S.J. 3D QSAR studies of dioxins and dioxin-like compounds using CoMFA and CoMSIA. Chemosphere 2006, 65, 521–529. [Google Scholar]
- Xue, C.X.; Cui, S.Y.; Liu, M.C.; Hu, Z.D.; Fan, B.T. 3D QSAR studies on antimalarial alkoxylated and hydroxylated chalcones by CoMFA and CoMSIA. Eur. J. Med. Chem 2004, 39, 745–753. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drugs a | R | IC50 | Log10IC50 (exp.) | CoMFA | CoMSIA | ||
|---|---|---|---|---|---|---|---|
| Log10IC50 (pre.) | Residual | Log10IC50 (pre.) | Residual | ||||
| SMR | 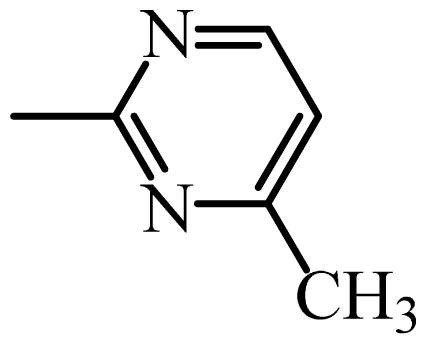 | 19 | 1.28 | 1.24 | 0.04 | 1.22 | 0.06 |
| SMZ | 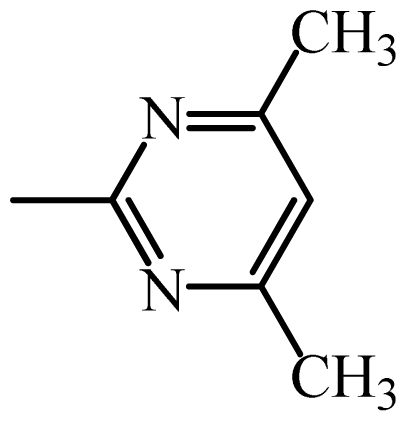 | 65.7 | 1.82 | 1.87 | −0.05 | 1.93 | −0.11 |
| SDZ |  | 137 | 2.14 | 2.10 | 0.04 | 2.17 | −0.03 |
| SDM | 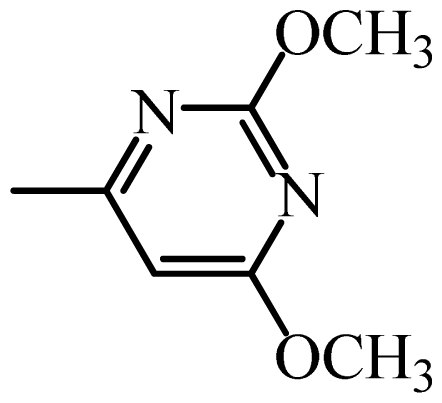 | 528 | 2.72 | 2.77 | −0.05 | 2.75 | −0.03 |
| SME |  | 760 | 2.88 | 2.87 | 0.01 | 2.88 | 0.00 |
| STZ | 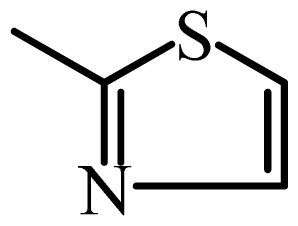 | 1000 | 3.00 | 3.03 | −0.03 | 3.00 | 0.00 |
| SMP |  | 1056 | 3.02 | 3.01 | 0.01 | 3.02 | 0.00 |
| SMO |  | 1118 | 3.05 | 3.01 | 0.04 | 2.89 | 0.16 |
| SPY | 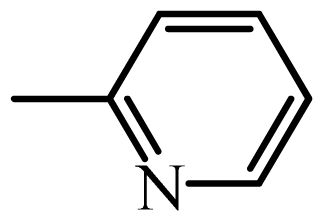 | 1189 | 3.07 | 3.10 | −0.03 | 3.11 | −0.04 |
| SQX |  | 1538 | 3.20 | 3.20 | 0.00 | 3.17 | 0.03 |
| SCP | 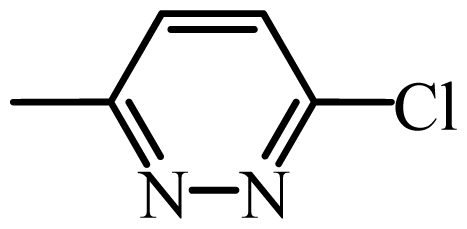 | 3800 | 3.58 | 3.55 | 0.03 | 3.60 | −0.02 |
| SMT | 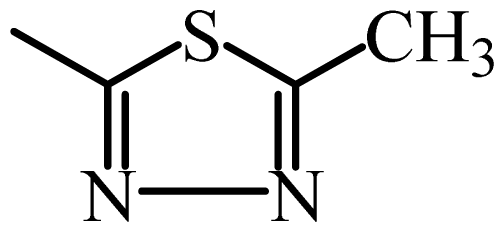 | 6333 | 3.80 | 3.83 | −0.03 | 3.82 | −0.02 |
| SMX | 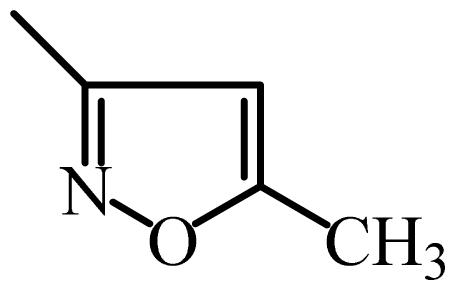 | 9048 | 3.96 | 3.94 | 0.02 | 3.88 | 0.08 |
| SMM | 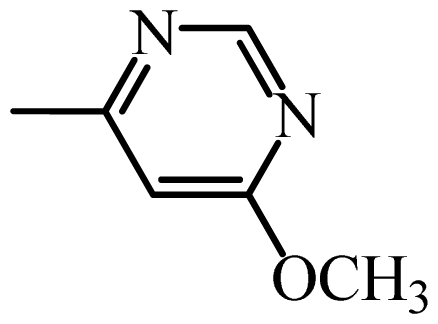 | 12,667 | 4.10 | 4.09 | 0.01 | 4.10 | 0.00 |
| SFX | 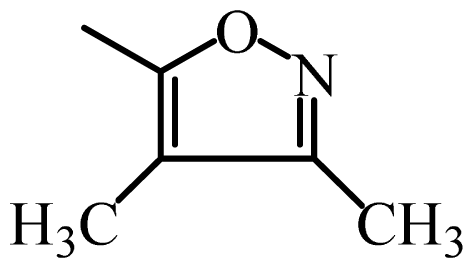 | 19,000 | 4.28 | 4.27 | 0.01 | 4.33 | −0.05 |
| SPA |  | >190,000 | - | - | - | - | - |
| SAM | H | >190,000 | - | - | - | - | - |
| No validation | ||||||
|---|---|---|---|---|---|---|
| Models | Delete compounds | Leave-one-out (LOO) q2 | Cross-validated q2 cv | Standard error of the estimate (SEE) | r2 | F value |
| M1 | - | 0.258 | 0.241 | - | - | - |
| M2 | SAM | 0.126 | 0.127 | - | - | - |
| M3 | SPA | 0.310 | 0.234 | - | - | - |
| M4 | SAM, SPA | 0.582 | 0.600 | 0.071 | 0.995 | 397.263 |
| Models | Steric and electrostatic | Hydrophobic | Donor and acceptor | Leaveone- out (LOO) q2 | Crossvalidated q2 cv | SEE | r2 | F value |
|---|---|---|---|---|---|---|---|---|
| N1 | √ | - | - | 0.558 | 0.678 | - | - | - |
| N2 | - | √ | - | −0.491 | −0.593 | - | - | - |
| N3 | - | - | √ | 0.407 | 0.431 | - | - | - |
| N4 | √ | √ | √ | 0.258 | 0.287 | - | - | - |
| N5 | √ | - | √ | 0.450 | 0.523 | 0.078 | 0.994 | 324.629 |
| N6 | √ | √ | - | 0.145 | 0.133 | - | - | - |
| N7 | - | √ | √ | 0.084 | 0.021 | - | - | - |
© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Wang, Z.; Kai, Z.; Beier, R.C.; Shen, J.; Yang, X. Investigation of Antigen-Antibody Interactions of Sulfonamides with a Monoclonal Antibody in a Fluorescence Polarization Immunoassay Using 3D-QSAR Models. Int. J. Mol. Sci. 2012, 13, 6334-6351. https://doi.org/10.3390/ijms13056334
Wang Z, Kai Z, Beier RC, Shen J, Yang X. Investigation of Antigen-Antibody Interactions of Sulfonamides with a Monoclonal Antibody in a Fluorescence Polarization Immunoassay Using 3D-QSAR Models. International Journal of Molecular Sciences. 2012; 13(5):6334-6351. https://doi.org/10.3390/ijms13056334
Chicago/Turabian StyleWang, Zhanhui, Zhenpeng Kai, Ross C. Beier, Jianzhong Shen, and Xinling Yang. 2012. "Investigation of Antigen-Antibody Interactions of Sulfonamides with a Monoclonal Antibody in a Fluorescence Polarization Immunoassay Using 3D-QSAR Models" International Journal of Molecular Sciences 13, no. 5: 6334-6351. https://doi.org/10.3390/ijms13056334