Aβ-40 Y10F Increases βfibrils Formation but Attenuates the Neurotoxicity of Amyloid-β Peptide

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
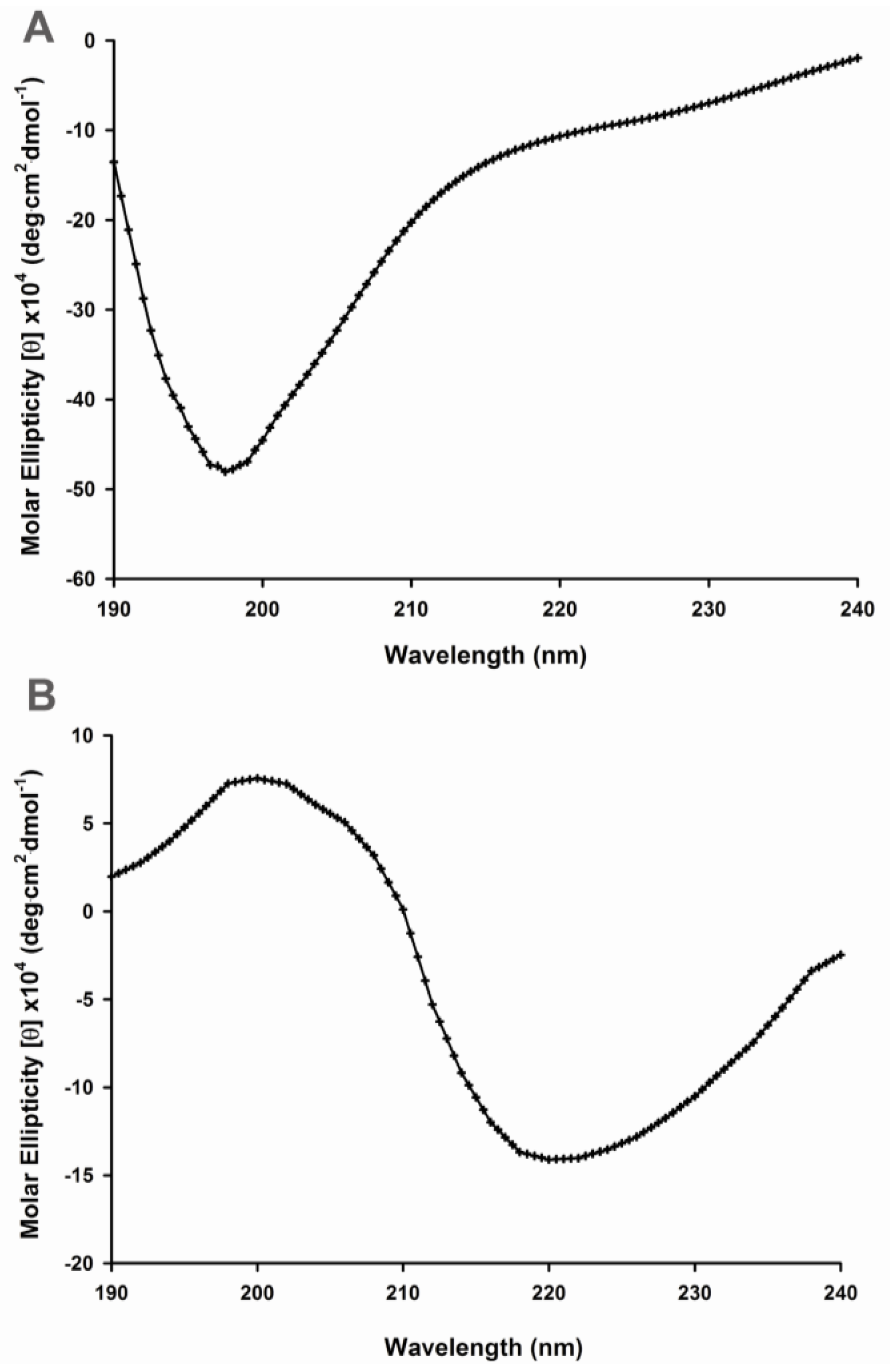
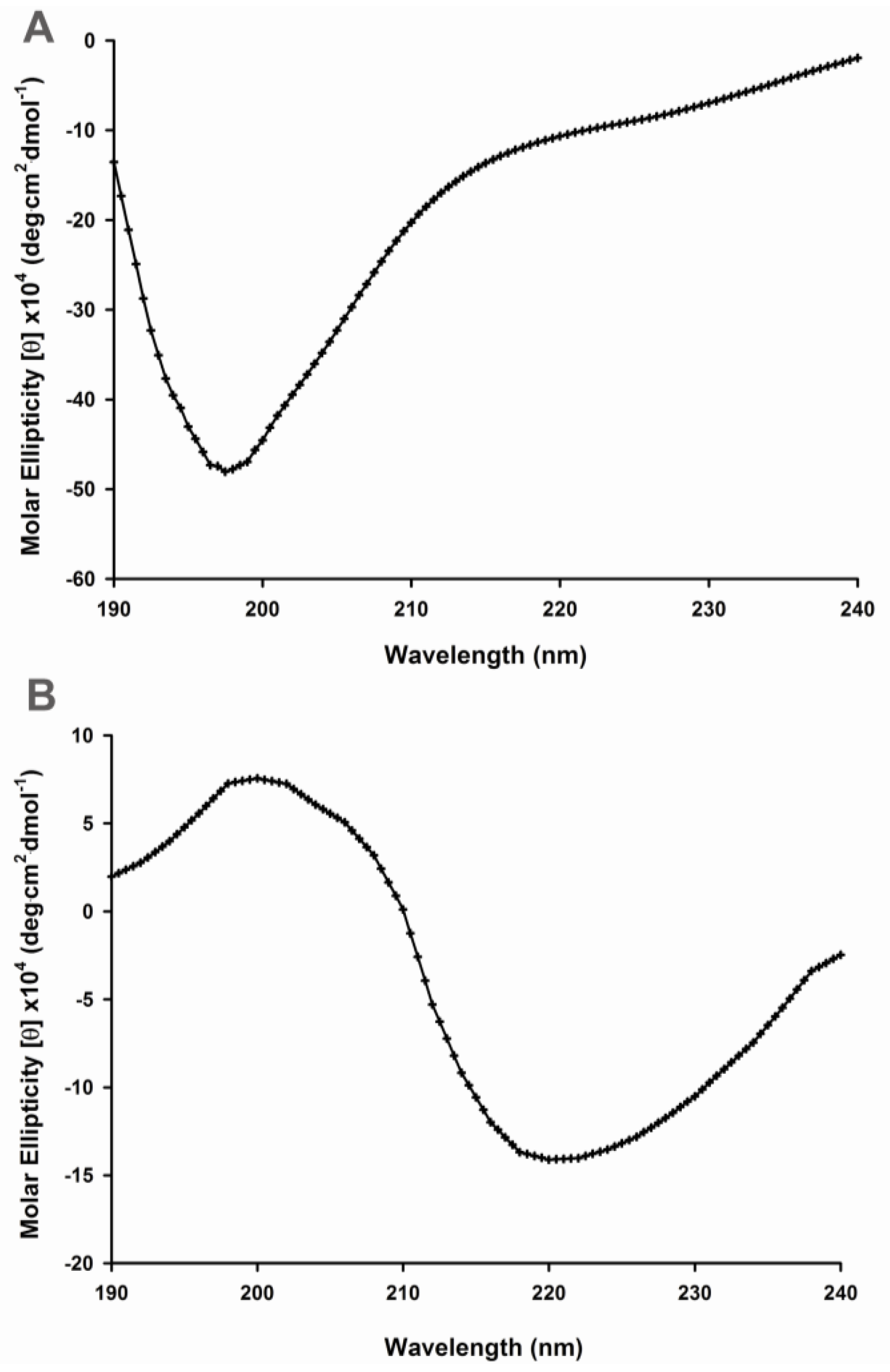
2.1. Secondary Structural Changes of Aβ (1–40) Y10F
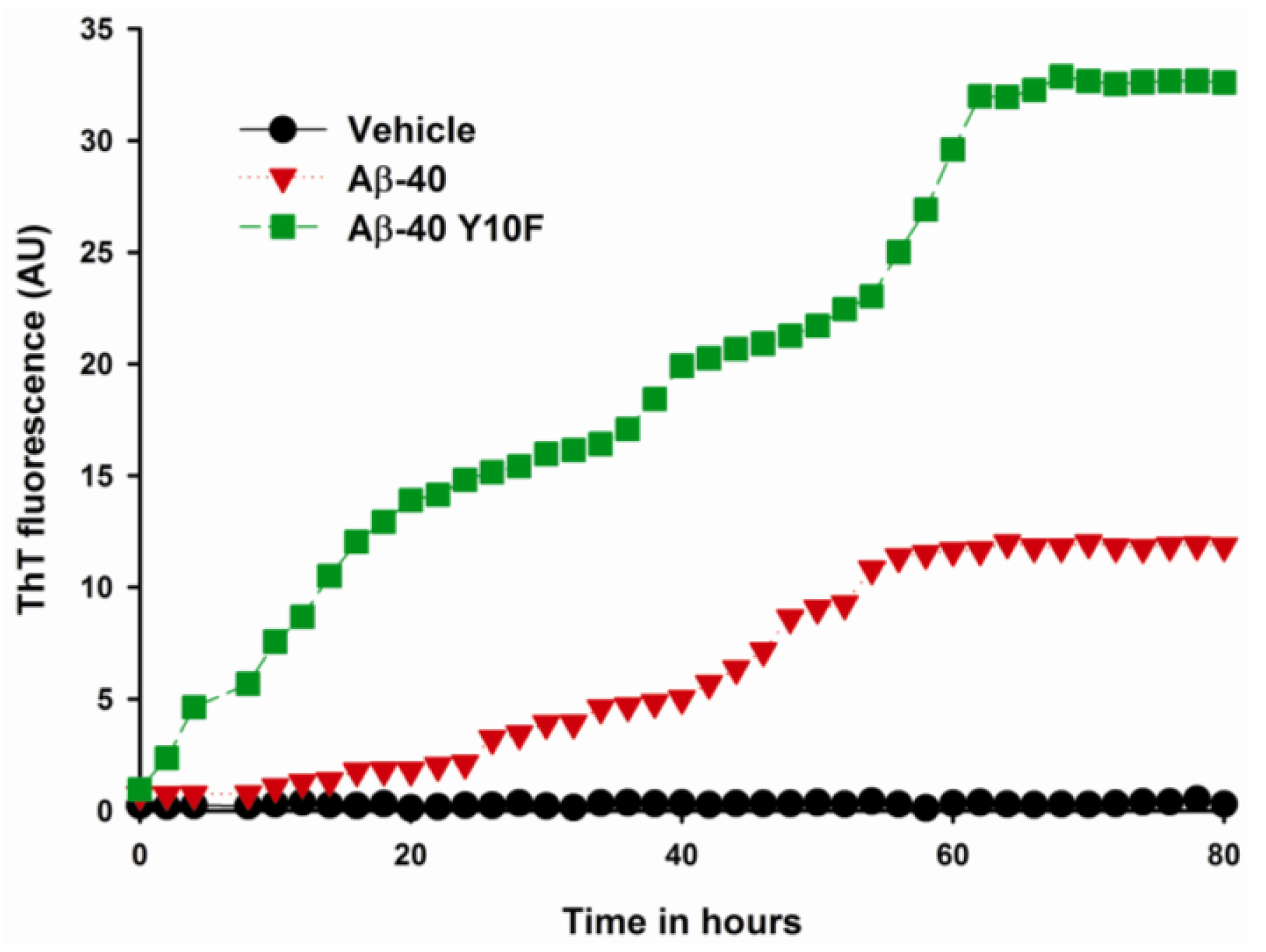
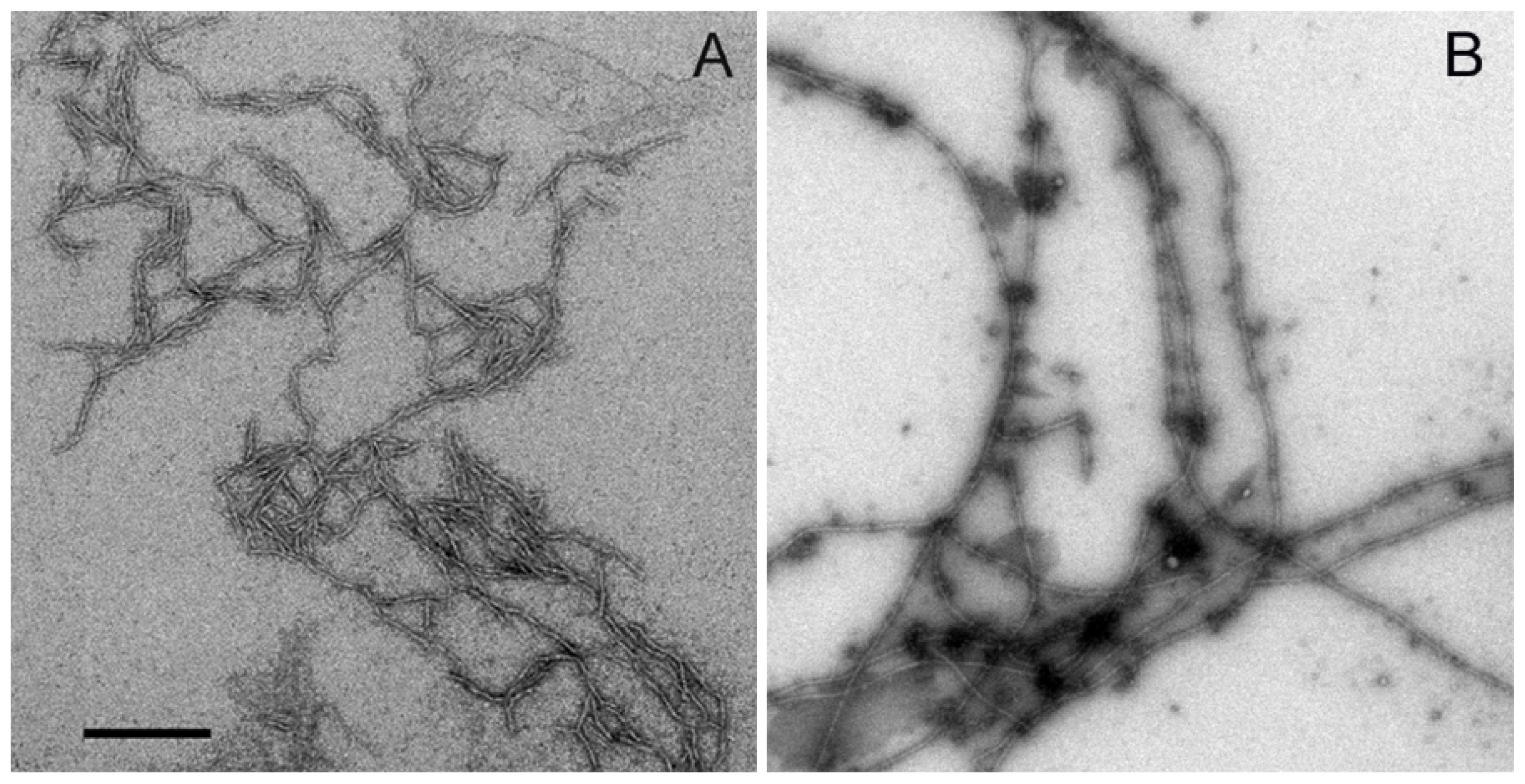
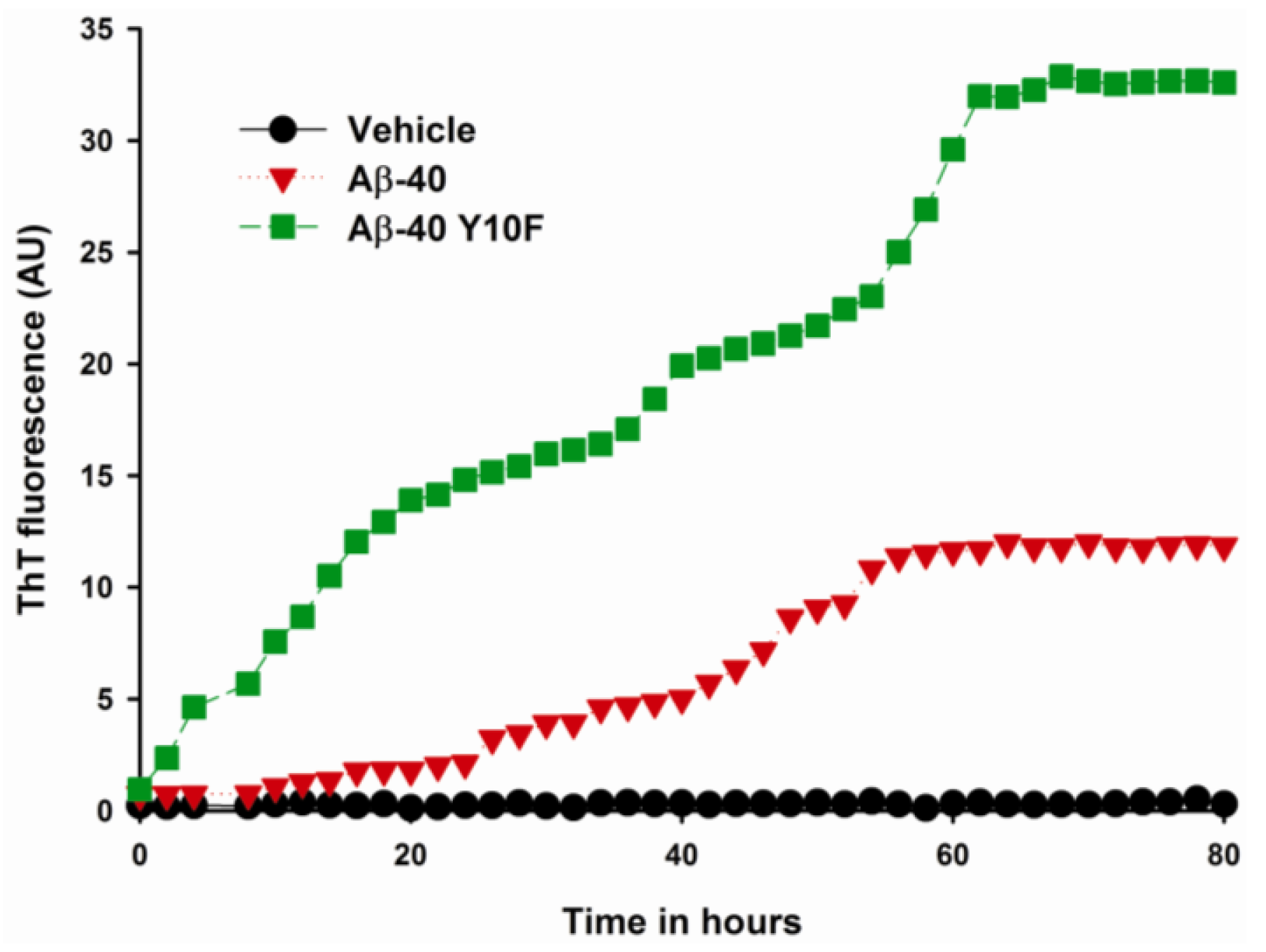
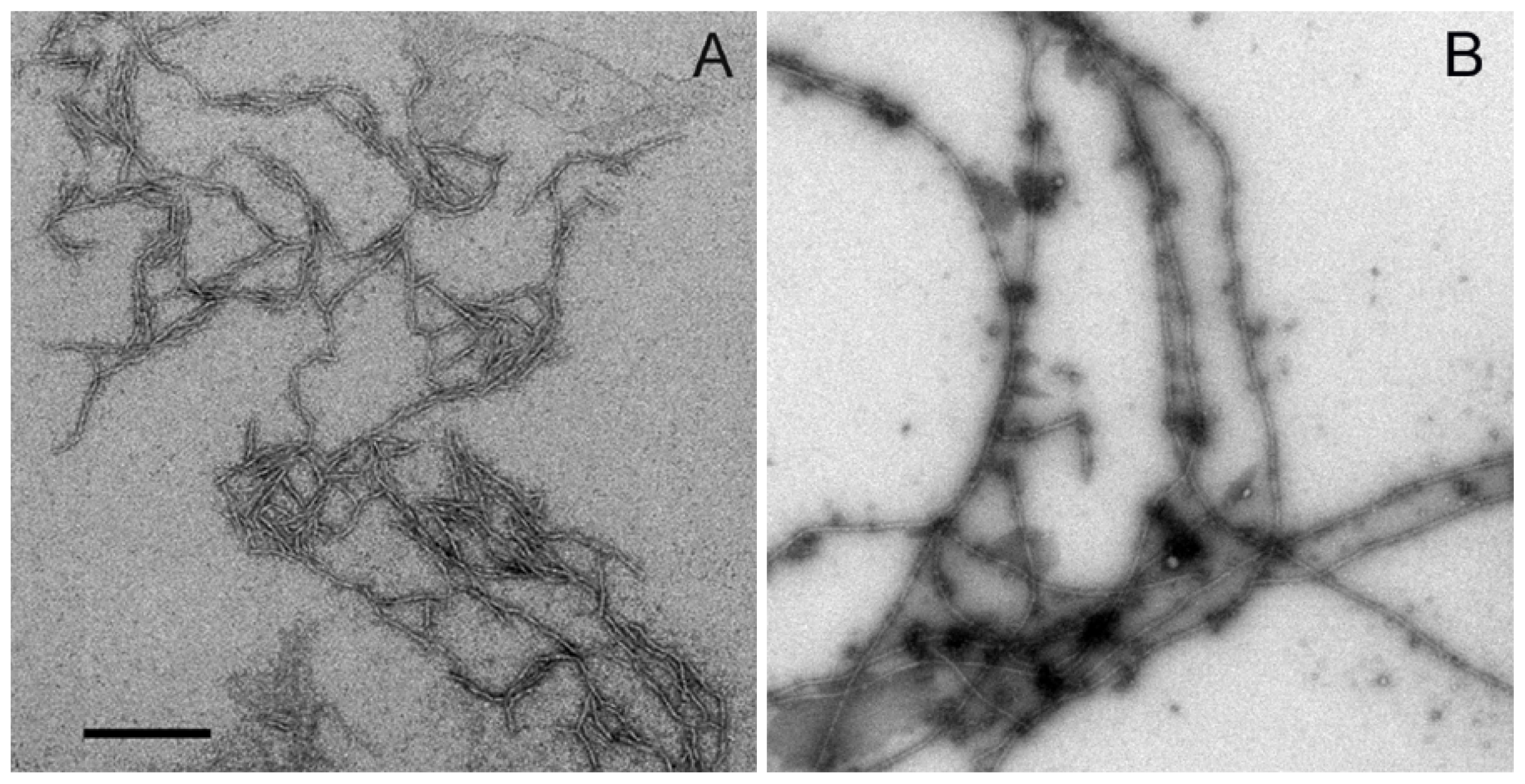
2.2. Aggregation of Wild Type Aβ (1–40) and Aβ-40 Y10F
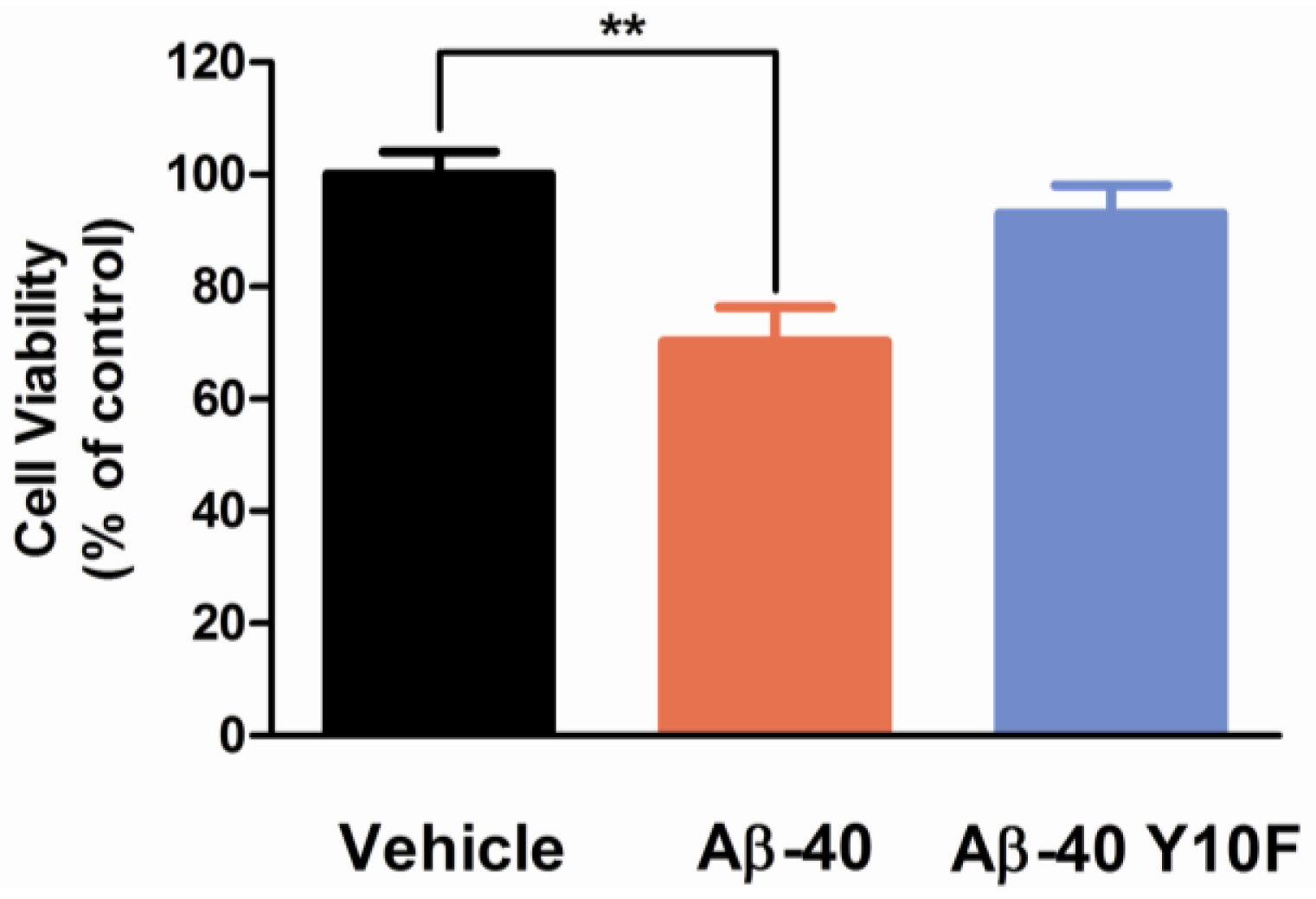
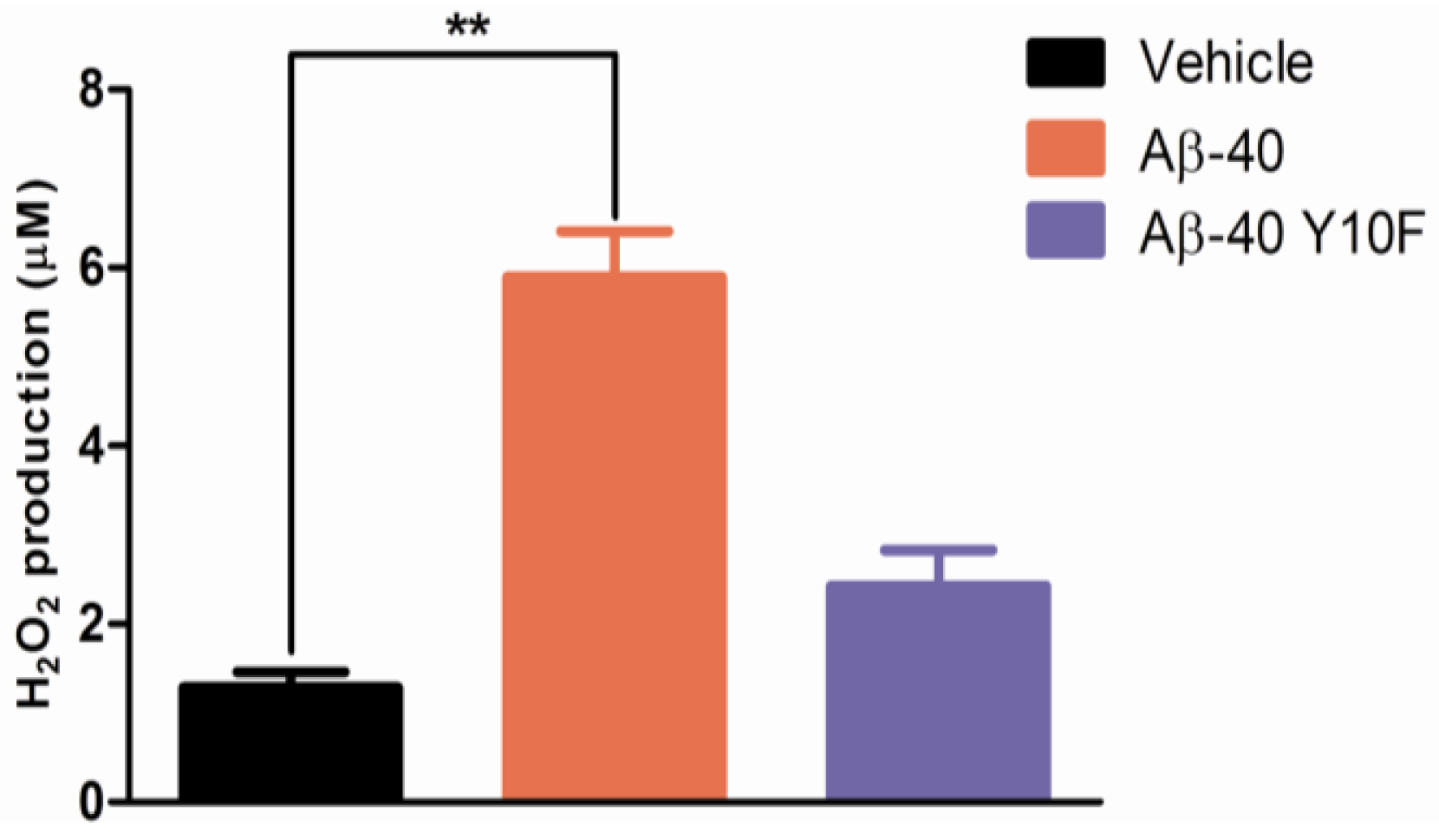
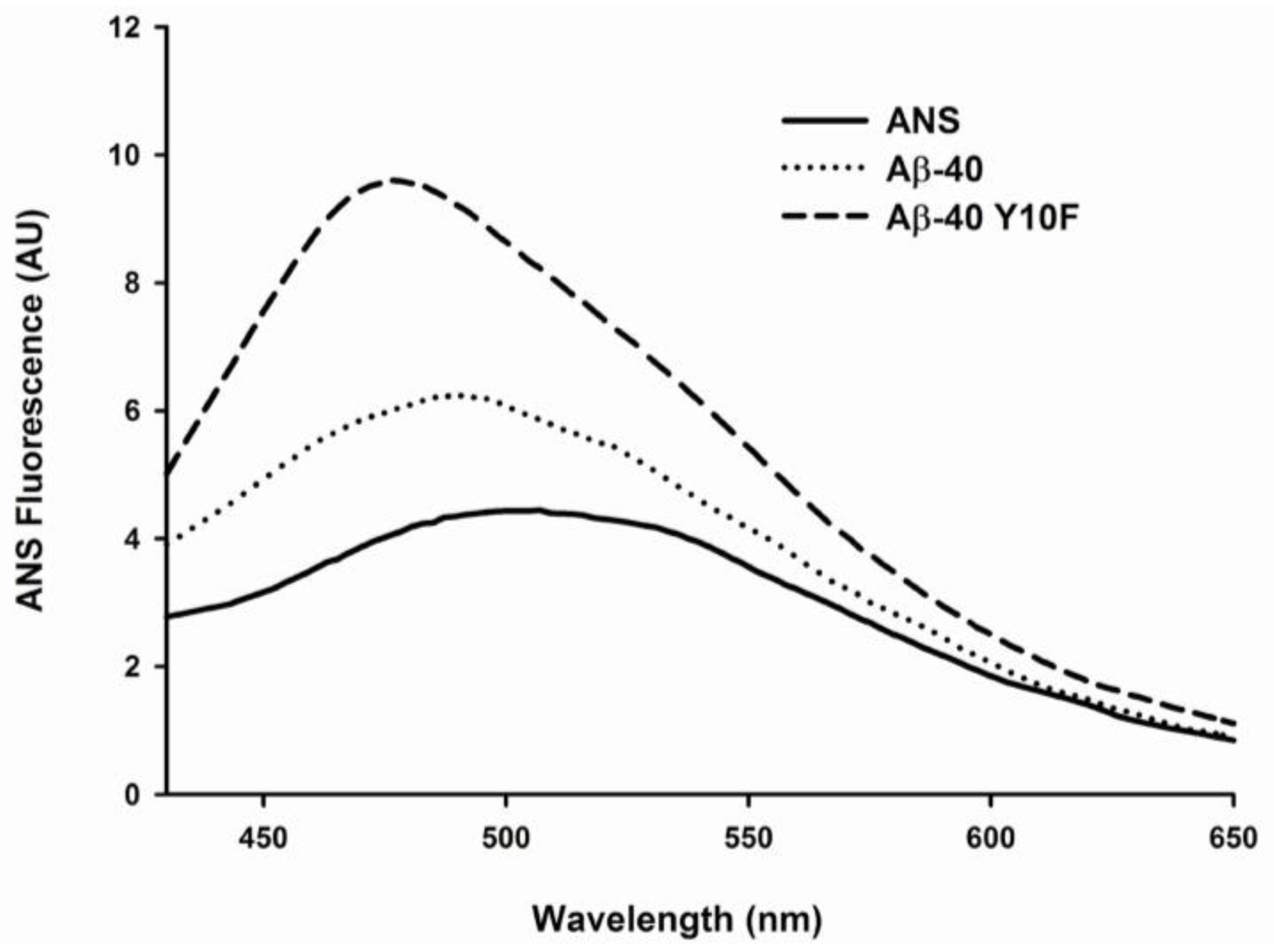
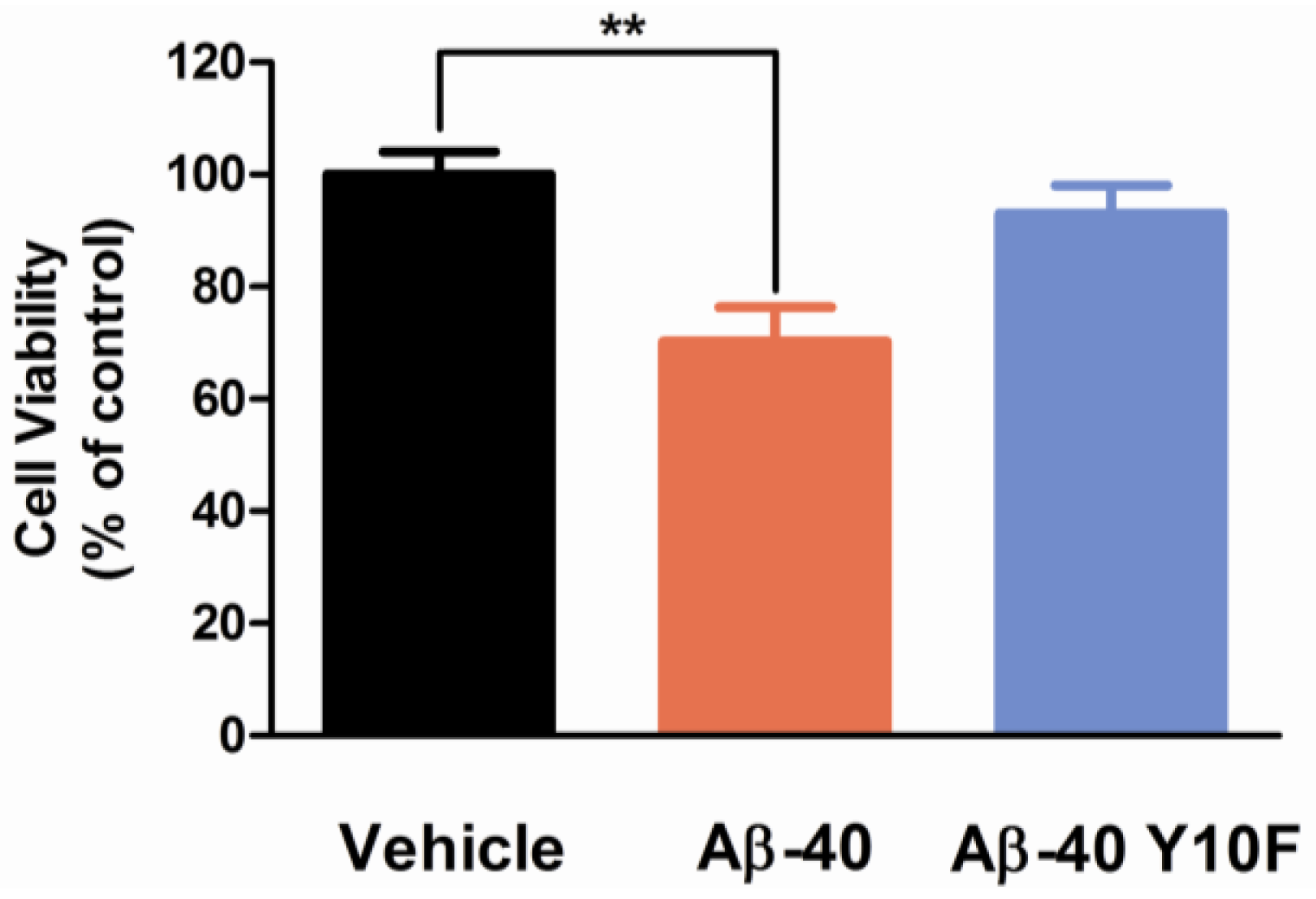
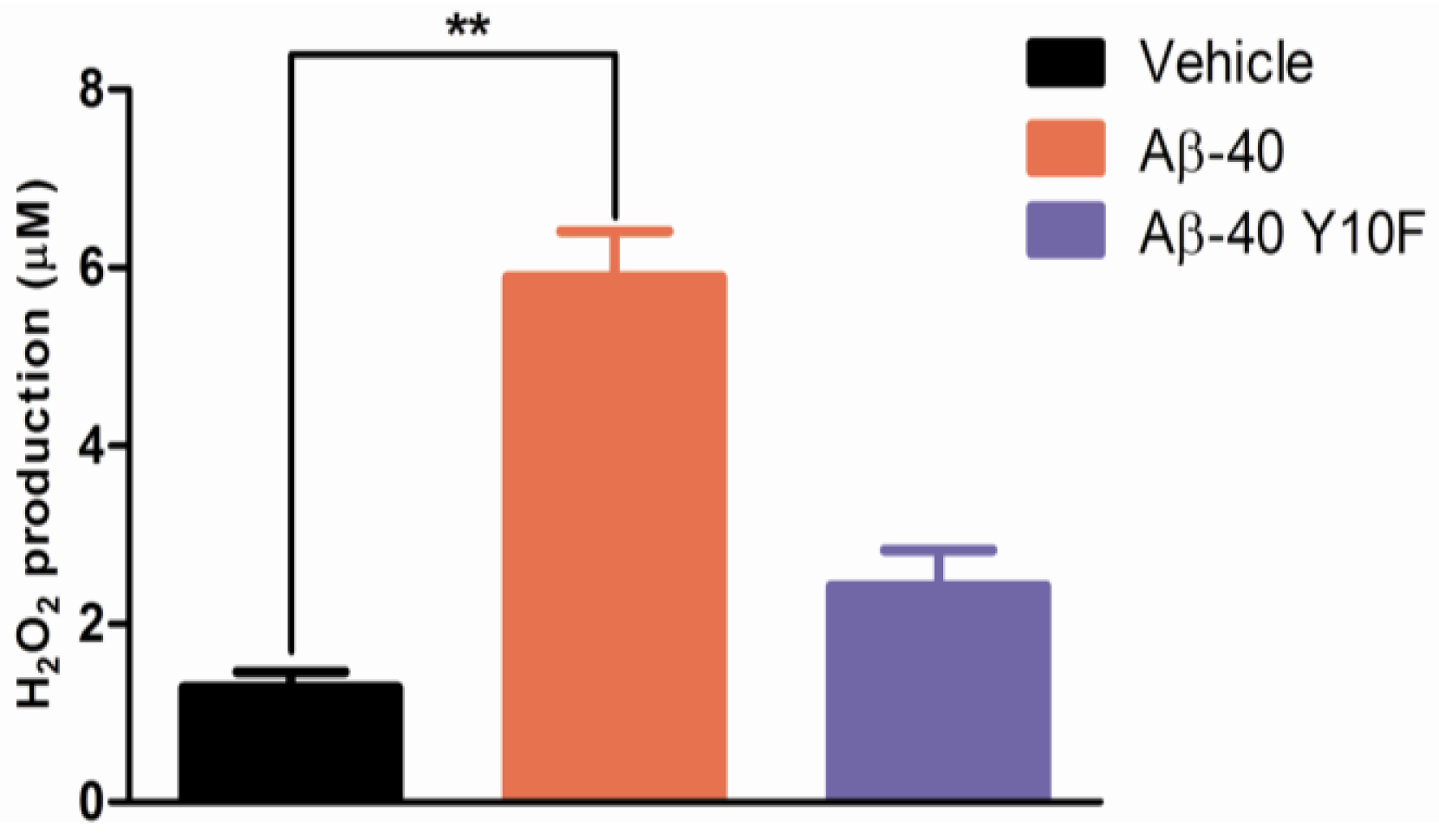
2.3. Tyrosine Substitution Rendered Aβ-40 Less Toxic via Reduced Generation of Hydrogen Peroxide
3. Experimental Section
3.1. Materials
3.2. Peptide Preparation
3.3. Circular Dichroism Spectroscopy
3.4. Fluorescence Spectroscopy (ThT Assay)
3.5. Transmission Electron Microscopy
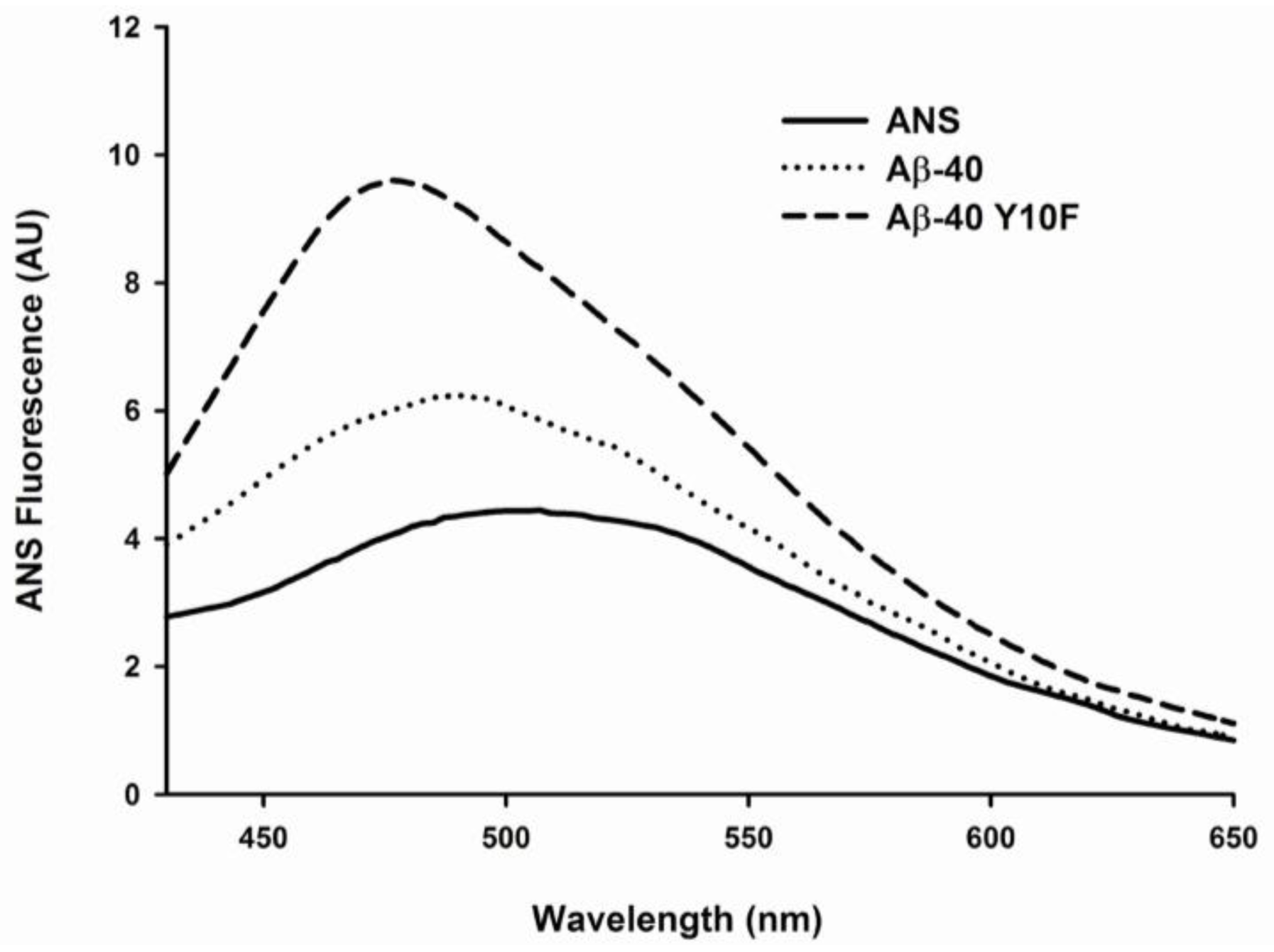
3.6. 1-Anilinonaphthaleine-8-Sulphonic Acid (ANS) Binding
3.7. Primary Neuronal Cultures
3.8. Cell Viability Assay
3.9. Hydrogen Peroxide Assay
4. Conclusions
Acknowledgments
References
- Selkoe, D.J. Alzheimer’s disease is a synaptic failure. Science 2002, 298, 789–791. [Google Scholar]
- Sisodia, S.S.; Koo, E.H.; Beyreuther, K.; Unterbeck, A.; Price, D.L. Evidence that beta-amyloid protein in Alzheimer’s disease is not derived by normal processing. Science 1990, 248, 492–495. [Google Scholar]
- Seubert, P.; Vigo-Pelfrey, C.; Esch, F.; Lee, M.; Dovey, H.; Davis, D.; Sinha, S.; Schiossmacher, M.; Whaley, J.; Swindlehurst, C. Isolation and quantification of soluble Alzheimer’s β-peptide from biological fluids. Nature 1992, 359, 325–327. [Google Scholar]
- Hardy, J.; Selkoe, D.J. The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeutics. Science 2002, 297, 353–356. [Google Scholar]
- Dahlgren, K.N.; Manelli, A.M.; Stine, W.B.; Baker, L.K.; Krafft, G.A.; LaDu, M.J. Oligomeric and fibrillar species of amyloid-β peptides differentially affect neuronal viability. J. Biol. Chem 2002, 277, 32046–32053. [Google Scholar]
- McLean, C.A.; Cherny, R.A.; Fraser, F.W.; Fuller, S.J.; Smith, M.J.; Beyreuther, K.; Bush, A.I.; Masters, C.L. Soluble pool of Aβ amyloid as a determinant of severity of neurodegeneration in Alzheimer’s disease. Ann. Neurol 1999, 46, 860–866. [Google Scholar]
- Davis, S.; Laroche, S. What can rodent models tell us about cognitive decline in Alzheimer’s disease? Mol. Neurobiol 2003, 27, 249–276. [Google Scholar]
- Dai, X.L.; Sun, Y.X.; Jiang, Z.F. Attenuated cytotoxicity but enhanced βfibril of a mutant amyloid β-peptide with a methionine to cysteine substitution. FEBS Lett 2007, 581, 1269–1274. [Google Scholar]
- Dai, X.; Sun, Y.; Gao, Z.; Jiang, Z. Copper enhances amyloid-β peptide neurotoxicity and non β-aggregation: A series of experiments conducted upon copper-bound and copper-free amyloid-β peptide. J. Mol. Neurosci 2010, 41, 66–73. [Google Scholar]
- Walsh, D.M.; Klyubin, I.; Fadeeva, J.V.; Cullen, W.K.; Anwyl, R.; Wolfe, M.S.; Rowan, M.J.; Selkoe, D.J. Naturally secreted oligomers of amyloid β protein potently inhibit hippocampal long-term potentiation in vivo. Nature 2002, 416, 535–539. [Google Scholar]
- Selkoe, D.J. Soluble oligomers of the amyloid β-protein impair synaptic plasticity and behavior. Behav. Brain Res 2008, 192, 106–113. [Google Scholar]
- Hensley, K.; Maidt, M.L.; Yu, Z.; Sang, H.; Markesbery, W.R.; Floyd, R.A. Electrochemical analysis of protein nitrotyrosine and dityrosine in the Alzheimer brain indicates region-specific accumulation. J. Neurosci 1998, 18, 8126–8132. [Google Scholar]
- Yoburn, J.C.; Tian, W.; Brower, J.O.; Nowick, J.S.; Glabe, C.G.; van Vranken, D.L. Dityrosine cross-linked Aβ peptides: Fibrillar β-structure in Aβ(1–40) is conducive to formation of dityrosine cross-links but a dityrosine cross-link in Aβ(8–14) does not induce β-structure. Chem. Res. Toxicol 2003, 16, 531–535. [Google Scholar]
- Barnham, K.J.; Haeffner, F.; Ciccotosto, G.D.; Curtain, C.C.; Tew, D.; Mavros, C.; Beyreuther, K.; Carrington, D.; Masters, C.L.; Cherny, R.A. Tyrosine gated electron transfer is key to the toxic mechanism of Alzheimer’s disease-amyloid. FASEB J 2004, 18, 1427–1429. [Google Scholar]
- Ali, F.E.; Leung, A.; Cherny, R.A.; Mavros, C.; Barnham, K.J.; Separovic, F.; Barrow, C.J. Dimerisation of N-acetyl-l-tyrosine ethyl ester and Aβ peptides via formation of dityrosine. Free Radic. Res 2006, 40, 1–9. [Google Scholar]
- Atwood, C.S.; Perry, G.; Zeng, H.; Kato, Y.; Jones, W.D.; Ling, K.Q.; Huang, X.; Moir, R.D.; Wang, D.; Sayre, L.M.; et al. Copper mediates dityrosine cross-linking of Alzheimer’s amyloid-β. Biochemistry 2004, 43, 560–568. [Google Scholar]
- Yankner, B.A.; Duffy, L.K.; Kirschner, D.A. Neurotrophic and neurotoxic effects of amyloid β protein: Reversal by tachykinin neuropeptides. Science 1990, 250, 279–282. [Google Scholar]
- Smith, D.G.; Cappai, R.; Barnham, K.J. The redox chemistry of the Alzheimer’s disease amyloid β peptide. Biochim. Biophys. Acta 2007, 1768, 1976–1990. [Google Scholar]
- Hardy, J. Amyloid, the presenilins and Alzheimer’s disease. Trends Neurosci 1997, 20, 154–159. [Google Scholar]
- Yoshiike, Y.; Tanemura, K.; Murayama, O.; Akagi, T.; Murayama, M.; Sato, S.; Sun, X.; Tanaka, N.; Takashima, A. New insights on how metals disrupt amyloid β-aggregation and their effects on amyloid-β cytotoxicity. J. Biol. Chem 2001, 276, 32293–32299. [Google Scholar]
- Murakami, K.; Irie, K.; Ohigashi, H.; Hara, H.; Nagao, M.; Shimizu, T.; Shirasawa, T. Formation and stabilization model of the 42-mer Aβ radical: Implications for the long-lasting oxidative stress in Alzheimer’s disease. J. Am. Chem. Soc 2005, 127, 15168–15174. [Google Scholar]
- Uversky, V.N.; Fink, A.L. Conformational constraints for amyloid fibrillation: The importance of being unfolded. Biochim. Biophys. Acta 2004, 1698, 131–153. [Google Scholar]
- Kheterpal, I.; Lashuel, H.A.; Hartley, D.M.; Walz, T.; Lansbury, P.T., Jr; Wetzel, R. Aβ protofibrils possess a stable core structure resistant to hydrogen exchange. Biochemistry 2003, 42, 14092–14098. [Google Scholar]
- Shivaprasad, S.; Wetzel, R. Scanning cysteine mutagenesis analysis of Aβ-(1–40) amyloid fibrils. J. Biol. Chem 2006, 281, 993–1000. [Google Scholar]
- Williams, A.D.; Sega, M.; Chen, M.; Kheterpal, I.; Geva, M.; Berthelier, V.; Kaleta, D.T.; Cook, K.D.; Wetzel, R. Structural properties of Aβ protofibrils stabilized by a small molecule. Proc. Natl. Acad. Sci. USA 2005, 102, 7115–7120. [Google Scholar]
- Jan, A.; Gokce, O.; Luthi-Carter, R.; Lashuel, H.A. The ratio of monomeric to aggregated forms of Aβ40 and Aβ42 is an important determinant of amyloid-β aggregation, fibrillogenesis, and toxicity. J. Biol. Chem 2008, 283, 28176–28189. [Google Scholar]
- Hegde, M.L.; Rao, K.S. DNA induces folding in alpha-synuclein: Understanding the mechanism using chaperone property of osmolytes. Arch. Biochem. Biophys 2007, 464, 57–69. [Google Scholar]
- Chamberlain, A.K.; MacPhee, C.E.; Zurdo, J.; Morozova-Roche, L.A.; Hill, H.A.; Dobson, C.M.; Davis, J.J. Ultrastructural organization of amyloid fibrils by atomic force microscopy. Biophys. J 2000, 79, 3282–3293. [Google Scholar]
- Jan, A.; Hartley, D.M.; Lashuel, H.A. Preparation and characterization of toxic Aβ aggregates for structural and functional studies in Alzheimer’s disease research. Nat. Protoc 2010, 5, 1186–1209. [Google Scholar]
- Qahwash, I.; Weiland, K.L.; Lu, Y.; Sarver, R.W.; Kletzien, R.F.; Yan, R. Identification of a mutant amyloid peptide that predominantly forms neurotoxic protofibrillar aggregates. J. Biol. Chem 2003, 278, 23187–23195. [Google Scholar]
- Butterfield, D.A.; Reed, T.; Newman, S.F.; Sultana, R. Roles of amyloid β-peptide-associated oxidative stress and brain protein modifications in the pathogenesis of Alzheimer’s disease and mild cognitive impairment. Free Radic. Biol. Med 2007, 43, 658–677. [Google Scholar]
- Walsh, D.M.; Selkoe, D.J. Aβ oligomers—A decade of discovery. J. Neurochem 2007, 101, 1172–1184. [Google Scholar]
- Necula, M.; Breydo, L.; Milton, S.; Kayed, R.; van der Veer, W.E.; Tone, P.; Glabe, C.G. Methylene blue inhibits amyloid Aβ oligomerization by promoting fibrillization. Biochemistry 2007, 46, 8850–8860. [Google Scholar]
- Boyd-Kimball, D.; Sultana, R.; Mohmmad-Abdul, H.; Butterfield, D.A. Rodent Aβ (1–42) exhibits oxidative stress properties similar to those of human Aβ (1–42): Implications for proposed mechanisms of toxicity. J. Alzheimers Dis 2004, 6, 515–525. [Google Scholar]
- Butterfield, D.A.; Boyd-Kimball, D. Amyloid β-peptide (1–42) contributes to the oxidative stress and neurodegeneration found in Alzheimer disease brain. Brain Pathol 2004, 14, 426–432. [Google Scholar]
- Butterfield, D.A.; Lauderback, C.M. Lipid peroxidation and protein oxidation in Alzheimer’s disease brain: Potential causes and consequences involving amyloid [β]-peptide-associated free radical oxidative stress1, 2. Free Radic. Biol. Med 2002, 32, 1050–1060. [Google Scholar]
- Castillo, G.M.; Ngo, C.; Cummings, J.; Wight, T.N.; Snow, A.D. Perlecan binds to the β-amyloid proteins (A β) of Alzheimer’s disease, accelerates A β fibril formation, and maintains A β fibril stability. J. Neurochem 1997, 69, 2452–2465. [Google Scholar]
- LeVine, H., 3rd. Quantification of β-sheet amyloid fibril structures with thioflavin T. Methods Enzymol 1999, 309, 274–284. [Google Scholar]
- Walsh, D.M.; Lomakin, A.; Benedek, G.B.; Condron, M.M.; Teplow, D.B. Amyloid β-protein fibrillogenesis: Detection of a protofibrillar intermediate. J. Biol. Chem 1997, 272, 22364–22372. [Google Scholar]
- Ory, J.J.; Banaszak, L.J. Studies of the ligand binding reaction of adipocyte lipid binding protein using the fluorescent probe 1, 8-anilinonaphthalene-8-sulfonate. Biophys. J 1999, 77, 1107–1116. [Google Scholar]
- Pastukhov, A.V.; Ropson, I.J. Fluorescent dyes as probes to study lipid-binding proteins. Proteins 2003, 53, 607–615. [Google Scholar]
- Malich, G.; Markovic, B.; Winder, C. The sensitivity and specificity of the MTS tetrazolium assay for detecting the in vitro cytotoxicity of 20 chemicals using human cell lines. Toxicology 1997, 124, 179–192. [Google Scholar]
- Syme, C.D.; Viles, J.H. Solution 1H NMR investigation of Zn2+ and Cd2+ binding to amyloid-β peptide (Aβ) of Alzheimer’s disease. Biochim. Biophys. Acta 2006, 1764, 246–256. [Google Scholar]
- Hartley, D.M.; Walsh, D.M.; Ye, C.P.; Diehl, T.; Vasquez, S.; Vassilev, P.M.; Teplow, D.B.; Selkoe, D.J. Protofibrillar intermediates of amyloid β-protein induce acute electrophysiological changes and progressive neurotoxicity in cortical neurons. J. Neurosci 1999, 19, 8876–8884. [Google Scholar]
- Lashuel, H.A.; Hartley, D.; Petre, B.M.; Walz, T.; Lansbury, P.T. Neurodegenerative disease: Amyloid pores from pathogenic mutations. Nature 2002, 418. [Google Scholar] [CrossRef]
- Maezawa, I.; Zimin, P.I.; Wulff, H.; Jin, L.-W. Amyloid-β protein oligomer at low nanomolar concentrations activates microglia and induces microglial neurotoxicity. J. Biol. Chem 2011, 286, 3693–3706. [Google Scholar]
© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Dai, X.; Chang, P.; Liu, W.; Xu, K.; Sun, Y.; Zhu, S.; Jiang, Z. Aβ-40 Y10F Increases βfibrils Formation but Attenuates the Neurotoxicity of Amyloid-β Peptide. Int. J. Mol. Sci. 2012, 13, 5324-5337. https://doi.org/10.3390/ijms13055324
Dai X, Chang P, Liu W, Xu K, Sun Y, Zhu S, Jiang Z. Aβ-40 Y10F Increases βfibrils Formation but Attenuates the Neurotoxicity of Amyloid-β Peptide. International Journal of Molecular Sciences. 2012; 13(5):5324-5337. https://doi.org/10.3390/ijms13055324
Chicago/Turabian StyleDai, Xueling, Ping Chang, Wenjuan Liu, Ke Xu, Yaxuan Sun, Shigong Zhu, and Zhaofeng Jiang. 2012. "Aβ-40 Y10F Increases βfibrils Formation but Attenuates the Neurotoxicity of Amyloid-β Peptide" International Journal of Molecular Sciences 13, no. 5: 5324-5337. https://doi.org/10.3390/ijms13055324