The Role of Protein Crystallography in Defining the Mechanisms of Biogenesis and Catalysis in Copper Amine Oxidase

Abstract
:
1. Introduction
2. Physiological Significance of CAOs
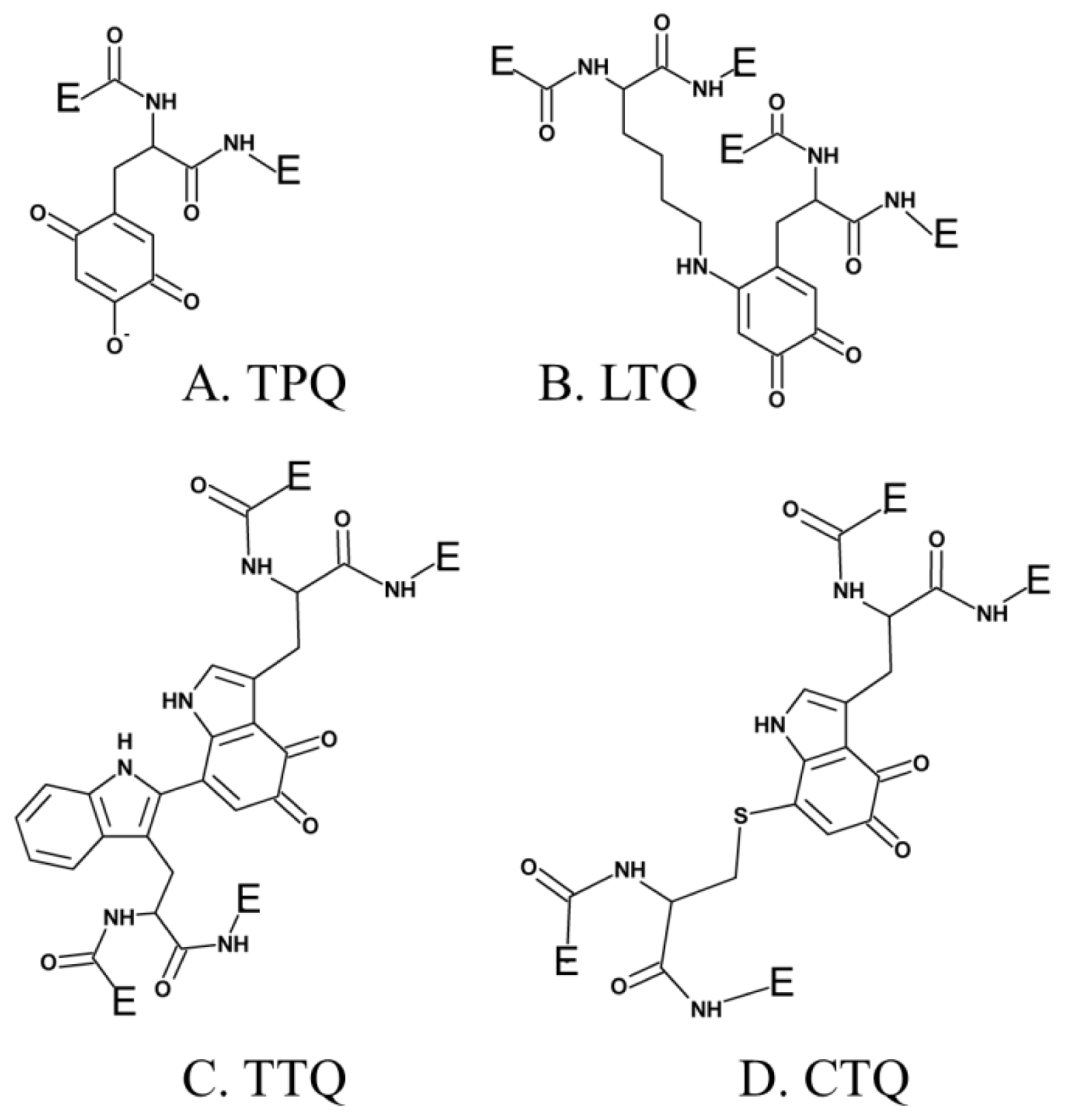
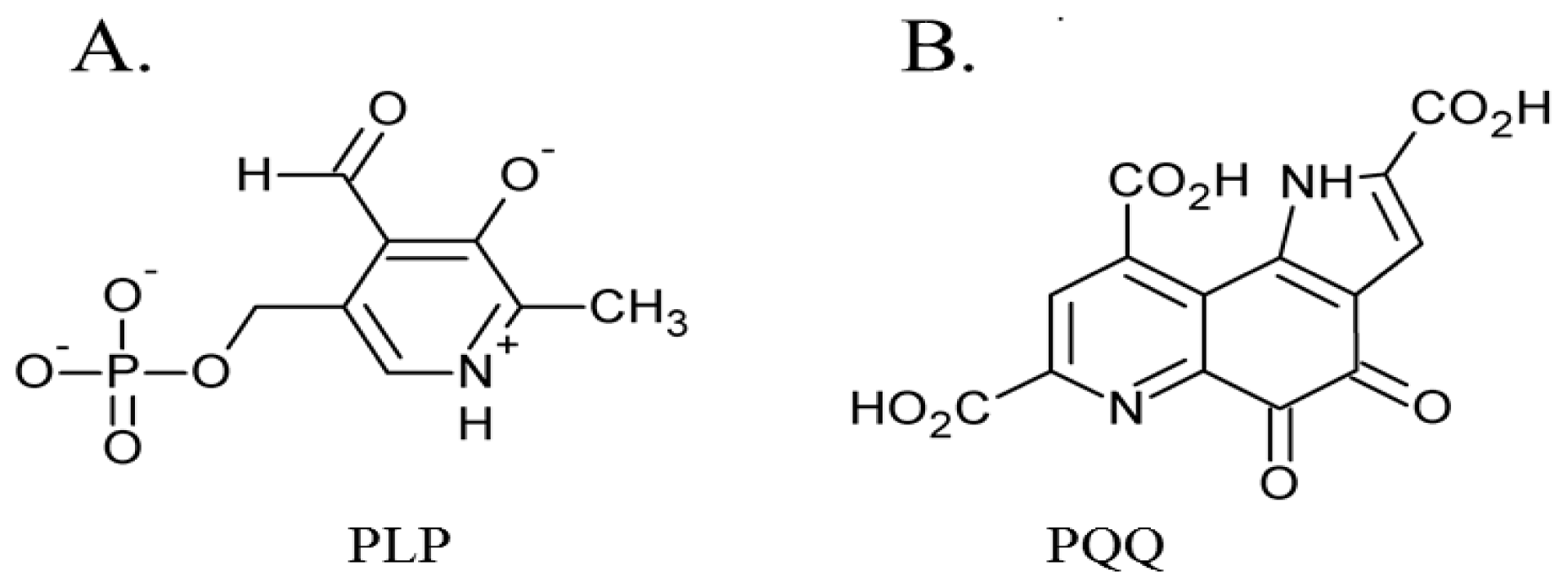
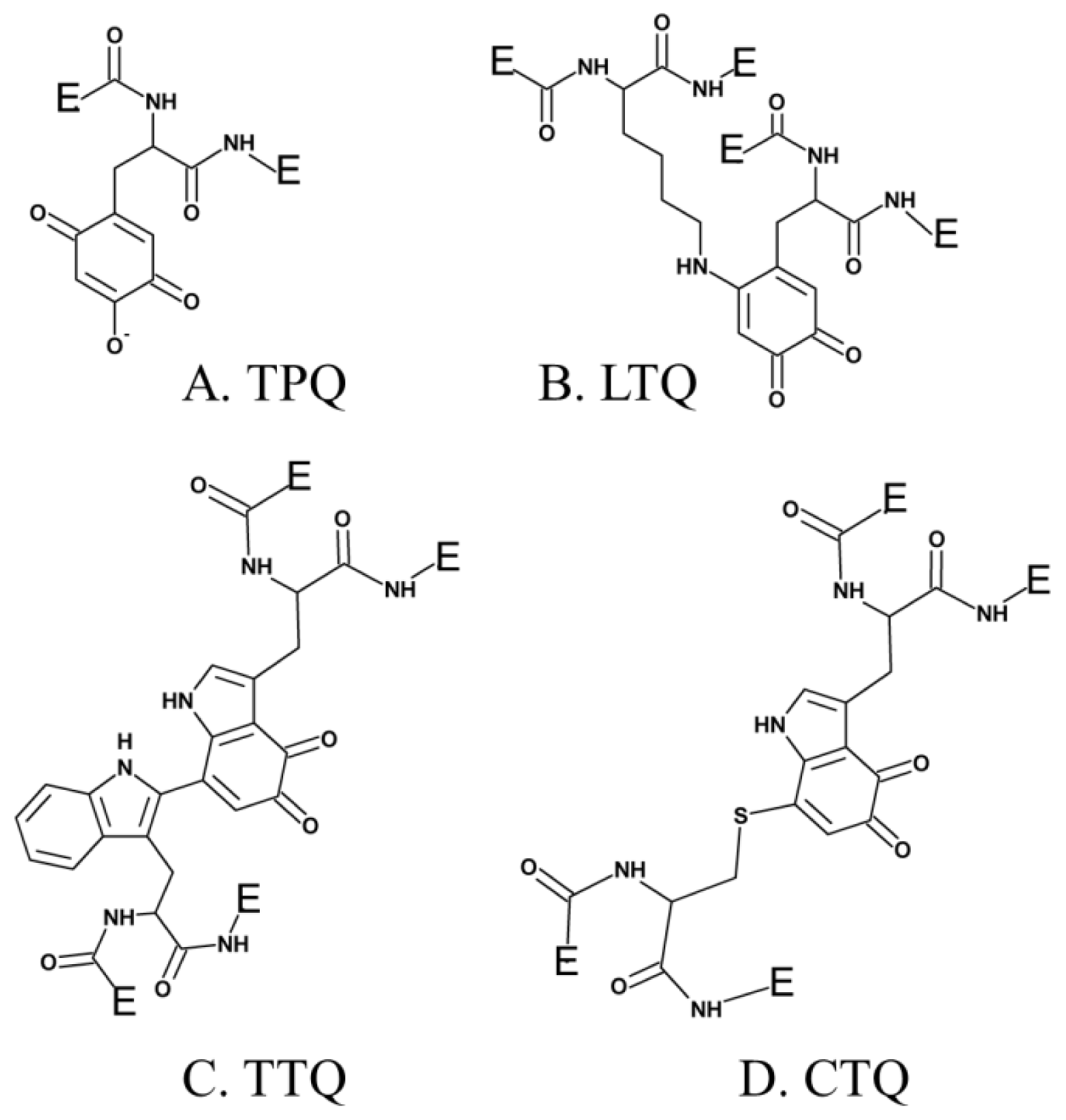
3. Post-Translationally Modified Amino Acid Cofactors
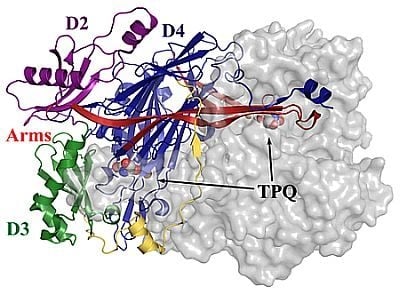
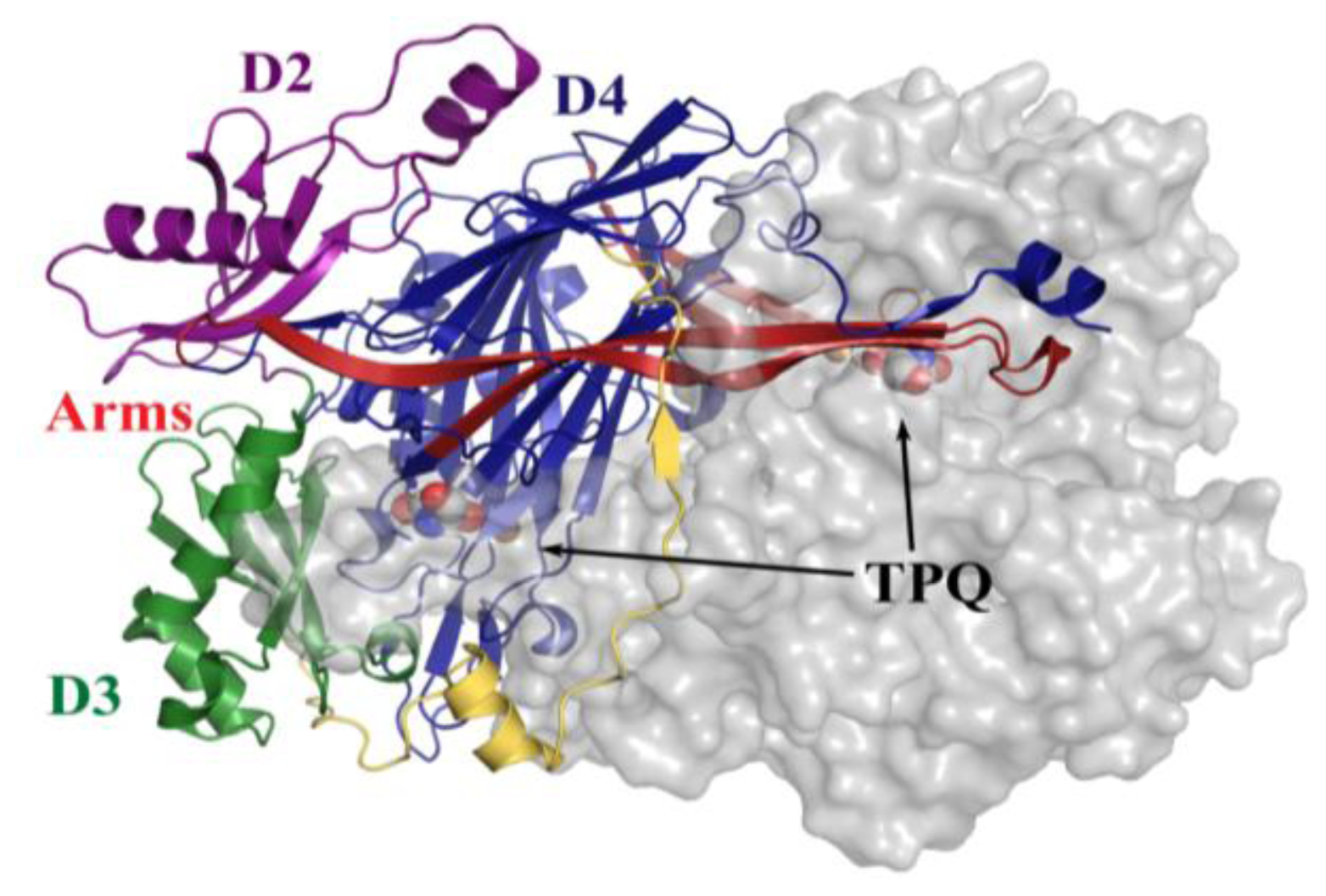
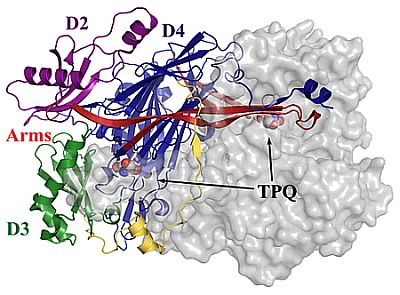
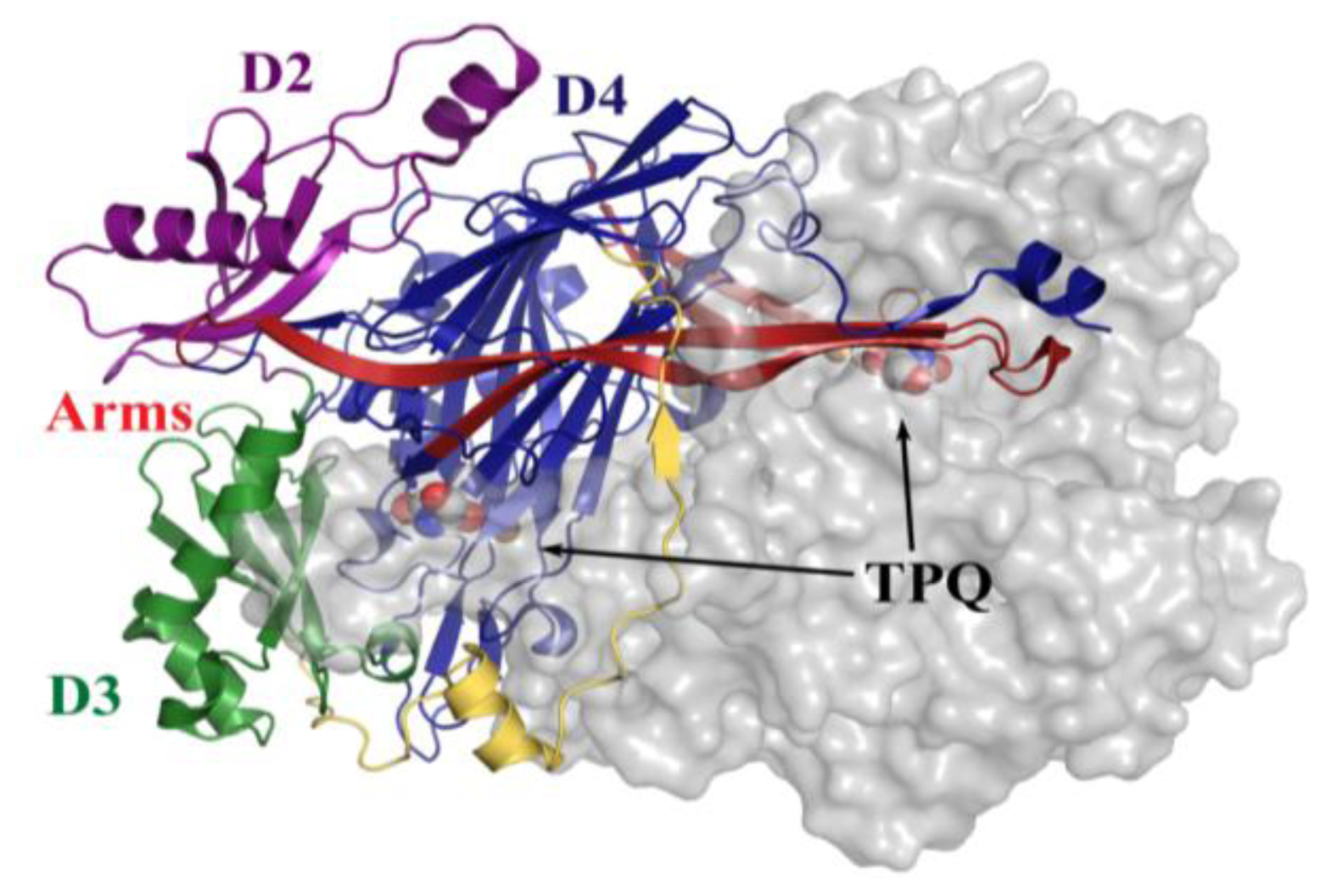
4. Overall CAO Fold
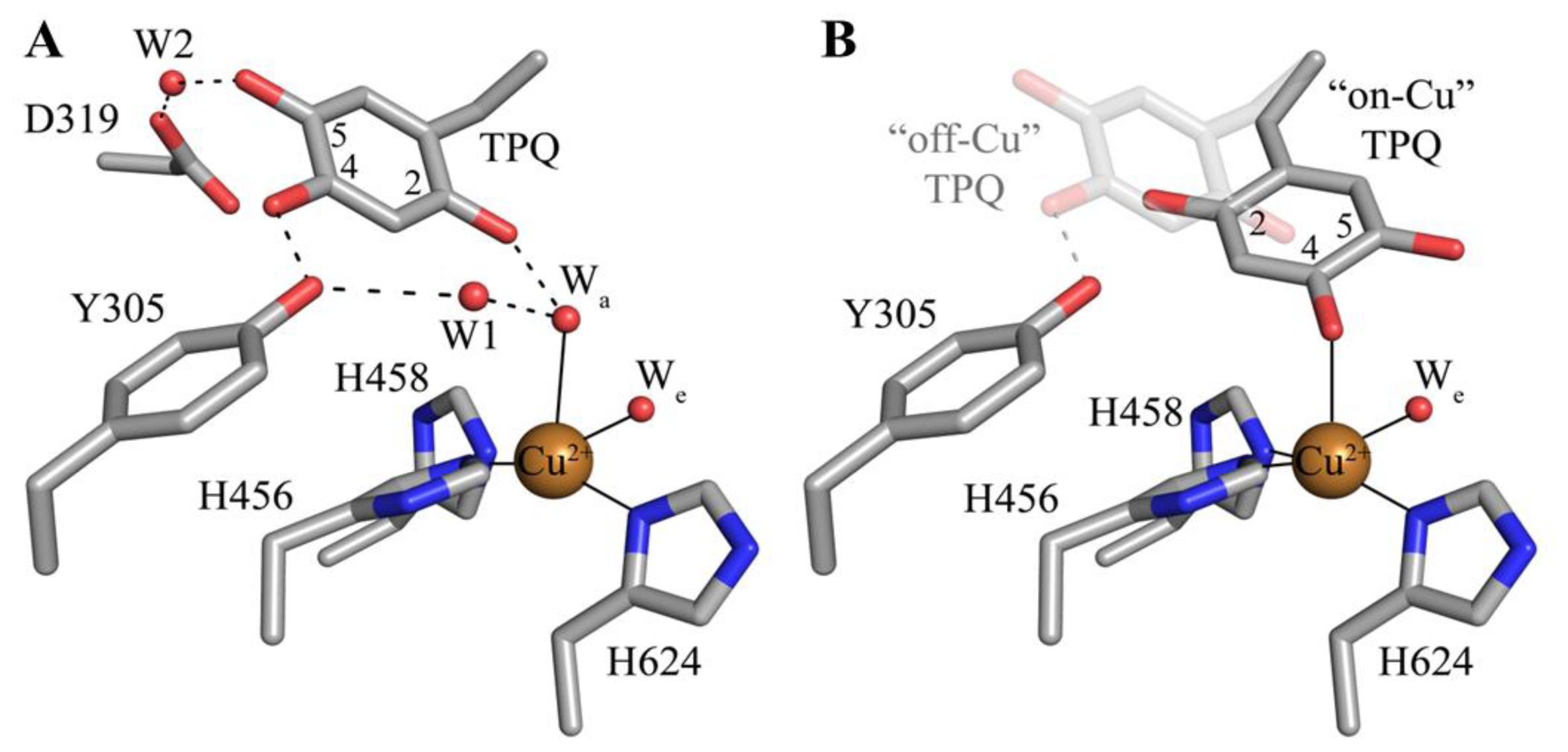
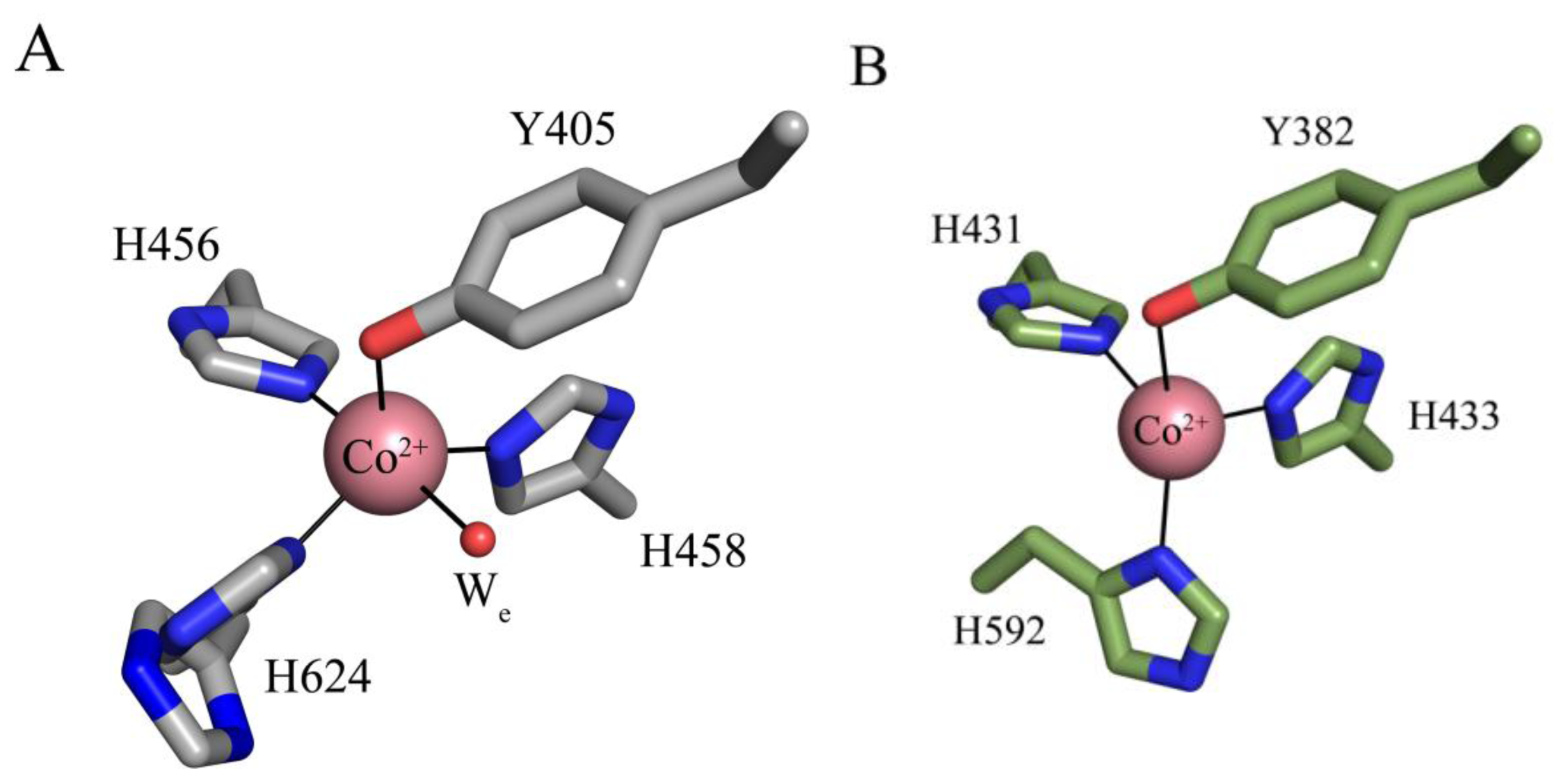
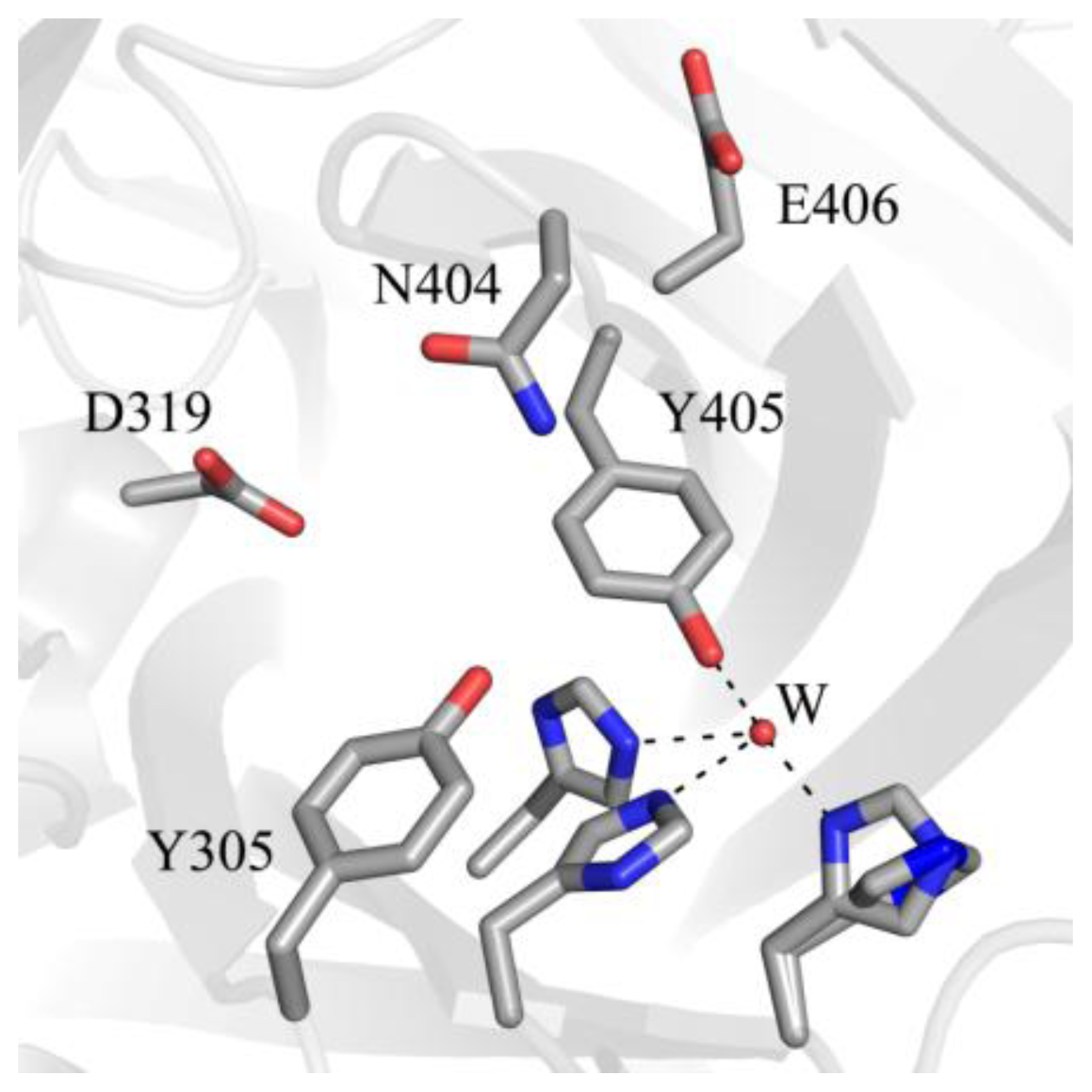
5. Structure of the CAO Active Site
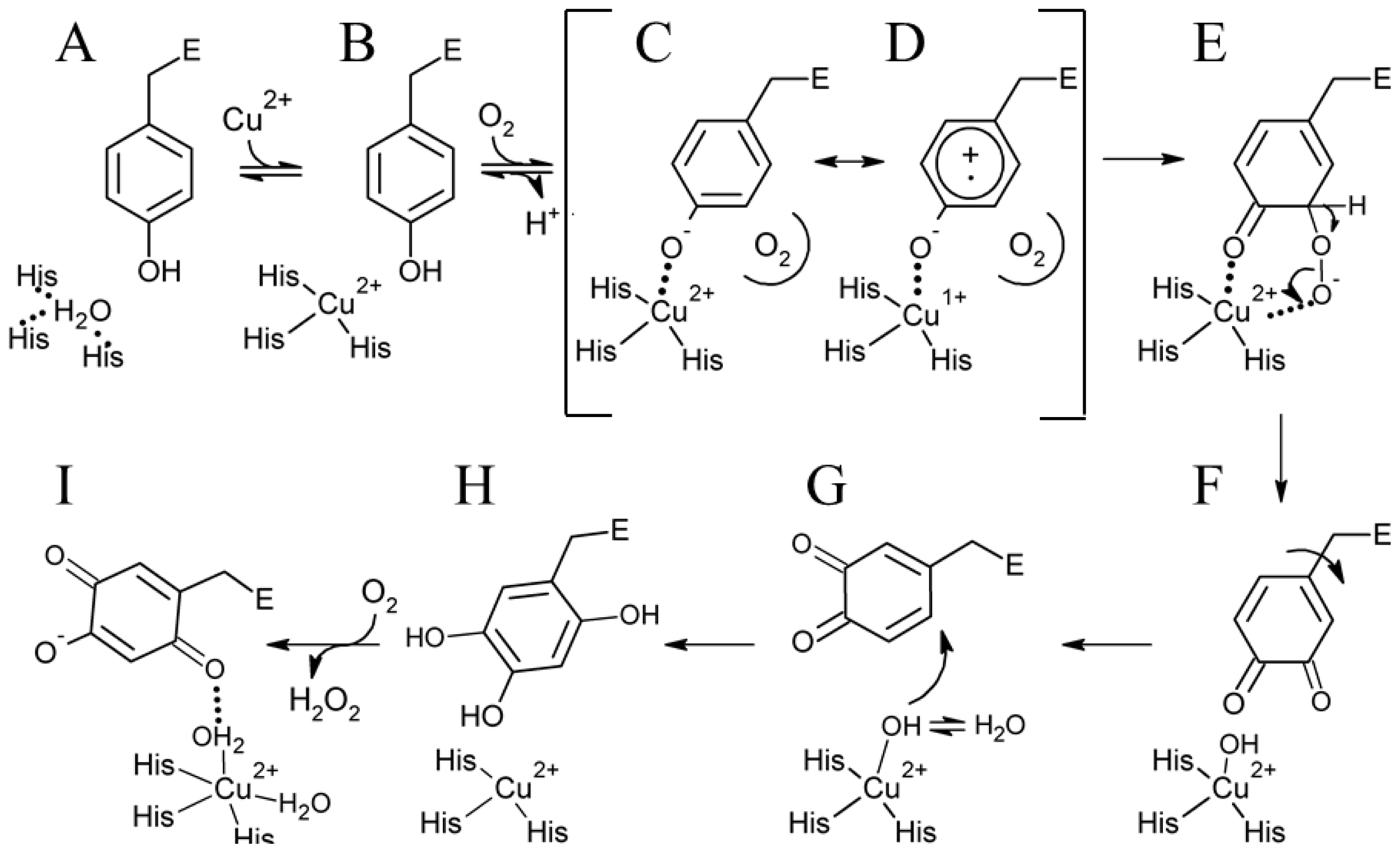
6. Biogenesis of TPQ
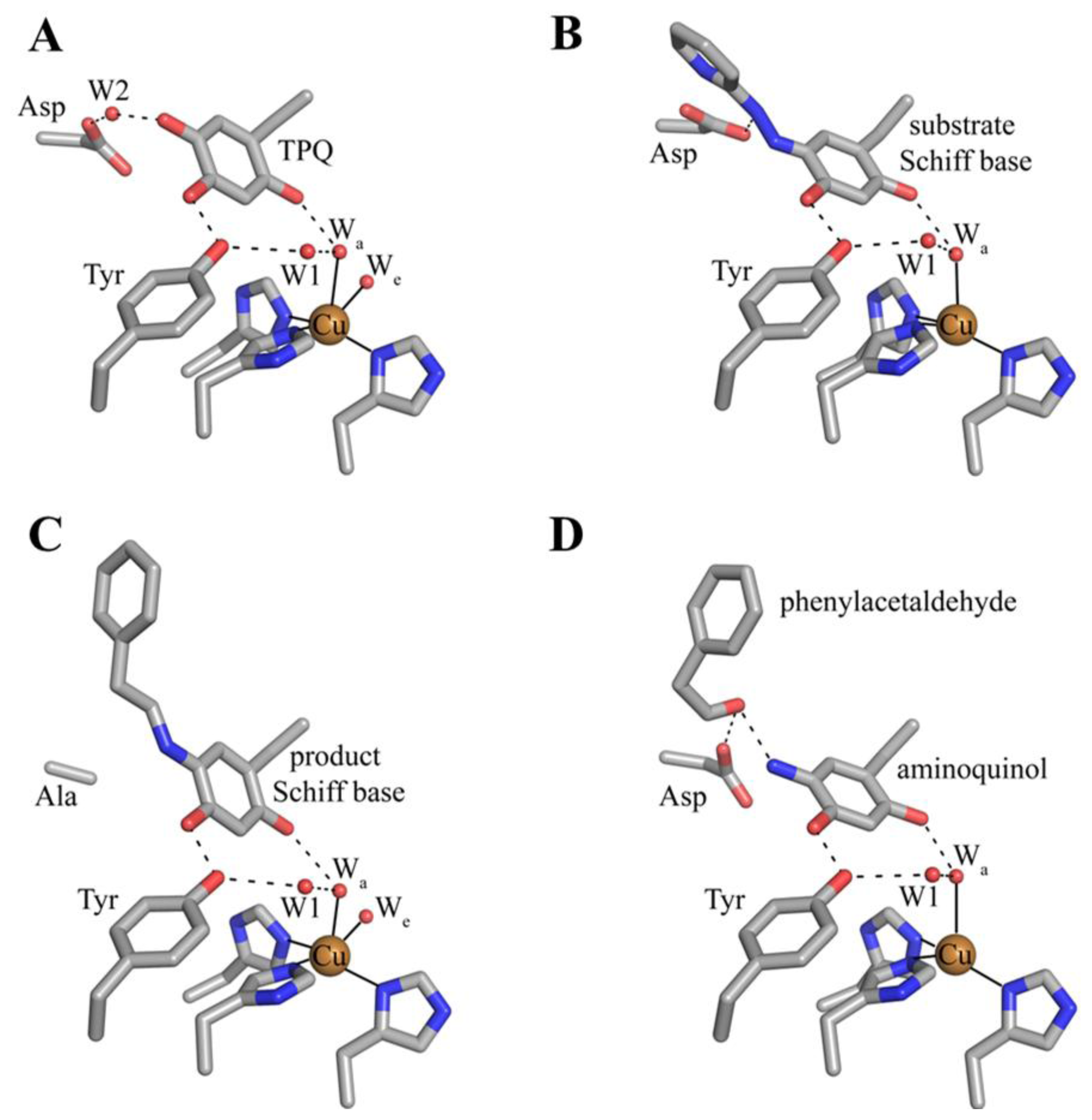
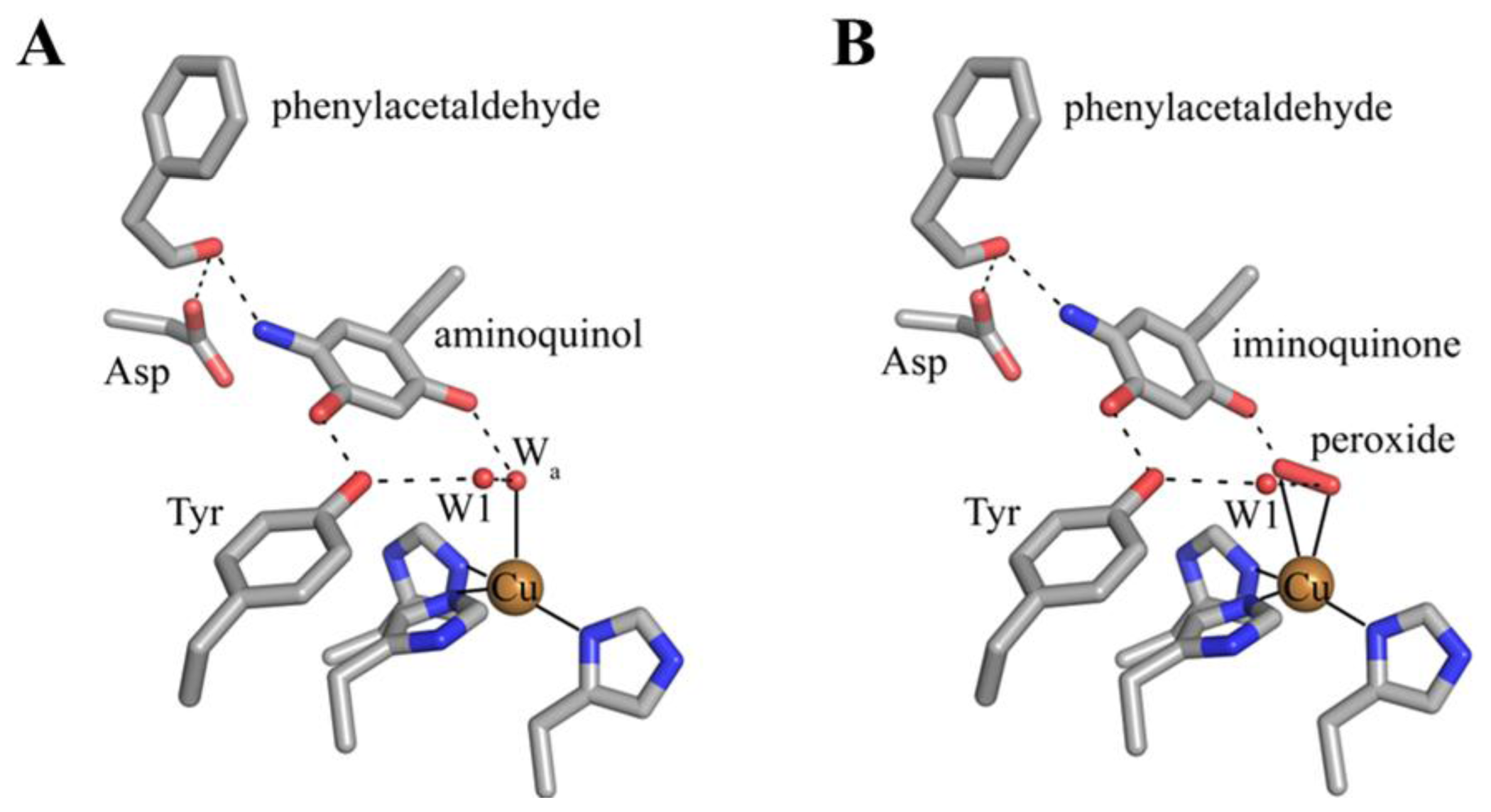
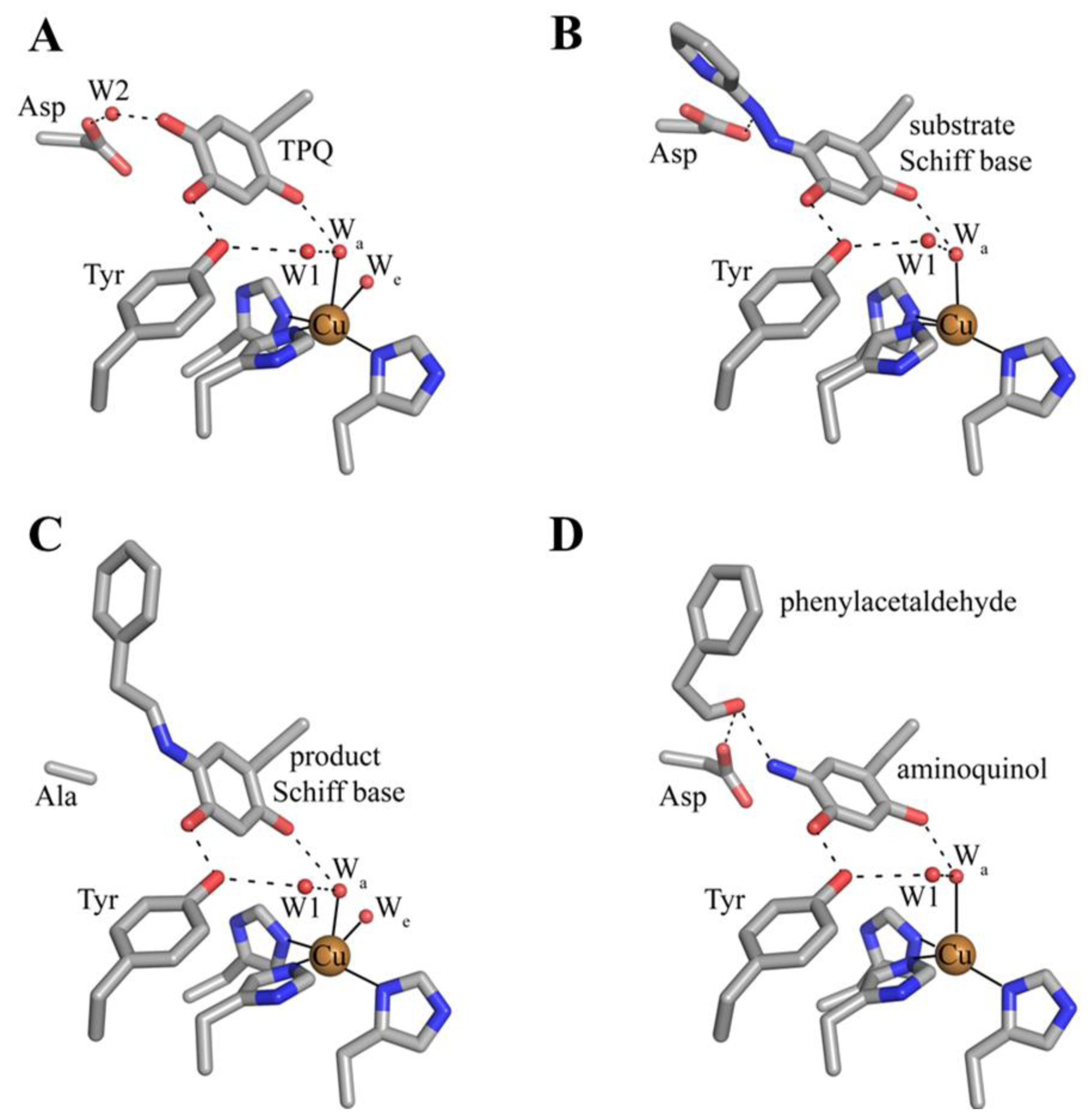
7. Catalysis in CAOs
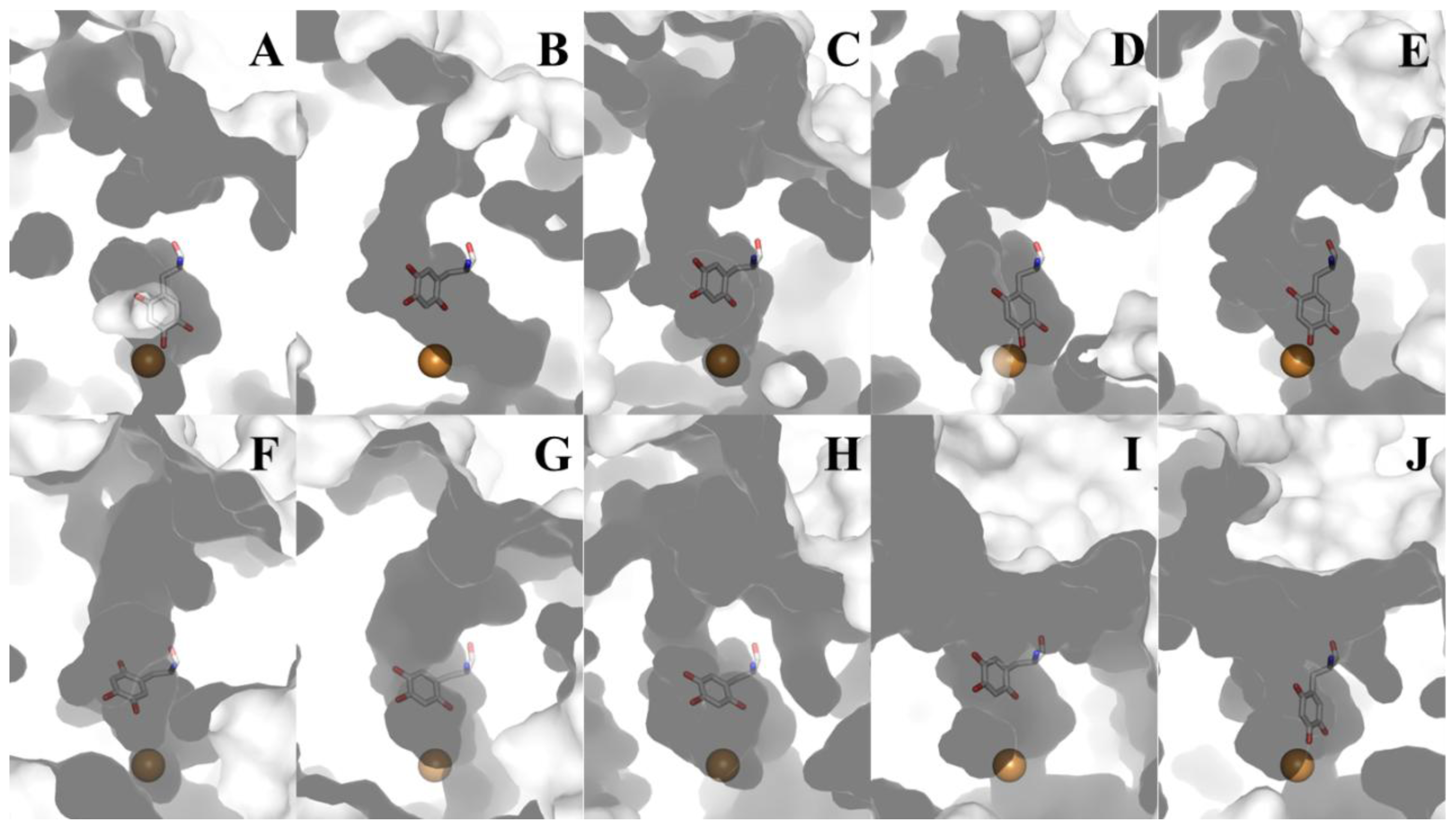
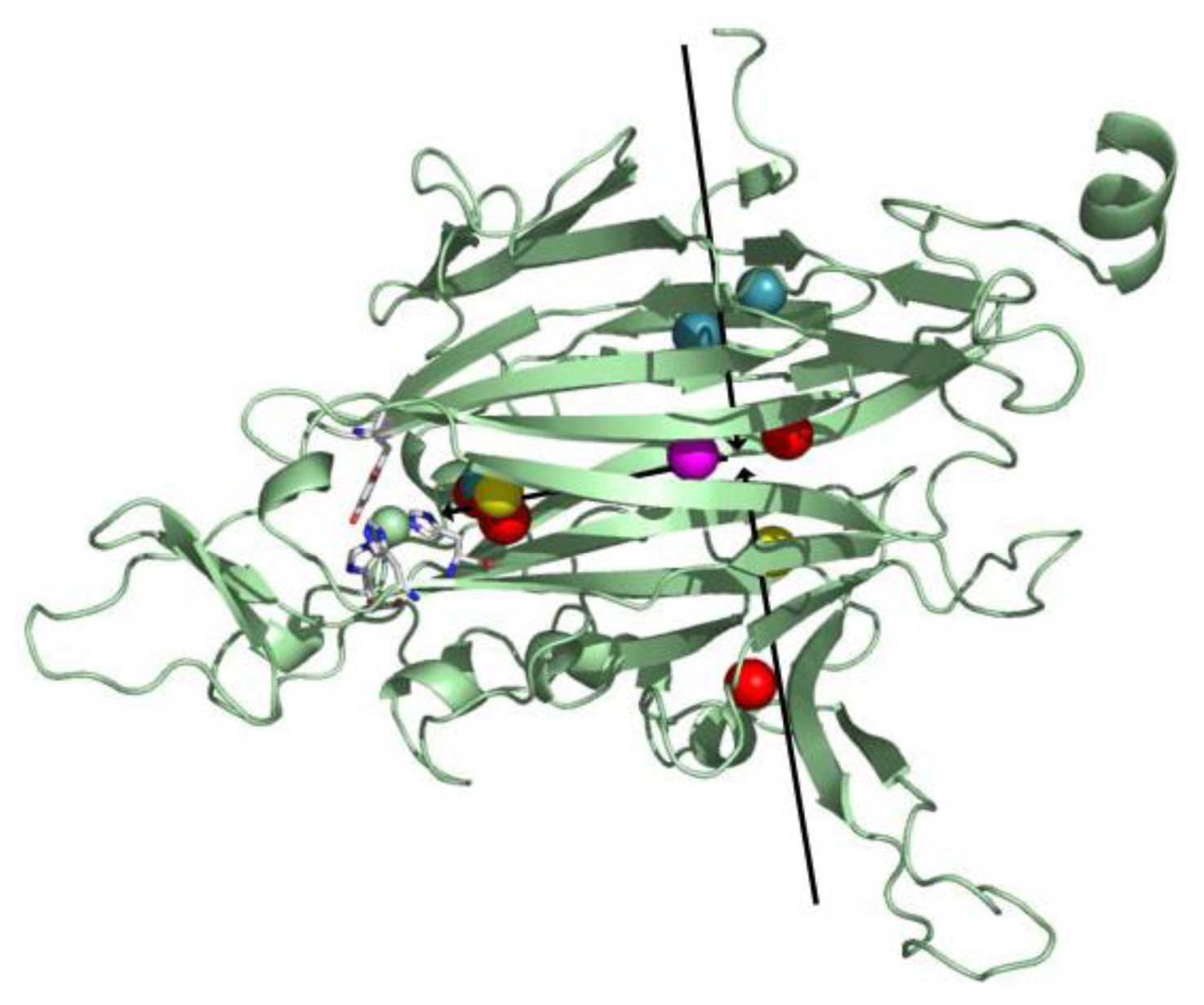
8. CAO Channels and Access to the Active Site
9. Summary
Acknowledgments
Abbreviations
| AGAO | Arthrobacter globiformis amine oxidase |
| ANAO | Aspergillus nidulans amine oxidase |
| apoCAO | precursor form of copper amine oxidase |
| BSAO | bovine serum amine oxidase |
| CAO | copper amine oxidase |
| CTQ | cysteine tryptophylquinone |
| DAO | diamine oxidase |
| DPQ | dopaquinone |
| ECAO | Escherichia coli amine oxidase |
| HPAO-1 | Hansenula polymorpha amine oxidase that prefers small aliphatic primary amines |
| HPAO-2 | Hansenula polymorpha amine oxidase that prefers aromatic primary amines |
| LMCT | ligand-metal charge transfer |
| LOX | lysyl oxidase |
| LOXL | lysyl oxidase-like proteins |
| LSAO | lentil seedling amine oxidase |
| LTQ | lysyl tyrosylquinone |
| MAO | monoamine oxidase |
| PDB | Protein Data Bank |
| PLP | pyridoxal phosphate |
| PMF | potential of mean force |
| PPLO | Pichia pastoris lysyl oxidase |
| PQQ | pyrroloquinoline quinone |
| PSAO | Pisum sativum amine oxidase |
| rmsd | root-mean-square deviation |
| SSAO | semicarbazide-sensitive amine oxidase |
| topaquinone or TPQ | 2,4,5-trihydroxyphenylalanine quinone |
| aminoquinol or TPQamq | 2,4-dihydroxy-5-aminophenylalanine |
| TPQred | 2,4,5-trihydroxyphenylalanine |
| TTQ | tryptophan tryptophylquinone |
| VAP-1 | vascular adhesion protein-1 |
References
- Shih, J.C.; Chen, K.; Ridd, M.J. Monoamine oxidase: From genes to behavior. Annu. Rev. Neurosci 1999, 22, 197–217. [Google Scholar]
- Seiler, N. Polyamine metabolism. Digestion 1990, 46, 319–330. [Google Scholar]
- Lucero, H.A.; Kagan, H.M. Lysyl oxidase: An oxidative enzyme and effector of cell function. Cell. Mol. Life Sci 2006, 63, 2304–2316. [Google Scholar]
- Okeley, N.M.; van der Donk, W.A. Novel cofactors via post-translational modifications of enzyme active sites. Chem. Biol 2000, 7, 159–171. [Google Scholar]
- Best, C.H. Disappearance of histamine from autolysing lung tissue. J. Physiol 1929, 67, 256. [Google Scholar]
- Parrott, S.; Jones, S.; Cooper, R.A. 2-phenylethylamine catabolism by Escherichia coli K12. J. Gen. Microbiol 1987, 133, 347–351. [Google Scholar]
- Van Dijken, J.P.; Bos, P. Utilization of amines in yeast. Arch. Microbiol 1981, 128, 320–324. [Google Scholar]
- Wimalasekera, R.; Villar, C.; Begum, T.; Scherer, G.F. Copper amine oxidase1 (CuAO1) of Arabidopsis thaliana contributes to abscisic acid- and polyamine-induced nitric oxide biosynthesis and abscisic acid signal transduction. Mol. Plant 2011, 4, 663–678. [Google Scholar]
- Cona, A.; Rea, G.; Angelini, R.; Federico, R.; Tavladoraki, P. Functions of amine oxidases in plant development and defence. Trends Plant Sci 2006, 11, 80–88. [Google Scholar]
- Angelini, R.; Cona, A.; Federico, R.; Fincato, P.; Tavladoraki, P.; Tisi, A. Plant amine oxidases “on the move”: An update. Plant Physiol. Biochem 2010, 48, 560–564. [Google Scholar]
- Campestre, M.P.; Bordenave, C.D.; Origone, A.C.; Menendez, A.B.; Ruiz, O.A.; Rodriguez, A.A.; Maiale, S.J. Polyamine catabolism is involved in response to salt stress in soybean hypocotyls. J. Plant Physiol 2011, 168, 1234–1240. [Google Scholar]
- Schwelberger, H.G. The origin of mammalian plasma amine oxidases. J. Neural Transm 2007, 114, 757–762. [Google Scholar]
- Novotny, W.F.; Chassande, O.; Baker, M.; Lazdunski, M.; Barbry, P. Diamine oxidase is the amiloride-binding protein and is inhibited by amiloride analogues. J. Biol. Chem 1994, 269, 9921–9925. [Google Scholar]
- Kaitaniemi, S.; Elovaara, H.; Grön, K.; Kidron, H.; Liukkonen, J.; Salminen, T.; Salmi, M.; Jalkanen, S.; Elima, K. The unique substrate specificity of human Aoc2, a semi-carbazide-sensitive amine oxidase. Cell. Mol. Life Sci 2009, 66, 2743–2757. [Google Scholar]
- Salmi, M.; Jalkanen, S. VAP-1: An adhesin and an enzyme. Trends Immunol 2001, 22, 211–216. [Google Scholar]
- Yraola, F.; Zorzano, A.; Albericio, F.; Royo, M. Structure-activity relationships of SSAO/VAP-1 arylalkylamine-based substrates. ChemMedChem 2009, 4, 495–503. [Google Scholar]
- Lyles, G.A. Substrate-specificity of mammalian tissue-bound semicarbazide-sensitive amine oxidase. Prog. Brain Res 1995, 106, 293–303. [Google Scholar]
- Gong, B.; Boor, P.J. The role of amine oxidases in xenobiotic metabolism. Expert Opin. Drug Metab. Toxicol 2006, 2, 559–571. [Google Scholar]
- Maintz, L.; Schwarzer, V.; Bieber, T.; van der ven, K.; Novak, N. Effects of histamine and diamine oxidase activities on pregnancy: A critical review. Hum. Reprod. Update 2008, 14, 485–495. [Google Scholar]
- McGrath, A.P.; Hilmer, K.M.; Collyer, C.A.; Shepard, E.M.; Elmore, B.O.; Brown, D.E.; Dooley, D.M.; Guss, J.M. Structure and inhibition of human diamine oxidase. Biochemistry 2009, 48, 9810–9822. [Google Scholar]
- Toninello, A.; Pietrangeli, P.; de Marchi, U.; Salvi, M.; Mondovi, B. Amine oxidases in apoptosis and cancer. Biochim. Biophys. Acta Rev. Cancer 2006, 1765, 1–13. [Google Scholar]
- Taksande, B.G.; Kotagale, N.R.; Patel, M.R.; Shelkar, G.P.; Ugale, R.R.; Chopde, C.T. Agmatine, an endogenous imidazoline receptor ligand modulates ethanol anxiolysis and withdrawal anxiety in rats. Eur. J. Pharmacol 2010, 637, 89–101. [Google Scholar]
- Dunkel, P.; Gelain, A.; Barlocco, D.; Haider, N.; Gyires, K.; Sperlagh, B.; Magyar, K.; Maccioni, E.; Fadda, A.; Matyus, P. Semicarbazide-sensitive amine oxidase/vascular adhesion protein 1: Recent developments concerning substrates and inhibitors of a promising therapeutic target. Curr. Med. Chem 2008, 15, 1827–1839. [Google Scholar]
- McDonald, A.; Tipton, K.; O'Sullivan, J.; Olivieri, A.; Davey, G.; Coonan, A.M.; Fu, W. Modelling the roles of MAO and SSAO in glucose transport. J. Neural Transm 2007, 114, 783–786. [Google Scholar]
- Kivi, E.; Elima, K.; Aalto, K.; Nymalm, Y.; Auvinen, K.; Koivunen, E.; Otto, D.M.; Crocker, P.R.; Salminen, T.A.; Salmi, M.; et al. Human Siglec-10 can bind to vascular adhesion protein-1 and serves as its substrate. Blood 2009, 114, 5385–5392. [Google Scholar]
- Stolen, C.M.; Marttila-Ichihara, F.; Koskinen, K.; Yegutkin, G.G.; Turja, R.; Bono, P.; Skurnik, M.; Hanninen, A.; Jalkanen, S.; Salmi, M. Absence of the endothelial oxidase Aoc3 leads to abnormal leukocyte traffic in vivo. Immunity 2005, 22, 105–115. [Google Scholar]
- Mathys, K.C.; Ponnampalam, S.N.; Padival, S.; Nagaraj, R.H. Semicarbazide-sensitive amine oxidase in aortic smooth muscle cells mediates synthesis of a methylglyoxal-AGE: Implications for vascular complications in diabetes. Biochem. Biophys. Res. Commun 2002, 297, 863–869. [Google Scholar]
- Jiang, Z.J.; Richardson, J.S.; Yu, P.H. The contribution of cerebral vascular semicarbazide-sensitive amine oxidase to cerebral amyloid angiopathy in Alzheimer’s disease. Neuropathol. Appl. Neurobiol 2008, 34, 194–204. [Google Scholar]
- Janes, S.M.; Mu, D.; Wemmer, D.; Smith, A.J.; Kaur, S.; Maltby, D.; Burlingame, A.L.; Klinman, J.P. A new redox cofactor in eukaryotic enzymes: 6-hydroxydopa at the active site of bovine serum amine oxidase. Science 1990, 248, 981–987. [Google Scholar]
- Klabunde, T.; Eicken, C.; Sacchettini, J.C.; Krebs, B. Crystal structure of a plant catechol oxidase containing a dicopper center. Nat. Struct. Biol 1998, 5, 1084–1090. [Google Scholar]
- Lerch, K. Primary structure of tyrosinase from Neurospora crassa: Purification and amino acid sequence and chemical structure of a tripeptide containing an unusual thioether. J. Biol. Chem 1982, 257, 6414–6419. [Google Scholar]
- Bravo, J.; Fita, I.; Ferrer, J.C.; Ens, W.; Hillar, A.; Switala, J.; Loewen, P.C. Identification of a novel bond between a histidine and the essential tyrosine in catalase HPII of Escherichia coli. Protein Sci 1997, 6, 1016–1023. [Google Scholar]
- Yoshikawa, S.; Shinzawa-Itoh, K.; Nakashima, R.; Yaono, R.; Yamashita, E.; Inoue, N.; Yao, M.; Fei, M.J.; Libeu, C.P.; Mizushima, T.; et al. Redox-coupled crystal structural changes in bovine heart cytochrome c oxidase. Science 1998, 280, 1723–1729. [Google Scholar]
- Ostermeier, C.; Harrenga, A.; Ermler, U.; Michel, H. Structure at 2.7 Angstrom resolution of the Paracoccus denitrificans two-subunit cytochrome c oxidase complexed with an antibody F-V fragment. Proc. Natl. Acad. Sci. USA 1997, 94, 10547–10553. [Google Scholar]
- Ito, N.; Phillips, S.E.V.; Stevens, C.; Ogel, Z.B.; McPherson, M.J.; Keen, J.N.; Yadav, K.D.S.; Knowles, P.F. Novel thioether bond revealed by a 1.7 Å crystal structure of galactose oxidase. Nature 1991, 350, 87–90. [Google Scholar]
- Gielens, C.; DeGeest, N.; Xin, X.Q.; Devreese, B.; van Beeumen, J.; Preaux, G. Evidence for a cysteine-histidine thioether bridge in functional units of molluscan haemocyanins and location of the disulfide bridges in functional units d and g of the beta(c)-haemocyanin of Helix pomatia. Eur. J. Biochem 1997, 248, 879–888. [Google Scholar]
- Taylor, T.C.; Andersson, I. The structure of the complex between rubisco and its natural substrate ribulose 1,5-bisphosphate. J. Mol. Biol 1997, 265, 432–444. [Google Scholar]
- Jabri, E.; Carr, M.B.; Hausinger, R.P.; Karplus, P.A. The crystal structure of urease from Klebsiella aerogenes. Science 1995, 268, 998–1004. [Google Scholar]
- Benning, M.M.; Kuo, J.M.; Raushel, F.M.; Holden, H.M. 3-dimensional structure of the binuclear metal center of phosphotriesterase. Biochemistry 1995, 34, 7973–7978. [Google Scholar]
- Yeh, J.I.; Claiborne, A.; Hol, W.G. Structure of the native cysteine-sulfenic acid redox center of enterococcal NADH peroxidase refined at 2.8 Å resolution. Biochemistry 1996, 35, 9951–9957. [Google Scholar]
- Schmidt, B.; Selmer, T.; Ingendoh, A.; von Figura, K. A novel amino acid modification in sulfatases that is defective in multiple sulfatase deficiency. Cell 1995, 82, 271–278. [Google Scholar]
- Nagashima, S.; Nakasako, M.; Dohmae, N.; Tsujimura, M.; Takio, K.; Odaka, M.; Yohda, M.; Kamiya, N.; Endo, I. Novel non-heme iron center of nitrile hydratase with a claw setting of oxygen atoms. Nat. Struct. Biol 1998, 5, 347–351. [Google Scholar]
- Claiborne, A.; Yeh, J.I.; Mallett, T.C.; Luba, J.; Crane, E.J.; Charrier, V.; Parsonage, D. Protein-sulfenic acids: Diverse roles for an unlikely player in enzyme catalysis and redox regulation. Biochemistry 1999, 38, 15407–15416. [Google Scholar]
- Wang, S.X.; Mure, M.; Medzihradszky, K.F.; Burlingame, A.L.; Brown, D.E.; Dooley, D.M.; Smith, A.J.; Kagan, H.M.; Klinman, J.P. A crosslinked cofactor in lysyl oxidase: Redox function for amino acid side chains. Science 1996, 273, 1078–1084. [Google Scholar]
- Mcintire, W.S.; Wemmer, D.E.; Chistoserdov, A.; Lidstrom, M.E. A new cofactor in a prokaryotic enzyme: Tryptophan tryptophylquinone as the redox prosthetic group in methylamine dehydrogenase. Science 1991, 252, 817–824. [Google Scholar]
- Vandenberghe, I.; Kim, J.K.; Devreese, B.; Hacisalihoglu, A.; Iwabuki, H.; Okajima, T.; Kuroda, S.; Adachi, O.; Jongejan, J.A.; Duine, J.A.; et al. The covalent structure of the small subunit from Pseudomonas putida amine dehydrogenase reveals the presence of three novel types of internal cross-linkages, all involving cysteine in a thioether bond. J. Biol. Chem 2001, 276, 42923–42931. [Google Scholar]
- Yasunobu, K.T.; Ishizaki, H.; Minamiura, N. The molecular mechanistic and immunological properties of amine oxidases. Mol. Cell. Biochem 1976, 13, 3–29. [Google Scholar]
- Ameyama, M.; Hayashi, M.; Matsushita, K.; Shinagawa, E.; Adachi, O. Microbial production of pyrroloquinoline quinone. Agric. Biol. Chem 1984, 48, 561–565. [Google Scholar]
- Lobenstein-Verbeek, C.L.; Jongejan, J.A.; Frank, J.; Duine, J.A. Bovine serum amine oxidase—A mammalian enzyme having covalently bound PQQ as prosthetic group. FEBS Lett 1984, 170, 305–309. [Google Scholar]
- Moog, R.S.; McGuirl, M.A.; Cote, C.E.; Dooley, D.M. Evidence for methoxatin (pyrroloquinolinequinone) as the cofactor in bovine plasma amine oxidase from resonance Raman spectroscopy. Proc. Natl. Acad. Sci. USA 1986, 83, 8435–8439. [Google Scholar]
- Knowles, P.F.; Pandeya, K.B.; Rius, F.X.; Spencer, C.M.; Moog, R.S.; McGuirl, M.A.; Dooley, D.M. The organic cofactor in plasma amine oxidase—Evidence for pyrroloquinoline quinone and against pyridoxal phosphate. Biochem. J 1987, 241, 603–608. [Google Scholar]
- Airenne, T.T.; Nymalm, Y.; Kidron, H.; Smith, D.J.; Pihlavisto, M.; Salmi, M.; Jalkanen, S.; Johnson, M.S.; Salminen, T.A. Crystal structure of the human vascular adhesion protein-1: Unique structural features with functional implications. Protein Sci 2005, 14, 1964–1974. [Google Scholar]
- Jakobsson, E.; Nilsson, J.; Ogg, D.; Kleywegt, G.J. Structure of human semicarbazide-sensitive amine oxidase/vascular adhesion protein-1. Acta Crystallogr. Sect. D Biol. Crytallogr 2005, 61, 1550–1562. [Google Scholar]
- Lunelli, M.; di Paolo, M.L.; Biadene, M.; Calderone, V.; Battistutta, R.; Scarpa, M.; Rigo, A.; Zanotti, G. Crystal structure of amine oxidase from bovine serum. J. Mol. Biol 2005, 346, 991–1004. [Google Scholar]
- Li, R.; Klinman, J.P.; Mathews, F.S. Copper amine oxidase from Hansenula polymorpha: The crystal structure determined at 2.4 Å resolution reveals the active conformation. Structure 1998, 6, 293–307. [Google Scholar]
- Chang, C.M.; Klema, V.J.; Johnson, B.J.; Mure, M.; Klinman, J.P.; Wilmot, C.M. Kinetic and structural analysis of substrate specificity in two copper amine oxidases from Hansenula polymorpha. Biochemistry 2010, 49, 2540–2550. [Google Scholar]
- Duff, A.P.; Cohen, A.E.; Ellis, P.J.; Kuchar, J.A.; Langley, D.B.; Shepard, E.M.; Dooley, D.M.; Freeman, H.C.; Guss, J.M. The crystal structure of Pichia pastoris lysyl oxidase. Biochemistry 2003, 42, 15148–15157. [Google Scholar]
- Wilce, M.C.; Dooley, D.M.; Freeman, H.C.; Guss, J.M.; Matsunami, H.; McIntire, W.S.; Ruggiero, C.E.; Tanizawa, K.; Yamaguchi, H. Crystal structures of the copper-containing amine oxidase from Arthrobacter globiformis in the holo and apo forms: Implications for the biogenesis of topaquinone. Biochemistry 1997, 36, 16116–1633. [Google Scholar]
- Parsons, M.R.; Convery, M.A.; Wilmot, C.M.; Yadav, K.D.; Blakeley, V.; Corner, A.S.; Phillips, S.E.; McPherson, M.J.; Knowles, P.F. Crystal structure of a quinoenzyme: copper amine oxidase of Escherichia coli at 2 Å resolution. Structure 1995, 3, 1171–1184. [Google Scholar]
- Kumar, V.; Dooley, D.M.; Freeman, H.C.; Guss, J.M.; Harvey, I.; McGuirl, M.A.; Wilce, M.C.; Zubak, V.M. Crystal structure of a eukaryotic (pea seedling) copper-containing amine oxidase at 2.2 Å resolution. Structure 1996, 4, 943–955. [Google Scholar]
- McGrath, A.P.; Mithieux, S.M.; Collyer, C.A.; Bakhuis, J.G.; van den Berg, M.; Sein, A.; Heinz, A.; Schmelzer, C.; Weiss, A.S.; Guss, J.M. Structure and activity of Aspergillus nidulans copper amine oxidase. Biochemistry 2011, 50, 5718–5730. [Google Scholar]
- Smith, M.A.; Pirrat, P.; Pearson, A.R.; Kurtis, C.R.; Trinh, C.H.; Gaule, T.G.; Knowles, P.F.; Phillips, S.E.; McPherson, M.J. Exploring the roles of the metal ions in Escherichia coli copper amine oxidase. Biochemistry 2010, 49, 1268–1280. [Google Scholar]
- Sebela, M.; Luhova, L.; Frebort, I.; Hirota, S.; Faulhammer, H.G.; Stuzka, V.; Pec, P. Confirmation of the presence of a Cu(II) topa quinone active site in the amine oxidase from fenugreek seedlings. J. Exp. Bot 1997, 48, 1897–1907. [Google Scholar]
- Johnson, B.J.; Cohen, J.; Welford, R.W.; Pearson, A.R.; Schulten, K.; Klinman, J.P.; Wilmot, C.M. Exploring molecular oxygen pathways in Hansenula polymorpha copper-containing amine oxidase. J. Biol. Chem 2007, 282, 17767–17776. [Google Scholar]
- Cai, D.; Klinman, J.P. Evidence of a self-catalytic mechanism of 2,4,5-trihydroxyphenylalanine quinone biogenesis in yeast copper amine oxidase. J. Biol. Chem 1994, 269, 32039–32042. [Google Scholar]
- Matsuzaki, R.; Fukui, T.; Sato, H.; Ozaki, Y.; Tanizawa, K. Generation of the topa quinone cofactor in bacterial monoamine oxidase by cupric ion-dependent autooxidation of a specific tyrosyl residue. FEBS Lett 1994, 351, 360–364. [Google Scholar]
- Ruggiero, C.E.; Dooley, D.M. Stoichiometry of the topa quinone biogenesis reaction in copper amine oxidases. Biochemistry 1999, 38, 9556–9556. [Google Scholar]
- Mu, D.; Janes, S.M.; Smith, A.J.; Brown, D.E.; Dooley, D.M.; Klinman, J.P. Tyrosine codon corresponds to topa quinone at the active site of copper amine oxidases. J. Biol. Chem 1992, 267, 7979–7982. [Google Scholar]
- Kim, M.; Okajima, T.; Kishishita, S.; Yoshimura, M.; Kawamori, A.; Tanizawa, K.; Yamaguchi, H. X-ray snapshots of quinone cofactor biogenesis in bacterial copper amine oxidase. Nat. Struct. Biol 2002, 9, 591–596. [Google Scholar]
- Schwartz, B.; Dove, J.E.; Klinman, J.P. Kinetic analysis of oxygen utilization during cofactor biogenesis in a copper-containing amine oxidase from yeast. Biochemistry 2000, 39, 3699–3707. [Google Scholar]
- Klema, V.J.; Johnson, B.J.; Klinman, J.P.; Wilmot, C.M. The precursor form of Hansenula polymorpha copper amine oxidase-1 in complex with Cu(I) and Co(II). Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun 2012, 68, 501–510. [Google Scholar]
- Dove, J.E.; Schwartz, B.; Williams, N.K.; Klinman, J.P. Investigation of spectroscopic intermediates during copper-binding and TPQ formation in wild-type and active-site mutants of a copper-containing amine oxidase from yeast. Biochemistry 2000, 39, 3690–3698. [Google Scholar]
- Goto, Y.; Klinman, J.P. Binding of dioxygen to non-metal sites in proteins: exploration of the importance of binding site size versus hydrophobicity in the copper amine oxidase from Hansenula polymorpha. Biochemistry 2002, 41, 13637–13643. [Google Scholar]
- Dubois, J.; Klinman, J.P. The nature of O2 reactivity leading to topa quinone in the copper amine oxidase from Hansenula polymoropha and its relationship to catalytic turnover. Biochemistry 2005, 44, 11381–11388. [Google Scholar]
- Chen, Z.; Schwartz, B.; Williams, N.K.; Li, R.; Klinman, J.P.; Mathews, F.S. Crystal structure at 2.5 Å resolution of zinc-substituted copper amine oxidase of Hansenula polymorpha expressed in Escherichia coli. Biochemistry 2000, 39, 9709–9717. [Google Scholar]
- McGrath, A.P.; Caradoc-Davies, T.; Collyer, C.A.; Guss, J.M. Correlation of active site metal content in human diamine oxidase with trihydroxyphenylalanine quinone cofactor biogenesis. Biochemistry 2010, 49, 8316–8324. [Google Scholar]
- Okajima, T.; Kishishita, S.; Chiu, Y.C.; Murakawa, T.; Kim, M.; Yamaguchi, H.; Hirota, S.; Kuroda, S.; Tanizawa, K. Reinvestigation of metal ion specificity for quinone cofactor biogenesis in bacterial copper amine oxidase. Biochemistry 2005, 44, 12041–12048. [Google Scholar]
- Samuels, N.M.; Klinman, J.P. 2,4,5-trihydroxyphenylalanine quinone biogenesis in the copper amine oxidase from Hansenula polymorpha with the alternate metal nickel. Biochemistry 2005, 44, 14308–14317. [Google Scholar]
- Samuels, N.M.; Klinman, J.P. Investigation of Cu(I)-dependent 2,4,5-trihydroxyphenylalanine quinone biogenesis in Hansenula polymorpha amine oxidase. J. Biol. Chem 2006, 281, 21114–21118. [Google Scholar]
- Drummond, J.T.; Matthews, R.G. Nitrous oxide degradation by cobalamin-dependent methionine synthase: Characterization of the reactants and products in the inactivation reaction. Biochemistry 1994, 33, 3732–4371. [Google Scholar]
- Gomes, L.; Pereira, E.; de Castro, B. Nickel(II) complexes with N2OS and N2S2 co-ordination spheres: Reduction and spectroscopic study of the corresponding Ni(I) complexes. J. Chem. Soc. Dalton Trans 2000, 8, 1373–1379. [Google Scholar]
- Chen, Z.W.; Datta, S.; Dubois, J.L.; Klinman, J.P.; Mathews, F.S. Mutation at a strictly conserved, active site tyrosine in the copper amine oxidase leads to uncontrolled oxygenase activity. Biochemistry 2010, 49, 7393–7402. [Google Scholar]
- Schwartz, B.; Green, E.L.; Sanders-Loehr, J.; Klinman, J.P. Relationship between conserved consensus site residues and the productive conformation for the TPQ cofactor in a copper-containing amine oxidase from yeast. Biochemistry 1998, 37, 16591–16600. [Google Scholar]
- DuBois, J.L.; Klinman, J.P. Methods for characterizing TPQ-containing proteins. Methods Enzymol 2004, 378, 17–31. [Google Scholar]
- Holm, R.H.; Kennepohl, P.; Solomon, E.I. Structural and functional aspects of metal sites in biology. Chem. Rev 1996, 96, 2239–2314. [Google Scholar]
- Wilmot, C.M.; Murray, J.M.; Alton, G.; Parsons, M.R.; Convery, V.B.; Corner, A.S.; Palcic, M.M.; Knowles, P.F.; McPherson, M.J.; Phillips, S.E. Catalytic mechanism of the quinoenzyme amine oxidase from Escherichia coli: Exploring the reductive half-reaction. Biochemistry 1997, 36, 1608–1620. [Google Scholar]
- Murray, J.M.; Saysell, C.G.; Wilmot, C.M.; Tambyrajah, W.S.; Jaeger, J.; Knowles, P.F.; Phillips, S.E.; McPherson, M.J. The active site base controls cofactor reactivity in Escherichia coli amine oxidase: X-ray crystallographic studies with mutational variants. Biochemistry 1999, 38, 8217–8227. [Google Scholar]
- Wilmot, C.M.; Hajdu, J.; McPherson, M.J.; Phillips, S.E. Visualization of dioxygen bound to copper during enzyme catalysis. Science 1999, 286, 1724–1728. [Google Scholar]
- Pietrangeli, P.; Nocera, S.; Mondovi, B.; Morpurgo, L. Is the catalytic mechanism of bacteria, plant, and mammal copper-TPQ amine oxidases identical? Biochim. Biophys. Acta 2003, 1647, 152–156. [Google Scholar]
- Dooley, D.M.; McGuirl, M.A.; Brown, D.E.; Turowski, P.N.; McIntire, W.S.; Knowles, P.F. A Cu(I)-semiquinone state in substrate-reduced amine oxidases. Nature 1991, 349, 262–264. [Google Scholar]
- Welford, R.W.; Lam, A.; Mirica, L.M.; Klinman, J.P. Partial conversion of Hansenula polymorpha amine oxidase into a “plant” amine oxidase: Implications for copper chemistry and mechanism. Biochemistry 2007, 46, 10817–10827. [Google Scholar]
- Medda, R.; Padiglia, A.; Bellelli, A.; Pedersen, J.Z.; Agro, A.F.; Floris, G. Cu(I)-semiquinone radical species in plant copper-amine oxidases. FEBS Lett 1999, 453, 1–5. [Google Scholar]
- Shepard, E.M.; Dooley, D.M. Intramolecular electron transfer rate between active-site copper and TPQ in Arthrobacter globiformis amine oxidase. J. Biol. Inorg. Chem 2006, 11, 1039–1048. [Google Scholar]
- Mukherjee, A.; Smirnov, V.V.; Lanci, M.P.; Brown, D.E.; Shepard, E.M.; Dooley, D.M.; Roth, J.P. Inner-sphere mechanism for molecular oxygen reduction catalyzed by copper amine oxidases. J. Am. Chem. Soc 2008, 130, 9459–9473. [Google Scholar]
- Turowski, P.N.; McGuirl, M.A.; Dooley, D.M. Intramolecular electron transfer rate between active-site copper and topa quinone in pea seedling amine oxidase. J. Biol. Chem 1993, 268, 17680–17682. [Google Scholar]
- Barker, R.; Boden, N.; Cayley, G.; Charlton, S.C.; Henson, R.; Holmes, M.C.; Kelly, I.D.; Knowles, P.F. Properties of cupric ions in benzylamine oxidase from pig plasma as studied by magnetic-resonance and kinetic methods. Biochem. J 1979, 177, 289–302. [Google Scholar]
- Juda, G.A.; Shepard, E.M.; Elmore, B.O.; Dooley, D.M. A comparative study of the binding and inhibition of four copper-containing amine oxidases by azide: Implications for the role of copper during the oxidative half-reaction. Biochemistry 2006, 45, 8788–8800. [Google Scholar]
- Su, Q.; Klinman, J.P. Probing the mechanism of proton coupled electron transfer to dioxygen: The oxidative half-reaction of bovine serum amine oxidase. Biochemistry 1998, 37, 12513–12525. [Google Scholar]
- Mills, S.A.; Klinman, J.P. Evidence against reduction of Cu2+ to Cu+ during dioxygen activation in a copper amine oxidase from yeast. J. Am. Chem. Soc 2000, 122, 9897–9904. [Google Scholar]
- Mills, S.A.; Goto, Y.; Su, Q.; Plastino, J.; Klinman, J.P. Mechanistic comparison of the cobalt-substituted and wild-type copper amine oxidase from Hansenula polymorpha. Biochemistry 2002, 41, 10577–10584. [Google Scholar]
- Padiglia, A.; Medda, R.; Pedersen, J.Z.; Finazzi, A.A.; Lorrai, A.; Murgia, B.; Floris, G. Effect of metal substitution in copper amine oxidase from lentil seedlings. J. Biol. Inorg. Chem 1999, 4, 608–613. [Google Scholar]
- Schwartz, B.; Olgin, A.K.; Klinman, J.P. The role of copper in topa quinone biogenesis and catalysis, as probed by azide inhibition of a copper amine oxidase from yeast. Biochemistry 2001, 40, 2954–2963. [Google Scholar]
- Dawkes, H.C.; Phillips, S.E. Copper amine oxidase: Cunning cofactor and controversial copper. Curr. Opin. Struct. Biol 2001, 11, 666–673. [Google Scholar]
- Mills, S.A.; Brown, D.E.; Dang, K.; Sommer, D.; Bitsimis, A.; Nguyen, J.; Dooley, D.M. Cobalt substitution supports an inner-sphere electron transfer mechanism for oxygen reduction in pea seedling amine oxidase. J. Biol. Inorg. Chem 2012, 17, 507–515. [Google Scholar]
- Mure, M.; Klinman, J.P. Synthesis and spectroscopic characterization of model compounds for the active site cofactor in copper amine oxidases. J. Am. Chem. Soc 1993, 115, 7117–7127. [Google Scholar]
- Hartmann, C.; Brzovic, P.; Klinman, J.P. Spectroscopic detection of chemical intermediates in the reaction of para-substituted benzylamines with bovine serum amine oxidase. Biochemistry 1993, 32, 2234–2241. [Google Scholar]
- Cai, D.; Dove, J.; Nakamura, N.; Sanders-Loehr, J.; Klinman, J.P. Mechanism-based inactivation of a yeast methylamine oxidase mutant: Implications for the functional role of the consensus sequence surrounding topaquinone. Biochemistry 1997, 36, 11472–11478. [Google Scholar]
- Hevel, J.M.; Mills, S.A.; Klinman, J.P. Mutation of a strictly conserved, active-site residue alters substrate specificity and cofactor biogenesis in a copper amine oxidase. Biochemistry 1999, 38, 3683–3693. [Google Scholar]
- Hirota, S.; Iwamoto, T.; Kishishita, S.; Okajima, T.; Yamauchi, O.; Tanizawa, K. Spectroscopic observation of intermediates formed during the oxidative half-reaction of copper/topa quinone-containing phenylethylamine oxidase. Biochemistry 2001, 40, 15789–15796. [Google Scholar]
- Mure, M.; Klinman, J.P. Model studies of topa quinone: Synthesis and characterization of topa quinone derivatives. Methods Enzymol 1995, 258, 39–52. [Google Scholar]
- Nakamura, N.; Matsuzaki, R.; Choi, Y.H.; Tanizawa, K.; Sanders-Loehr, J. Biosynthesis of topa quinone cofactor in bacterial amine oxidases. Solvent origin of C-2 oxygen determined by Raman spectroscopy. J. Biol. Chem 1996, 271, 4718–4724. [Google Scholar]
- Murakawa, T.; Hayashi, H.; Taki, M.; Yamamoto, Y.; Kawano, Y.; Tanizawa, K.; Okajima, T. Structural insights into the substrate specificity of bacterial copper amine oxidase obtained by using irreversible inhibitors. J. Biochem 2012, 151, 167–178. [Google Scholar]
- Chiu, Y.C.; Okajima, T.; Murakawa, T.; Uchida, M.; Taki, M.; Hirota, S.; Kim, M.; Yamaguchi, H.; Kawano, Y.; Kamiya, N.; et al. Kinetic and structural studies on the catalytic role of the aspartic acid residue conserved in copper amine oxidase. Biochemistry 2006, 45, 4105–4120. [Google Scholar]
- Frebort, I.; Toyama, H.; Matsushita, K.; Adachi, O. Half-site reactivity with p-nitrophenylhydrazine and subunit separation of the dimeric copper-containing amine oxidase from Aspergillus niger Biochem. Mol. Biol. Int 1995, 36, 1207–1216. [Google Scholar]
- Morpurgo, L.; Agostinelli, E.; Mondovi, B.; Avigliano, L.; Silvestri, R.; Stefancich, G.; Artico, M. Bovine serum amine oxidase: Half-site reactivity with phenylhydrazine, semicarbazide, and aromatic hydrazides. Biochemistry 1992, 31, 2615–2621. [Google Scholar]
- Agostinelli, E.; Morpurgo, L.; Wang, C.; Giartosio, A.; Mondovi, B. Properties of cobalt-substituted bovine serum amine oxidase. Eur. J. Biochem 1994, 222, 727–732. [Google Scholar]
- Morpurgo, L.; Agostinelli, E.; Muccigrosso, J.; Martini, F.; Mondovi, B.; Avigliano, L. Benzylhydrazine as a pseudo-substrate of bovine serum amine oxidase. Biochem. J 1989, 260, 19–25. [Google Scholar]
- de Biase, D.; Agostinelli, E.; de Matteis, G.; Mondovi, B.; Morpurgo, L. Half-of-the-sites reactivity of bovine serum amine oxidase: Reactivity and chemical identity of the second site. Eur. J. Biochem 1996, 237, 93–99. [Google Scholar]
- Takahashi, K.; Klinman, J.P. Relationship of stopped flow to steady state parameters in the dimeric copper amine oxidase from Hansenula polymorpha and the role of zinc in inhibiting activity at alternate copper-containing subunits. Biochemistry 2006, 45, 4683–4694. [Google Scholar]
- Coleman, A.A.; Hindsgaul, O.; Palcic, M.M. Stereochemistry of copper amine oxidase reactions. J. Biol. Chem 1989, 264, 19500–19505. [Google Scholar]
- Scaman, C.H.; Palcic, M.M. Stereochemical course of tyramine oxidation by semicarbazide-sensitive amine oxidase. Biochemistry 1992, 31, 6829–6841. [Google Scholar]
- Uchida, M.; Ohtani, A.; Kohyama, N.; Okajima, T.; Tanizawa, K.; Yamamoto, Y. Stereochemistry of 2-phenylethylamine oxidation catalyzed by bacterial copper amine oxidase. Biosci. Biotechnol. Biochem 2003, 67, 2664–2667. [Google Scholar]
- Taki, M.; Murakawa, T.; Nakamoto, T.; Uchida, M.; Hayashi, H.; Tanizawa, K.; Yamamoto, Y.; Okajima, T. Further insight into the mechanism of stereoselective proton abstraction by bacterial copper amine oxidase. Biochemistry 2008, 47, 7726–7733. [Google Scholar]
- Duff, A.P.; Trambaiolo, D.M.; Cohen, A.E.; Ellis, P.J.; Juda, G.A.; Shepard, E.M.; Langley, D.B.; Dooley, D.M.; Freeman, H.C.; Guss, J.M. Using xenon as a probe for dioxygen-binding sites in copper amine oxidases. J. Mol. Biol 2004, 344, 599–607. [Google Scholar]
- Pirrat, P.; Smith, M.A.; Pearson, A.R.; McPherson, M.J.; Phillips, S.E. Structure of a xenon derivative of Escherichia coli copper amine oxidase: Confirmation of the proposed oxygen-entry pathway. Acta. Crystallogr. Sect. F Struct. Biol. Cryst. Commun 2008, 64, 1105–1109. [Google Scholar]
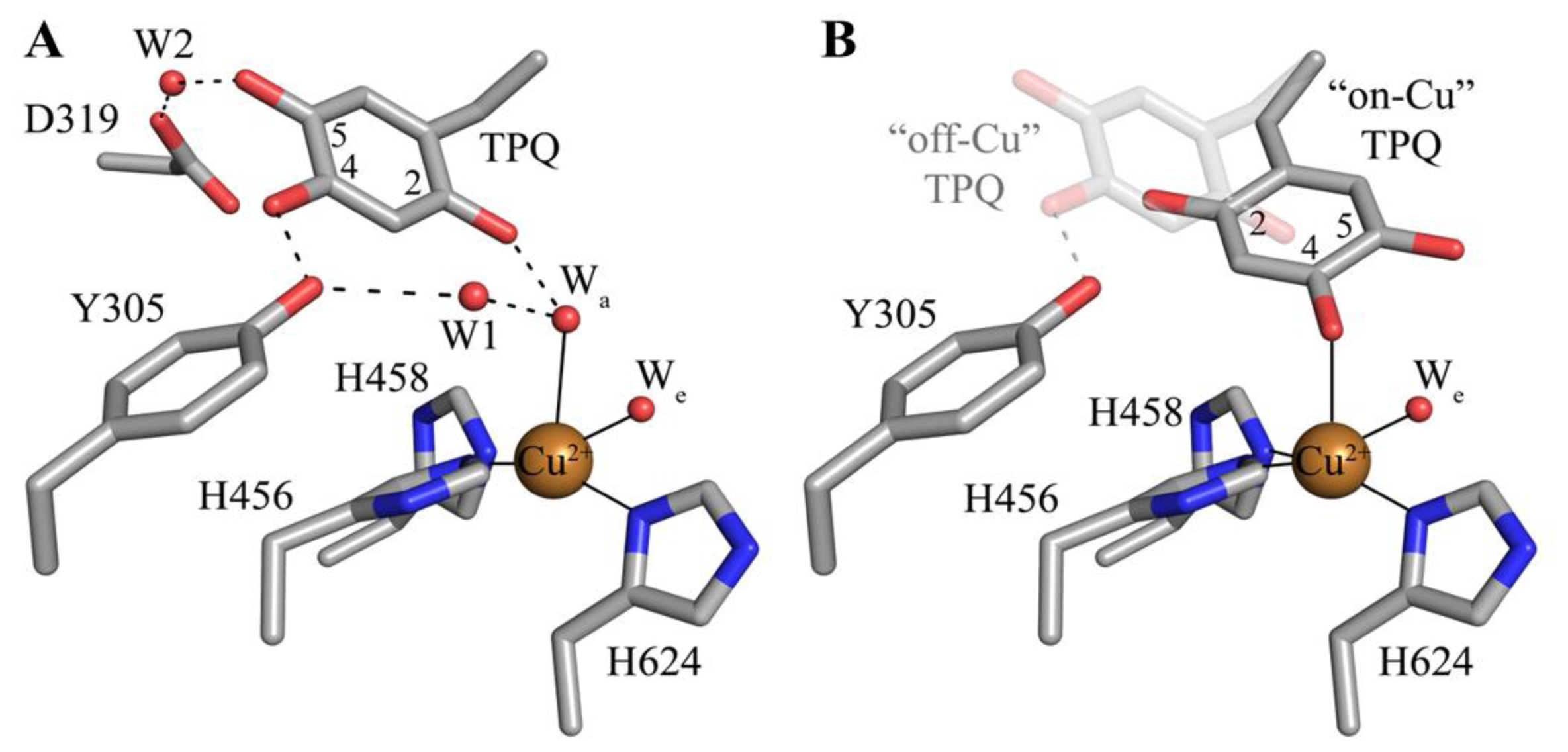
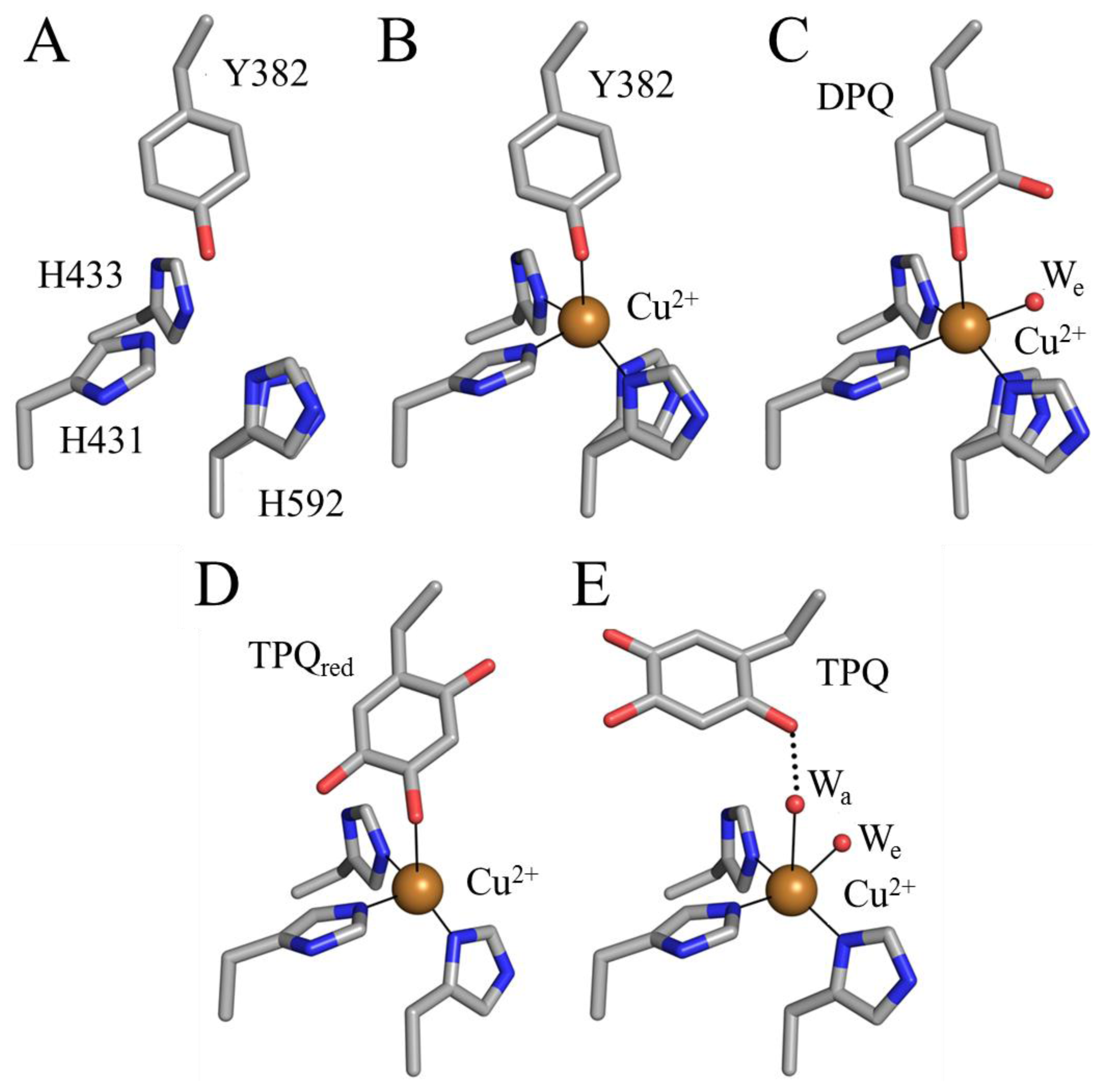
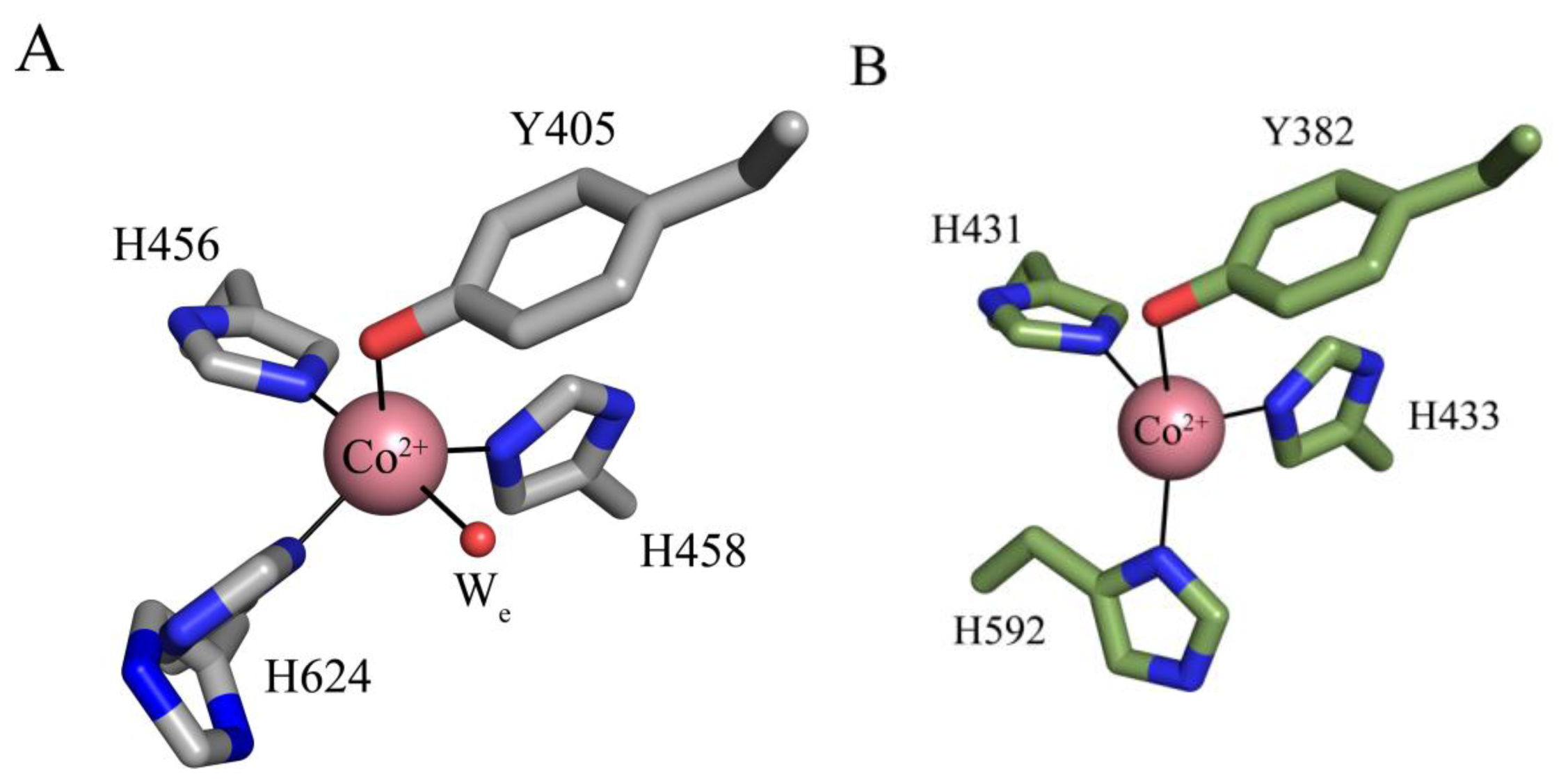
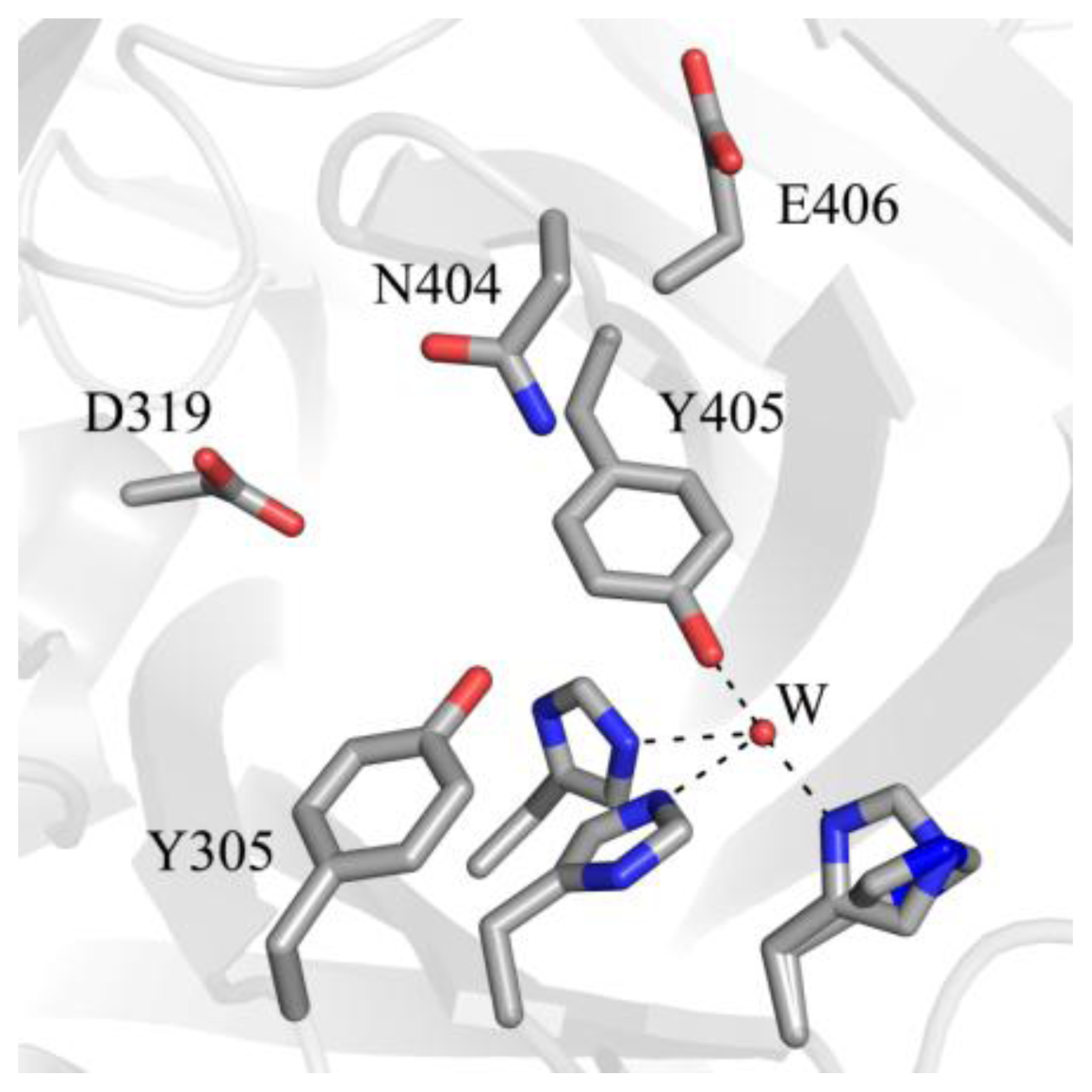

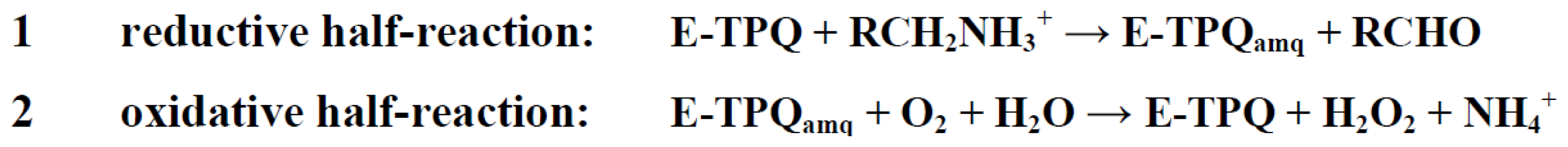


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Source | Organism | Reference |
|---|---|---|
| Mammalian | Homo sapiens | VAP-1/SSAO [52,53]; DAO [20]; |
| Bos taurus | BSAO [54] | |
| Yeast | Hansenula polymorpha | HPAO-1 [55]; HPAO-2 [56] |
| Pichia pastoris | PPLO [57] | |
| Bacterial | Arthrobacter globiformis | AGAO [58] |
| Escherichia coli | ECAO [59] | |
| Plant | Pisum savitum | PSAO [60] |
| Fungal | Aspergillus nidulans | ANAO [61] |
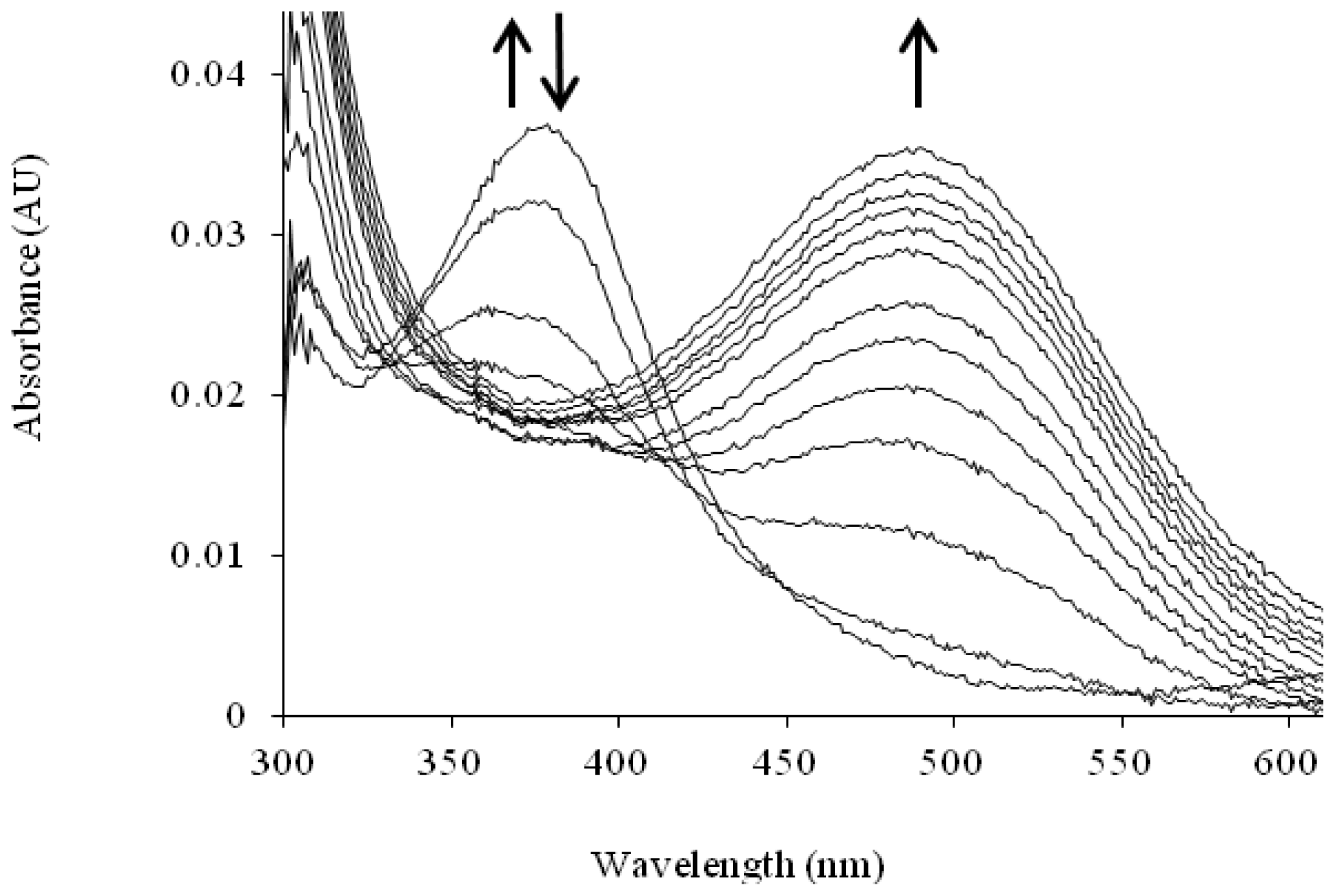
| Catalytic Intermediate | λmax |
|---|---|
| TPQ (quinone) [105] | 480 |
| Substrate Schiff base [106] | 340 |
| Product Schiff base [83,107,108] | 380 |
| Aminoquinol [106] | 310 |
| Semiquinone [90] | 360, 435, 465 |
| Iminoquinone [109,110] | 450 or 350 (if charge is delocalized, as in HPAO-1) |
| Cu(II)-peroxy [109] | 410 |
© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Klema, V.J.; Wilmot, C.M. The Role of Protein Crystallography in Defining the Mechanisms of Biogenesis and Catalysis in Copper Amine Oxidase. Int. J. Mol. Sci. 2012, 13, 5375-5405. https://doi.org/10.3390/ijms13055375
Klema VJ, Wilmot CM. The Role of Protein Crystallography in Defining the Mechanisms of Biogenesis and Catalysis in Copper Amine Oxidase. International Journal of Molecular Sciences. 2012; 13(5):5375-5405. https://doi.org/10.3390/ijms13055375
Chicago/Turabian StyleKlema, Valerie J., and Carrie M. Wilmot. 2012. "The Role of Protein Crystallography in Defining the Mechanisms of Biogenesis and Catalysis in Copper Amine Oxidase" International Journal of Molecular Sciences 13, no. 5: 5375-5405. https://doi.org/10.3390/ijms13055375