Rational Mutagenesis of Cyclodextrin Glucanotransferase at the Calcium Binding Regions for Enhancement of Thermostability


Abstract
:1. Introduction
2. Results and Discussion
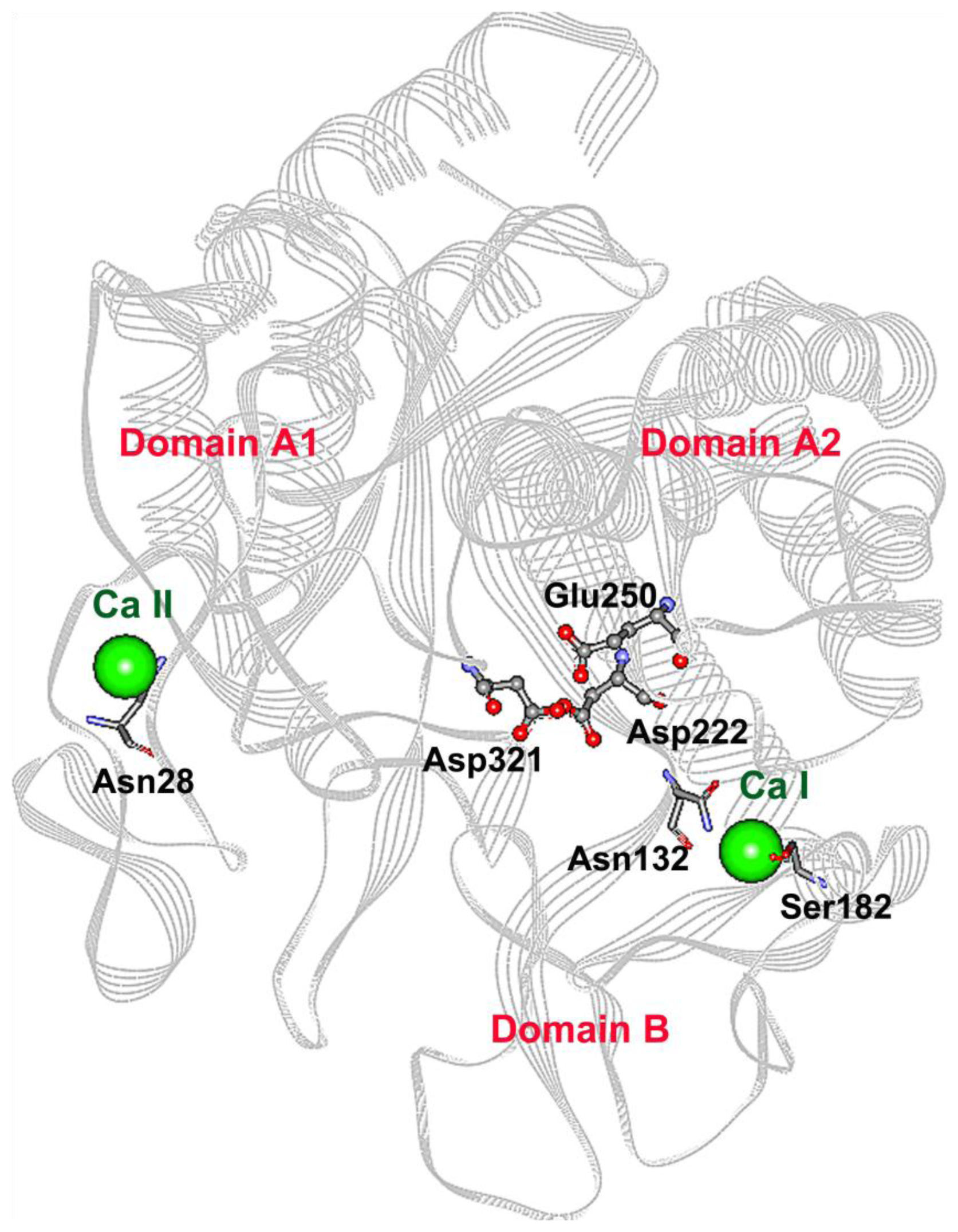
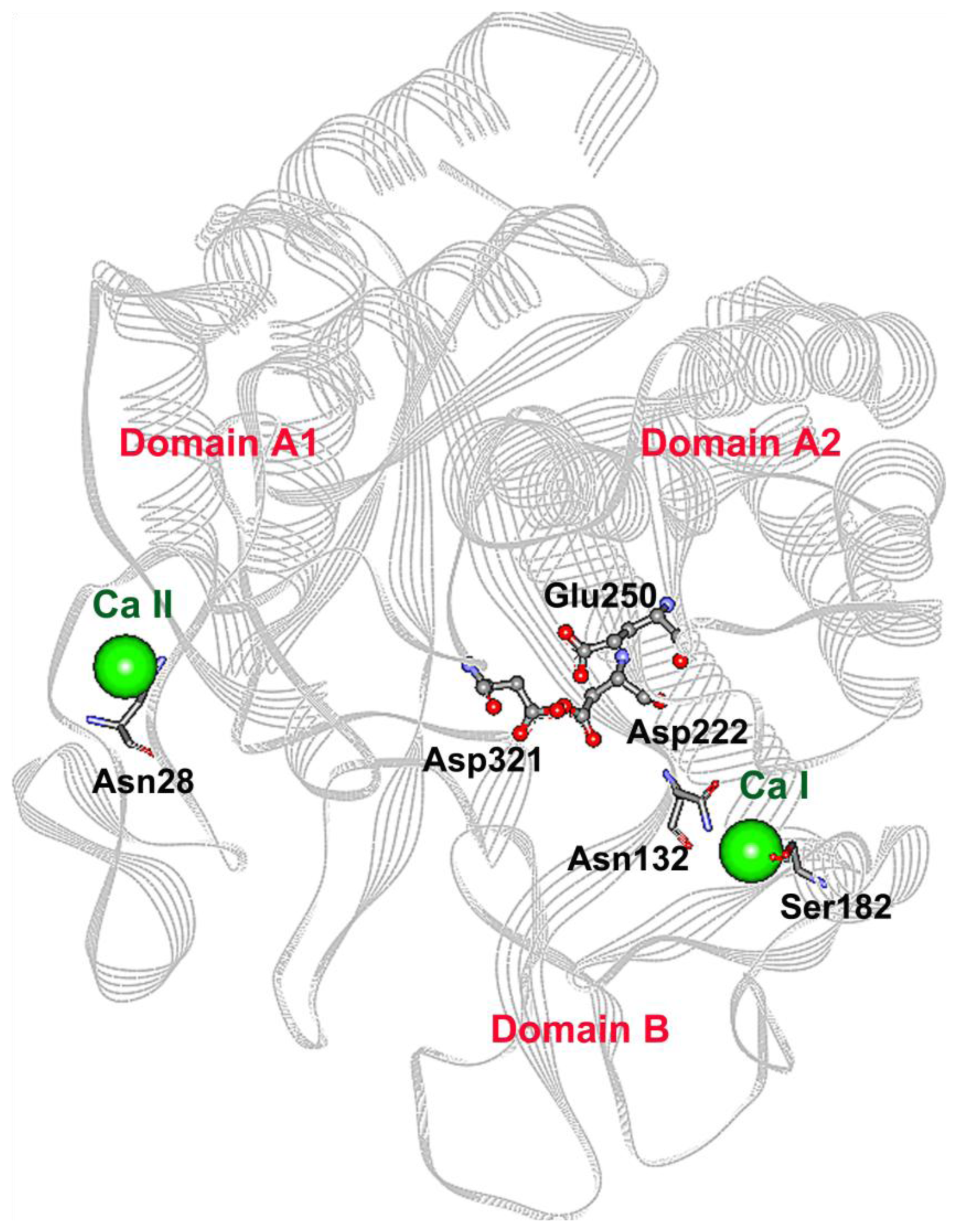
2.1. Mutagenesis Design
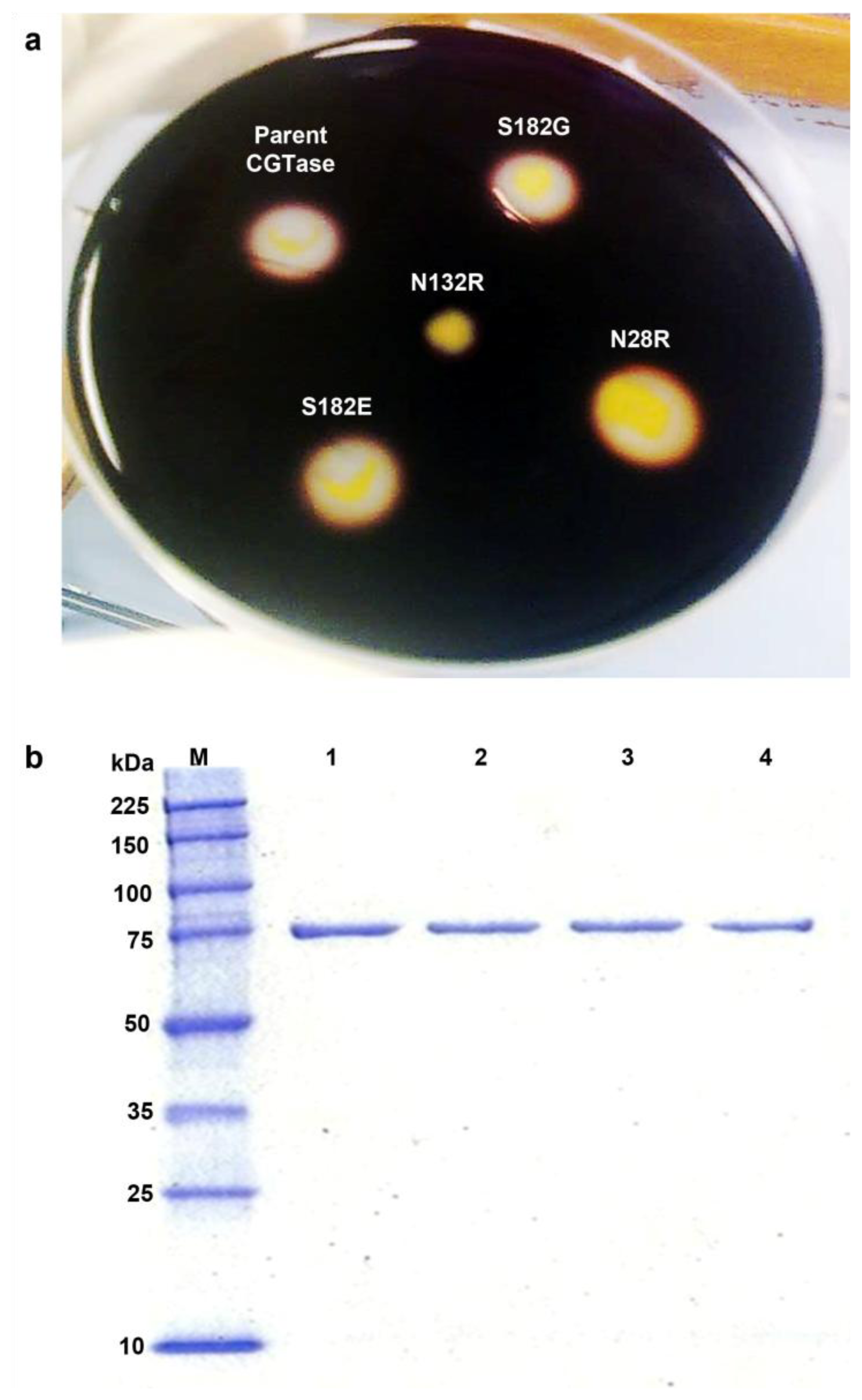
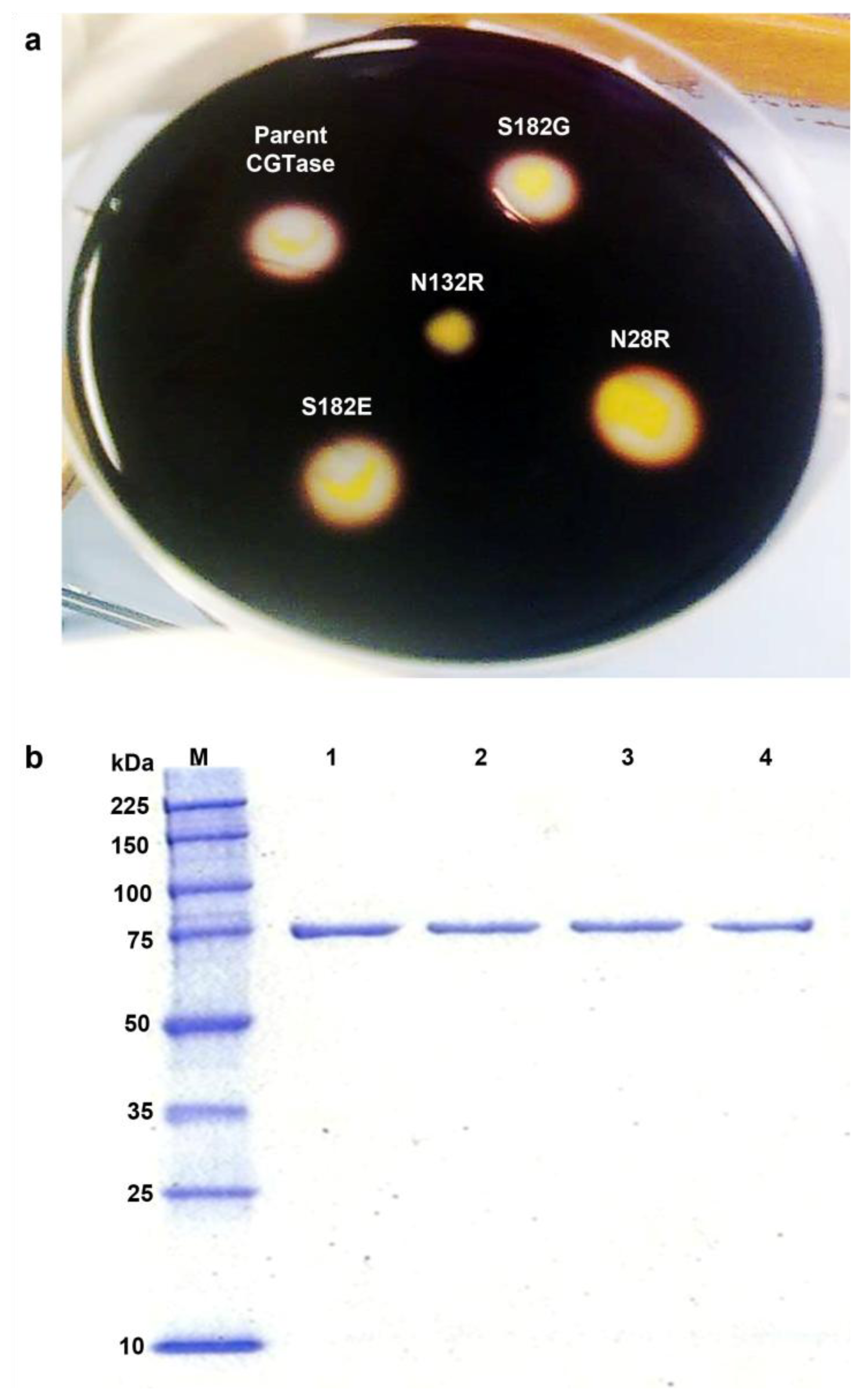
2.2. Activity Screening and Purification of Parent and Mutant CGTases
2.3. Mutations Adjacent to Calcium Binding Pocket I, S182G and S182E, Affected CGTase Thermostability
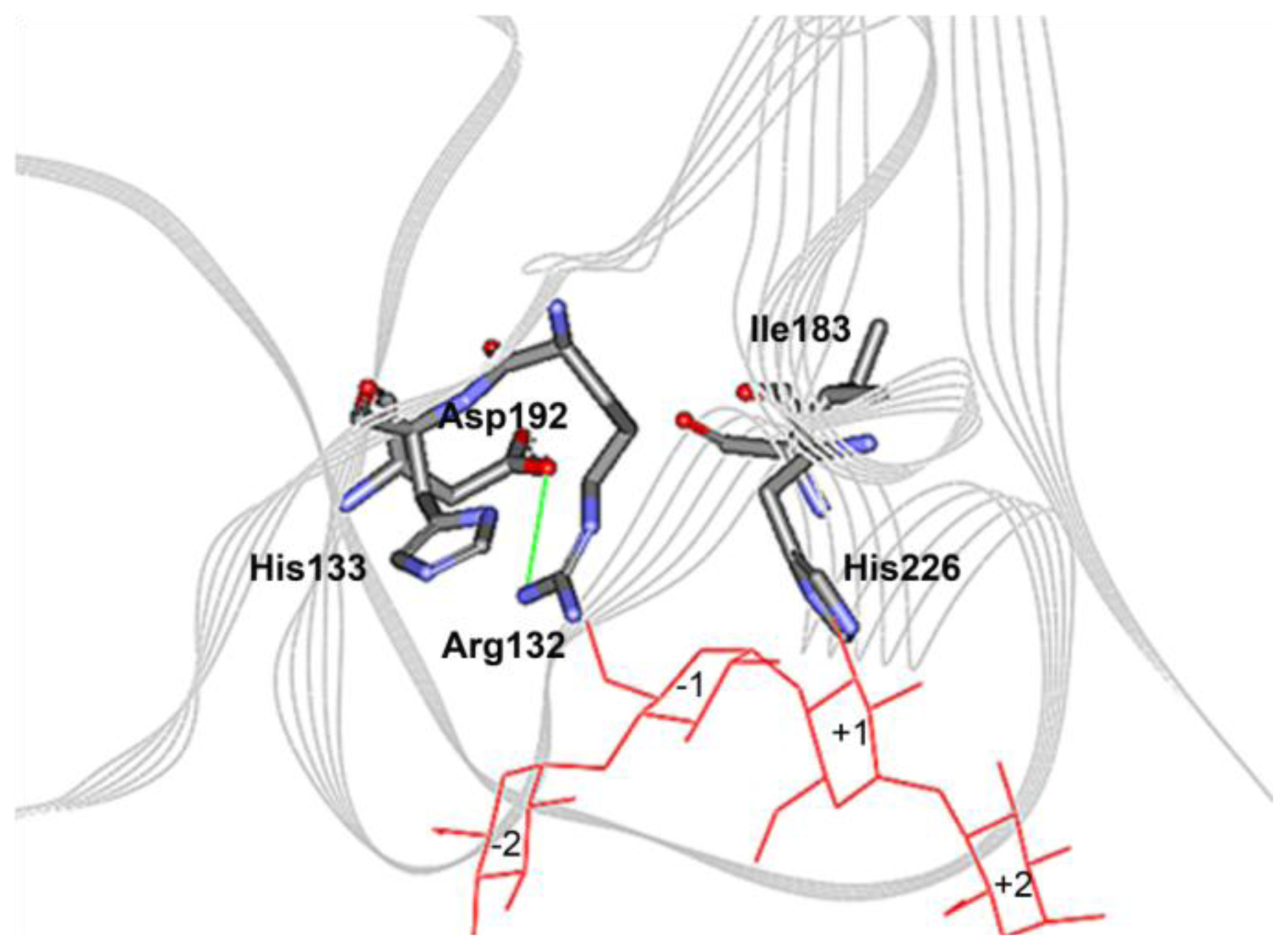
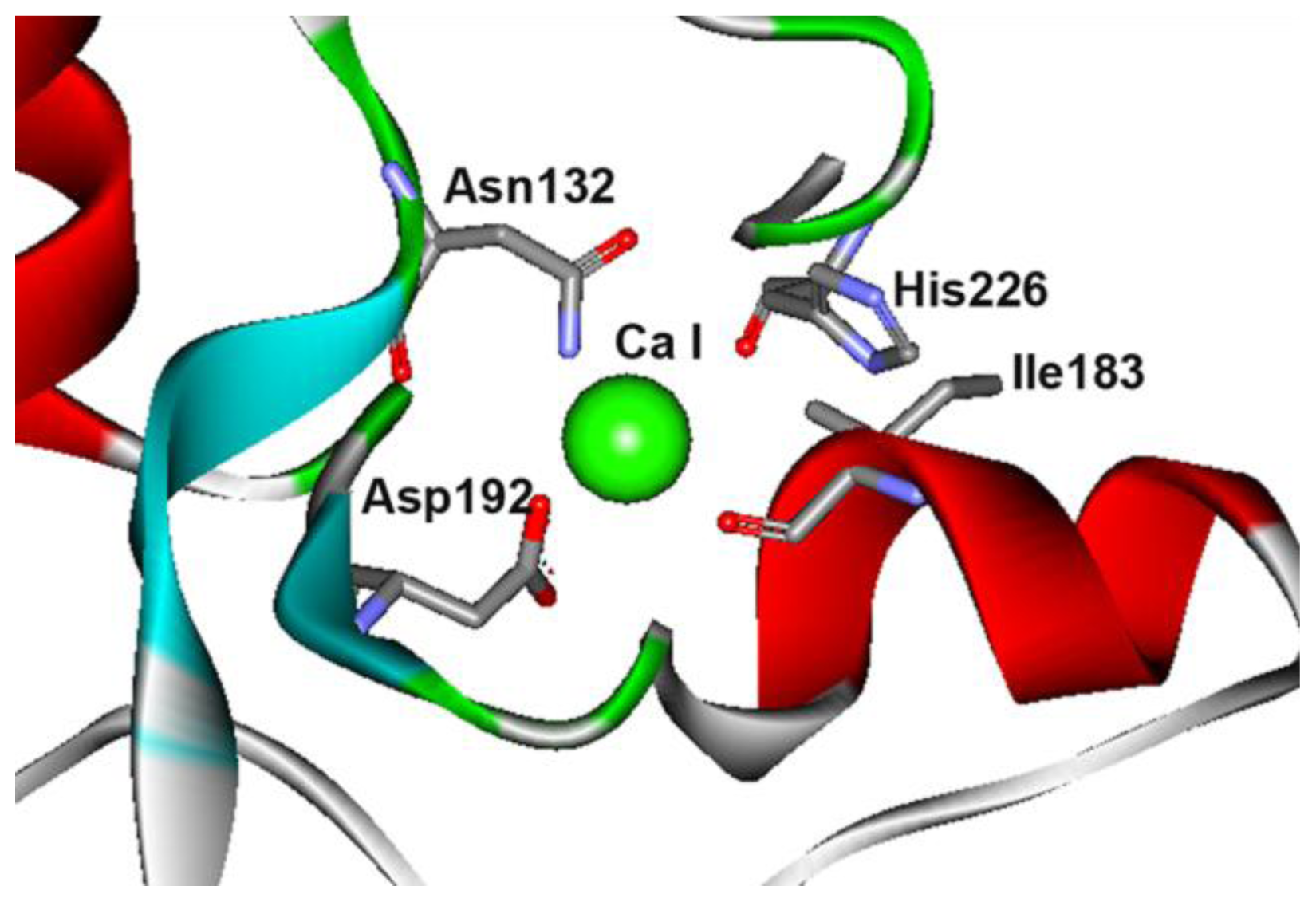
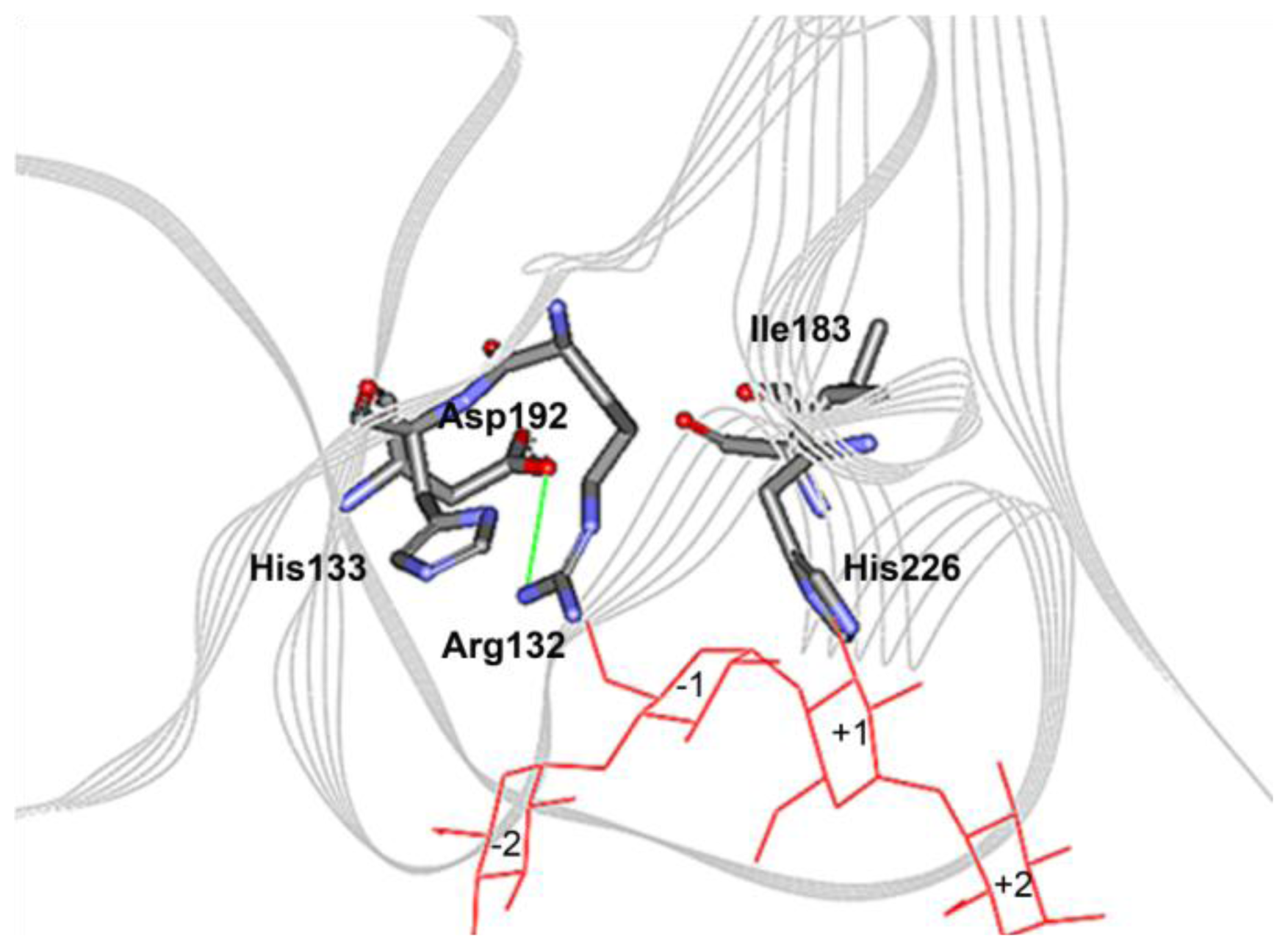
2.4. The N132R Mutation at the Calcium Binding Site I Caused Enzyme Dysfunction
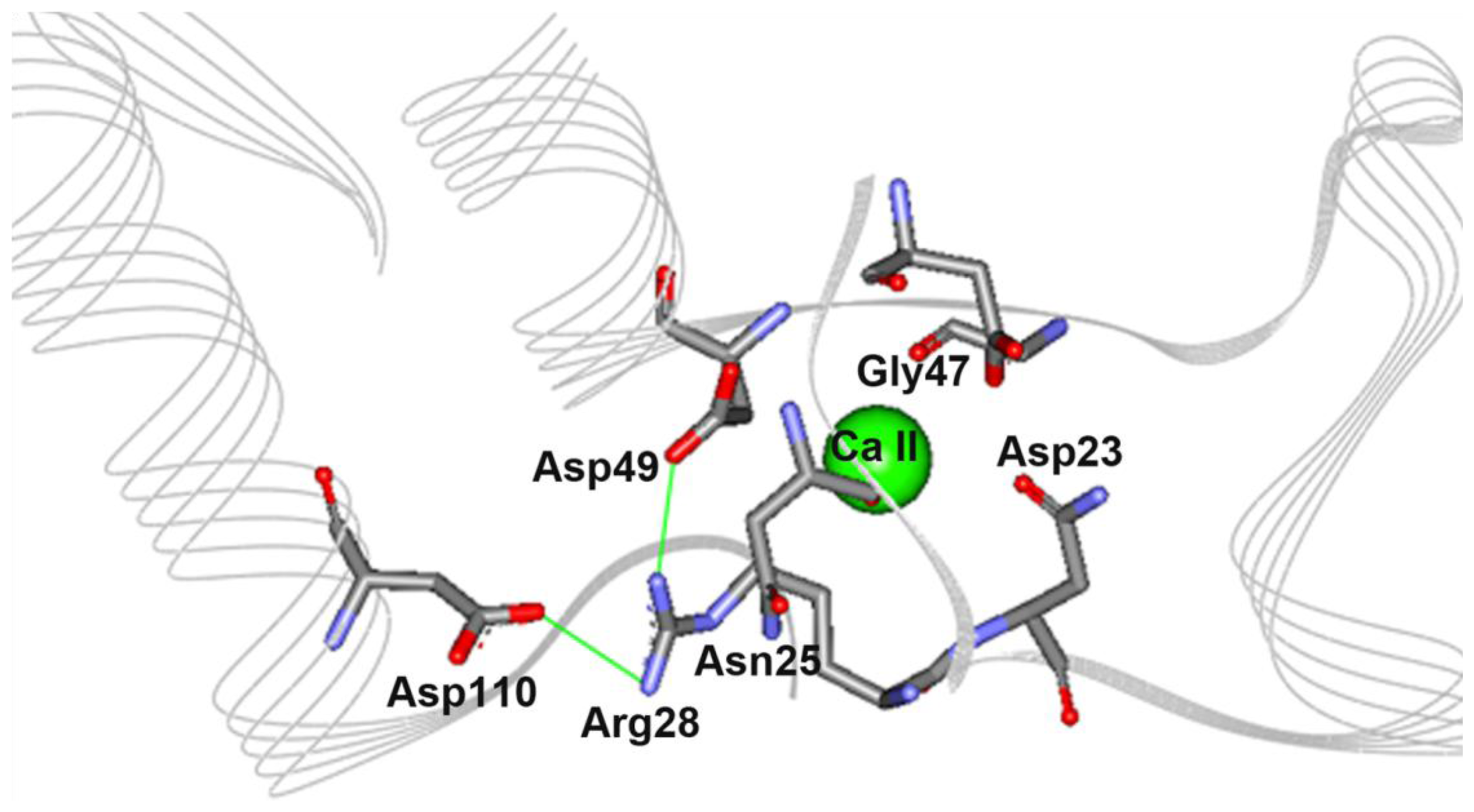
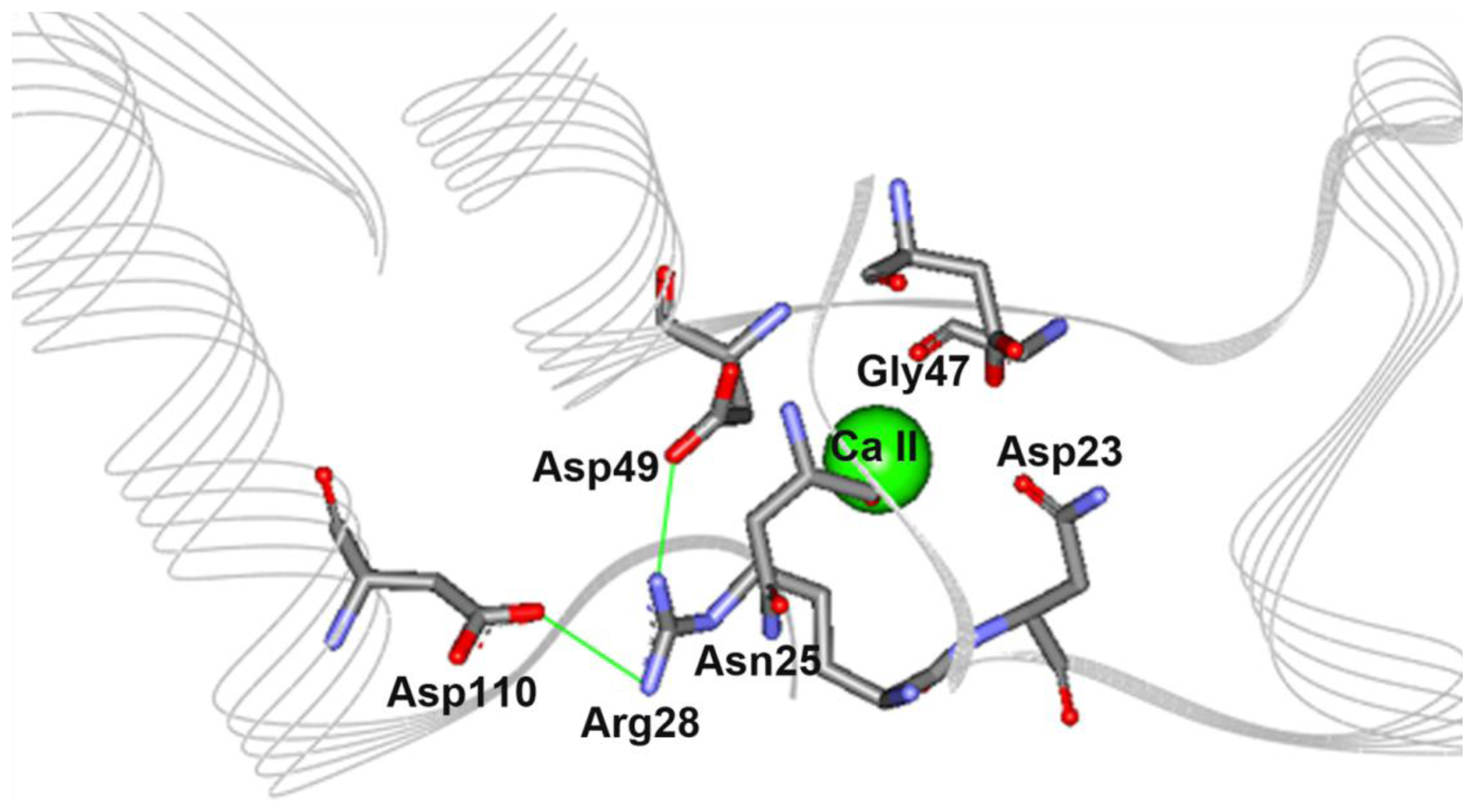
2.5. The N28R Mutation at the Calcium Binding Site II Affected CGTase Activity
2.6. Product Analysis by HPLC
3. Experimental Section
3.1. Bacterial Strains and Plasmids
3.2. Site-Directed Mutagenesis
3.3. Production and Purification of Recombinant Enzymes
3.4. SDS-PAGE
3.5. Protein Determination
3.6. CGTase Assays
3.7. Effects of Temperature on Enzyme Activity and Stability
3.8. Kinetic Studies
3.9. HPLC Product Analysis
3.10. Tertiary Structure Modeling
3.11. Protein Sequence Alignment
4. Conclusions
Acknowledgments
References
- Leemhuis, H.; Kelly, R.M.; Dijkhuizen, L. Engineering of cyclodextrin glucanotransferase and the impact for biotechnological applications. Appl. Microbiol. Biotechnol 2010, 85, 823–835. [Google Scholar]
- Fujiwara, S.; Kakihara, H.; Sakaguchi, K.; Imanaka, T. Analysis of mutations in cyclodextrin glucanotransferase from Bacillus stearothermophilus which affect cyclization characteristics and thermostability. J. Bacteriol 1992, 174, 7478–7481. [Google Scholar]
- Kelly, R.; Dijkhuizen, L.; Leemhuis, H. The evolution of cyclodextrin glucanotransferase product specificity. Appl. Microbiol. Biotechnol 2009, 84, 119–133. [Google Scholar]
- Li, Z.; Zhang, J.; Wang, M.; Gu, Z.; Du, G.; Li, J.; Wu, J.; Chen, J. Mutations at subsite −3 in cyclodextrin glycosyltransferase from Paenibacillus macerans enhancing α-cyclodextrin specificity. Appl. Microbiol. Biotechnol 2009, 83, 483–490. [Google Scholar]
- Costa, H.; Distéfano, A.J.; Marino-Buslje, C.; Hidalgo, A.; Berenguer, J.; Biscoglio de Jiménez Bonino, M.; Ferrarotti, S.A. The residue 179 is involved in product specificity of the Bacillus circulans DF 9R cyclodextrin glycosyltransferase. Appl. Microbiol. Biotechnol 2011, 94, 123–130. [Google Scholar]
- Goh, K.M.; Mahadi, N.M.; Hassan, O.; Rahman, R.N.Z.R.A.; Illias, R.M. A predominant β-CGTase G1 engineered to elucidate the relationship between protein structure product specificity. J. Mol. Catal. B Enzym 2009, 57, 270–277. [Google Scholar]
- Wind, R.D.; Liebl, W.; Buitelaar, R.M.; Penninga, D.; Spreinat, A.; Dijkhuizen, L.; Bahl, H. Cyclodextrin formation by the thermostable alpha-amylase of Thermoanaerobacterium thermosulfurigenes EM1 and reclassification of the enzyme as a cyclodextrin glycosyltransferase. Appl. Environ. Microbiol 1995, 61, 1257–1265. [Google Scholar]
- Sian, H.K.; Said, M.; Hassan, O.; Kamaruddin, K.; Ismail, A.F.; Rahman, R.A.; Mahmood, N.A.N.; Illias, R.M. Purification and characterization of cyclodextrin glucanotransferase from alkalophilic Bacillus sp. G1. Process Biochem 2005, 40, 1101–1111. [Google Scholar]
- Ferrarotti, S.A.; Bolivar, J.M.; Mateo, C.; Wilson, L.; Guisan, J.M.; Fernandez-Lafuente, R. Immobilization and stabilization of a cyclodextrin glycosyltransferase by covalent attachment on highly activated glyoxyl-agarose supports. Biotechnol. Prog 2006, 22, 1140–1145. [Google Scholar]
- Martín, M.T.; Plou, F.J.; Alcalde, M.; Ballesteros, A. Immobilization on Eupergit C of cyclodextrin glucosyltransferase (CGTase) and properties of the immobilized biocatalyst. J. Mol. Catal. B Enzym 2003, 21, 299–308. [Google Scholar]
- Leemhuis, H.; Rozeboom, H.J.; Dijkstra, B.W.; Dijkhuizen, L. Improved thermostability of Bacillus circulans cyclodextrin glycosyltransferase by the introduction of a salt bridge. Proteins 2004, 54, 128–134. [Google Scholar]
- Fu, Y.; Ding, Y.; Wang, Z.; Sun, J.; Fang, W.; Xu, W. Study on the relationship between cyclodextrin glycosyltransferase thermostability and salt bridge formation by molecular dynamics simulation. Protein Pept. Lett 2011, 17, 1403–1411. [Google Scholar]
- Declerck, N.; Machius, M.; Joyet, P.; Wiegand, G.; Huber, O.; Gaillardin, C. Engineering the thermostability of Bacillus licheniformis α-amylase. Biologia 2002, 57, 203–211. [Google Scholar]
- Priyadharshini, R.; Gunasekaran, P. Site-directed mutagenesis of the calcium-binding site of α-amylase of Bacillus licheniformis. Biotechnol. Lett 2007, 29, 1493–1499. [Google Scholar]
- Liu, Y.; Shen, W.; Shi, G.-Y.; Wang, Z.-X. Role of the calcium-binding residues Asp231, Asp233, and Asp438 in alpha-amylase of Bacillus amyloliquefaciens as revealed by mutational analysis. Curr. Microbiol 2010, 60, 162–166. [Google Scholar]
- Kuriki, T.; Imanaka, T. The concept of the α-amylase family: Structural similarity and common catalytic mechanism. J. Biosci. Bioeng 1999, 87, 557–565. [Google Scholar]
- Lawson, C.L.; van Montfort, R.; Strokopytov, B.; Rozeboom, H.J.; Kalk, K.H.; de Vries, G.E.; Penninga, D.; Dijkhuizen, L.; Dijkstra, B.W. Nucleotide sequence and X-ray structure of cyclodextrin glycosyltransferase from Bacillus circulans strain 251 in a maltose-dependent crystal form. J. Mol. Biol 1994, 236, 590–600. [Google Scholar]
- Khemakhem, B.; Ali, M.B.; Aghajari, N.; Juy, M.; Haser, R.; Bejar, S. The importance of an extra loop in the B-domain of an α-amylase from B. stearothermophilus US100. Biochem. Biophys. Res. Commun 2009, 385, 78–83. [Google Scholar]
- Ong, R.M.; Goh, K.M.; Mahadi, N.M.; Hassan, O.; Rahman, R.N.Z.R.A.; Illias, R.M. Cloning, extracellular expression and characterization of a predominant β-CGTase from Bacillus sp. G1 in E. coli. J. Ind. Mirobiol 2008, 35, 1705–1714. [Google Scholar]
- Hirano, K.; Ishihara, T.; Ogasawara, S.; Maeda, H.; Abe, K.; Nakajima, T.; Yamagata, Y. Molecular cloning and characterization of a novel γ-CGTase from alkalophilic Bacillus sp. Appl. Microbiol. Biotechnol 2006, 70, 193–201. [Google Scholar]
- Chung, H.J.; Yoon, S.Y.; Lee, M.J.; Lim, M.J.; Kweon, K.S.; Lee, I.W.; Kim, J.W.; Oh, B.H.; Lee, H.S.; Spiridonova, V.A.; Park, K.H. Characterization of a thermostable cyclodextrin glucanotransferase isolated from Bacillus stearothermophilus ET1. J. Agric. Food Chem 1998, 46, 952–959. [Google Scholar]
- Norman, B.E.; Jørgensen, S. Thermoanaerobacter sp. CGTase: Its properties and application. J. Jpn. Soc. Starch Sci 1992, 39, 101–108. [Google Scholar]
- Anbar, M.; Lamed, R.; Bayer, E.A. Thermostability enhancement of Clostridium thermocellum cellulosomal endoglucanase Cel8A by a single glycine substitution. ChemCatChem 2010, 2, 997–1003. [Google Scholar]
- Trevino, S.R.; Schaefer, S.; Scholtz, J.M.; Pace, C.N. Increasing protein conformational stability by optimizing β-turn sequence. J. Mol. Biol 2007, 373, 211–218. [Google Scholar]
- Nakamura, A.; Haga, K.; Yamane, K. Three histidine residues in the active center of cyclodextrin glucanotransferase from alkalophilic Bacillus sp. 1011: Effects of the replacement on pH dependence and transition-state stabilization. Biochemistry 1993, 32, 6624–6631. [Google Scholar]
- Goh, K.M.; Mahadi, N.M.; Hassan, O.; Abdul Rahman, R.N.Z.R.; Illias, R.M. The effects of reaction conditions on the production of γ-cyclodextrin from tapioca starch by using a novel recombinant engineered CGTase. J. Mol. Catal. B Enzym 2007, 49, 118–126. [Google Scholar]
- Knegtel, R.M.; Strokopytov, B.; Penninga, D.; Faber, O.G.; Rozeboom, H.J.; Kalk, K.H.; Dijkhuizen, L.; Dijkstra, B.W. Crystallographic studies of the interaction of cyclodextrin glycosyltransferase from Bacillus circulans strain 251 with natural substrates and products. J. Biol. Chem 1995, 270, 29256–29264. [Google Scholar]
- Larkin, M.A.; Blackshields, G.; Brown, N.P.; Chenna, R.; McGettigan, P.A.; McWilliam, H.; Valentin, F.; Wallace, I.M.; Wilm, A.; Lopez, R.; Thompson, J.D.; Gibson, T.J.; Higgins, D.G. Clustal W and Clustal X version 2.0. Bioinformatics 2007, 23, 2947–2948. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CGTase | Optimum temperature (°C) | Half-life, t1/2 (min) | Reference |
|---|---|---|---|
| Parent CGTase (this study) | 60 | 22 min at 60 °C | |
| Bacillus circulans 251 | 60 | 9.7 min at 60 °C | [11] |
| Bacillus sp. strain G-825-6 | 50–55 | nd * | [20] |
| Bacillus stearothermophilus ET1 | 80 | 10 min at 80 °C | [21] |
| Thermoanaerobacterium thermosulfurigenes EM1 | 80–95 | 30 min at 100 °C | [7] |
| Thermoanaerobacter sp. ATCC 53627 | 95 | nd * | [22] |
| CGTase | Specific activity (U mg−1) | ||
|---|---|---|---|
| β-CD cyclization | γ-CD cyclization | Coupling | |
| Parent | 216 (8.11) | 1.08 (0.01) | 0.67 (0.01) |
| N28R | 359 (23.46) | 2.13 (0.04) | 0.67 (0.01) |
| S182G | 162 (10.59) | 1.08 (0.02) | 0.70 (0.03) |
| S182E | 178 (22.29) | 0.76 (0.14) | 0.77 (0.04) |
| Enzyme | kcat (s−1) | Efficiency, kcat/Km (mL·mg−1·s−1) |
|---|---|---|
| Parent | 3.13 ± 0.13 | 1.82 ± 0.001 |
| N28R | 3.52 ± 0.17 | 2.52 ± 0.35 |
| S182G | 2.62 ± 0.12 | 2.00 ± 0.05 |
| S182E | 2.57 ± 0.22 | 1.82 ±0.27 |
| Enzyme | Conversion of starch into cyclodextrins (%) | ||||
|---|---|---|---|---|---|
| Tapioca starch | Soluble starch | Rice starch | Corn starch | Potato starch | |
| Parent | 9.01 | 6.73 | 7.86 | 3.31 | 4.11 |
| N28R | 10.26 | 7.84 | 9.74 | 4.78 | 5.48 |
| S182G | 14.01 | 10.75 | 13.31 | 6.48 | 7.41 |
| S182E | 12.01 | 9.62 | 10.81 | 5.49 | 6.73 |
| Name of primer | Sequence * |
|---|---|
| EFP_BamHI | 5′-CTCGGATCCGGACGTAACAAACAAAGTCAATTACTC-3′ |
| ERP_HindIII | 5′-GCCAAGCTTCCAATTAATCATAACCGTATCTGTTCCGG-3′ |
| IFP_N28R | 5′-CTGGCcgCAATCCTTCAGGCGCTATCTTTAG-3′ |
| IRP_N28R | 5′-AAGGATTGcgGCCAG GATTCCCGTCAGAGAATCG-3′ |
| IFP_N132R | 5′-CACGCCAcgcCATTCATCACCGGCACTTGAAACG-3′ |
| IRP_N132R | 5′-GATGAATGgcgTGGCGTGAAATCCATGATTACC-3′ |
| IFP_S182G | 5′-GATgGCATTTACAGAAACTTATATGATCTGGCAG-3′ |
| IRP_S182G | 5′-GTTTCTGTAAATGCcATCTTCATATGAAGAGAAATCTG-3′ |
| IFP_S182E | 5′-GAAGATgaaATTTACAGAAACTTATATGATCTGGCAG-3′ |
| IRP_S182E | 5′-TCTGTAAATttcATCTTCATATGAAGAGAAATCTG-3′ |
© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Goh, P.H.; Illias, R.M.; Goh, K.M. Rational Mutagenesis of Cyclodextrin Glucanotransferase at the Calcium Binding Regions for Enhancement of Thermostability. Int. J. Mol. Sci. 2012, 13, 5307-5323. https://doi.org/10.3390/ijms13055307
Goh PH, Illias RM, Goh KM. Rational Mutagenesis of Cyclodextrin Glucanotransferase at the Calcium Binding Regions for Enhancement of Thermostability. International Journal of Molecular Sciences. 2012; 13(5):5307-5323. https://doi.org/10.3390/ijms13055307
Chicago/Turabian StyleGoh, Poh Hong, Rosli Md. Illias, and Kian Mau Goh. 2012. "Rational Mutagenesis of Cyclodextrin Glucanotransferase at the Calcium Binding Regions for Enhancement of Thermostability" International Journal of Molecular Sciences 13, no. 5: 5307-5323. https://doi.org/10.3390/ijms13055307