Identification of Novel Potential β-N-Acetyl-D-Hexosaminidase Inhibitors by Virtual Screening, Molecular Dynamics Simulation and MM-PBSA Calculations

Abstract
:1. Introduction
2. Results and Discussion
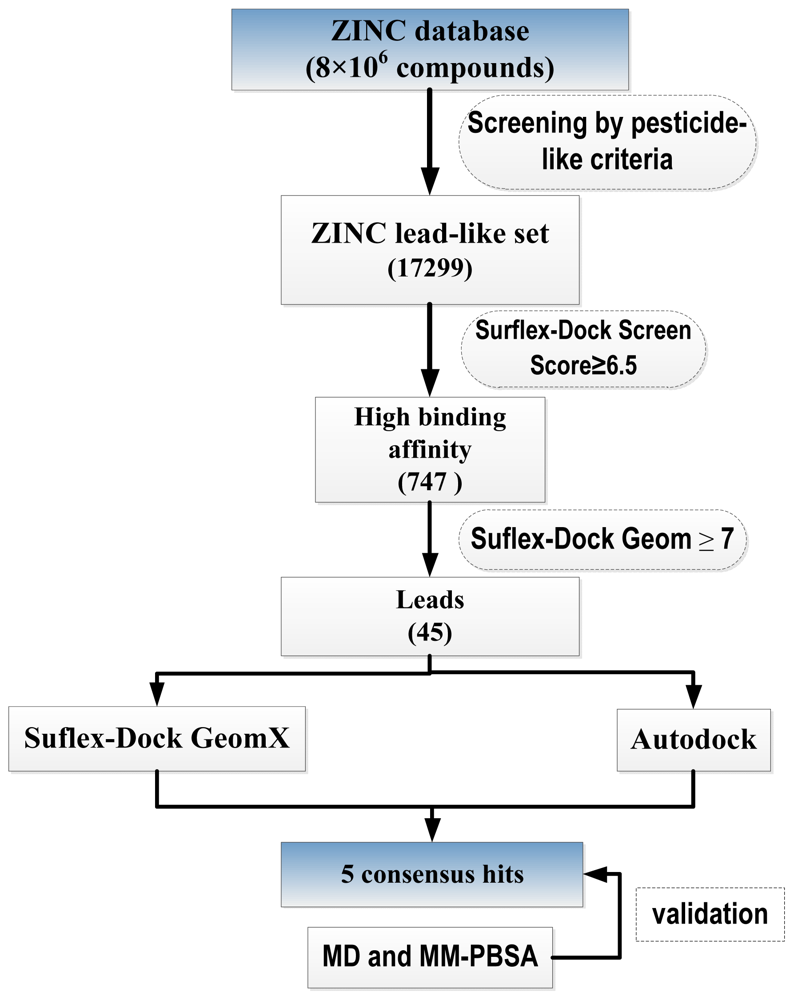
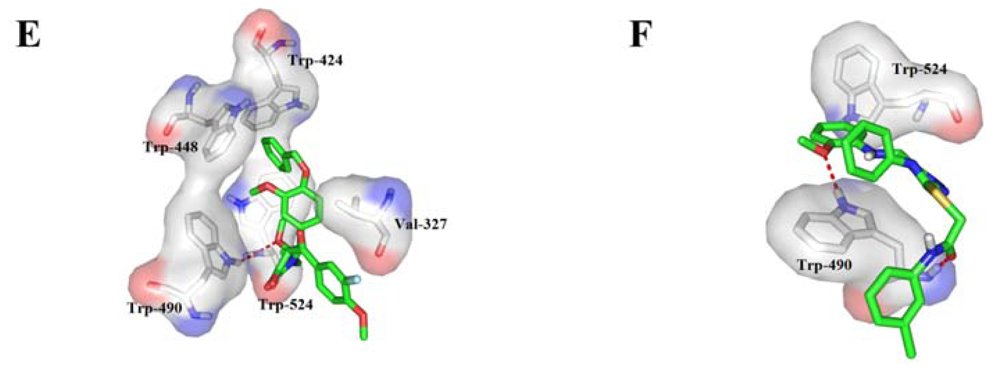
2.1. Virtual Screening
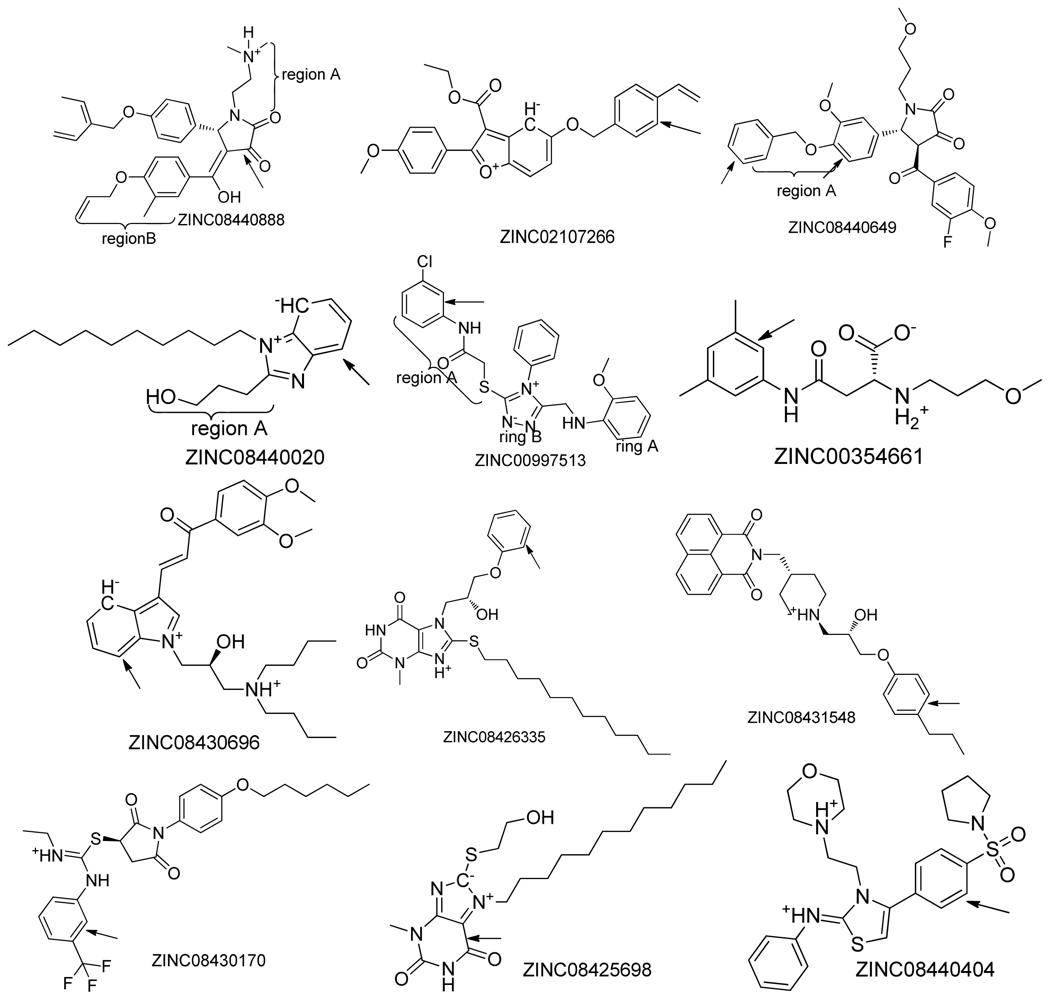
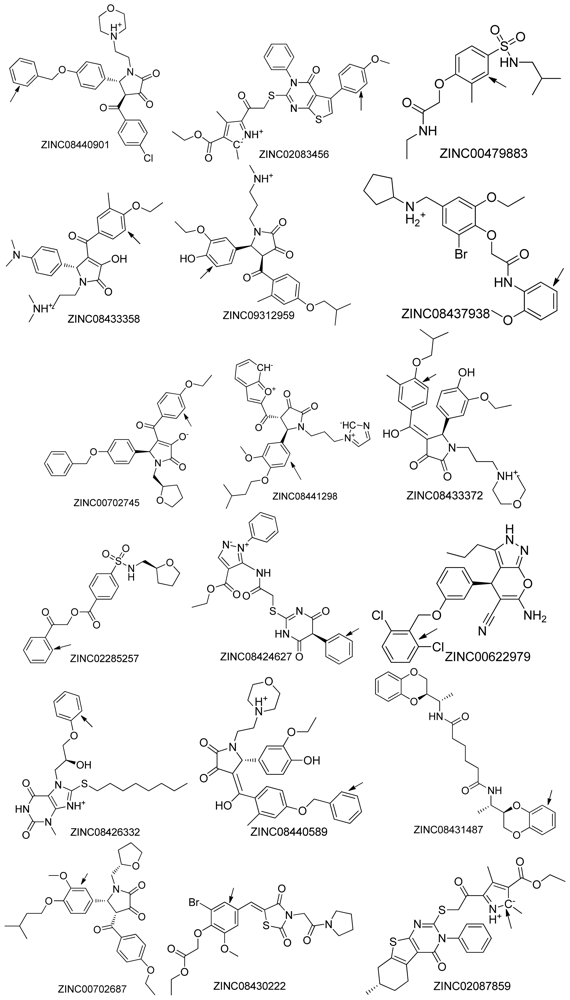
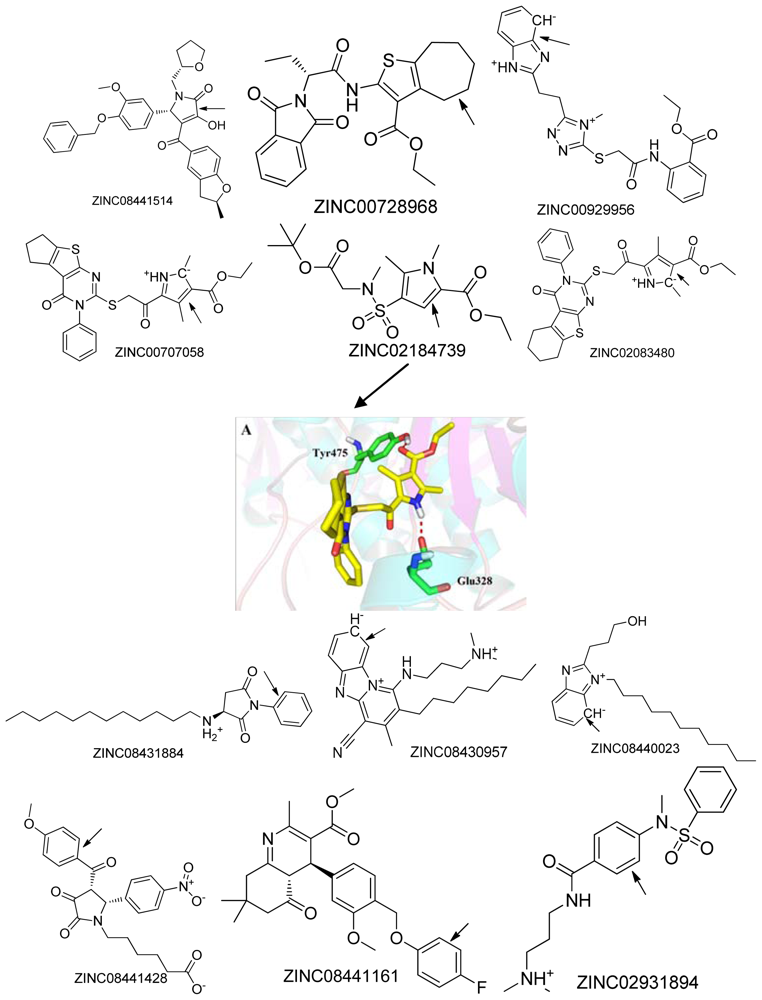
2.2. Top Scoring Compounds
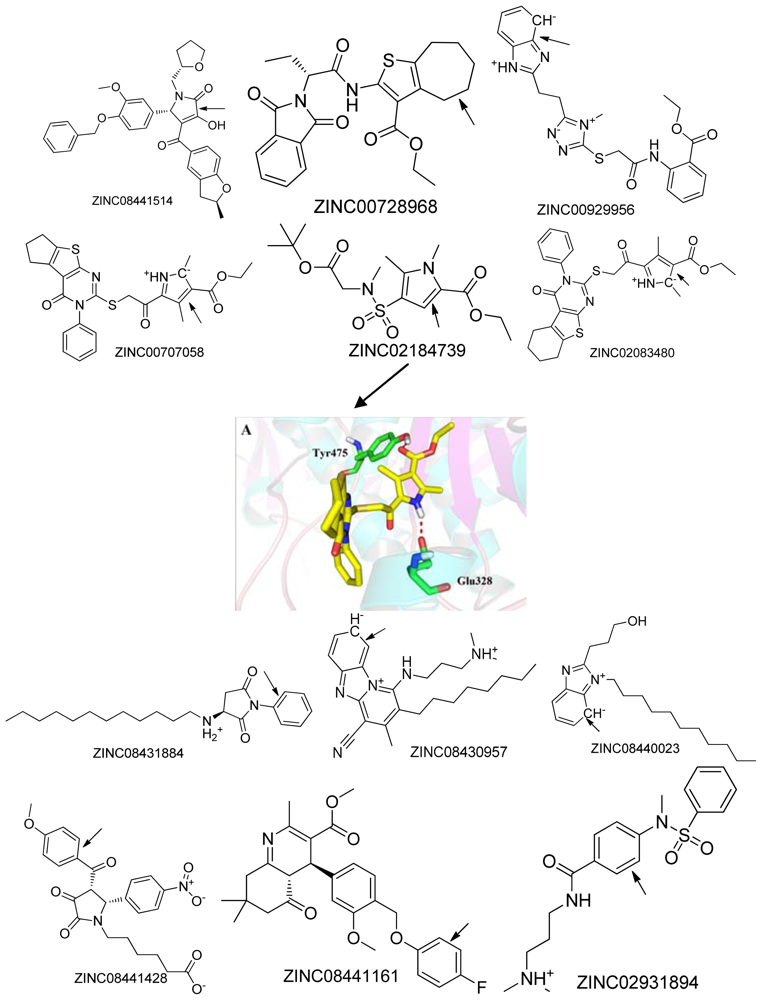
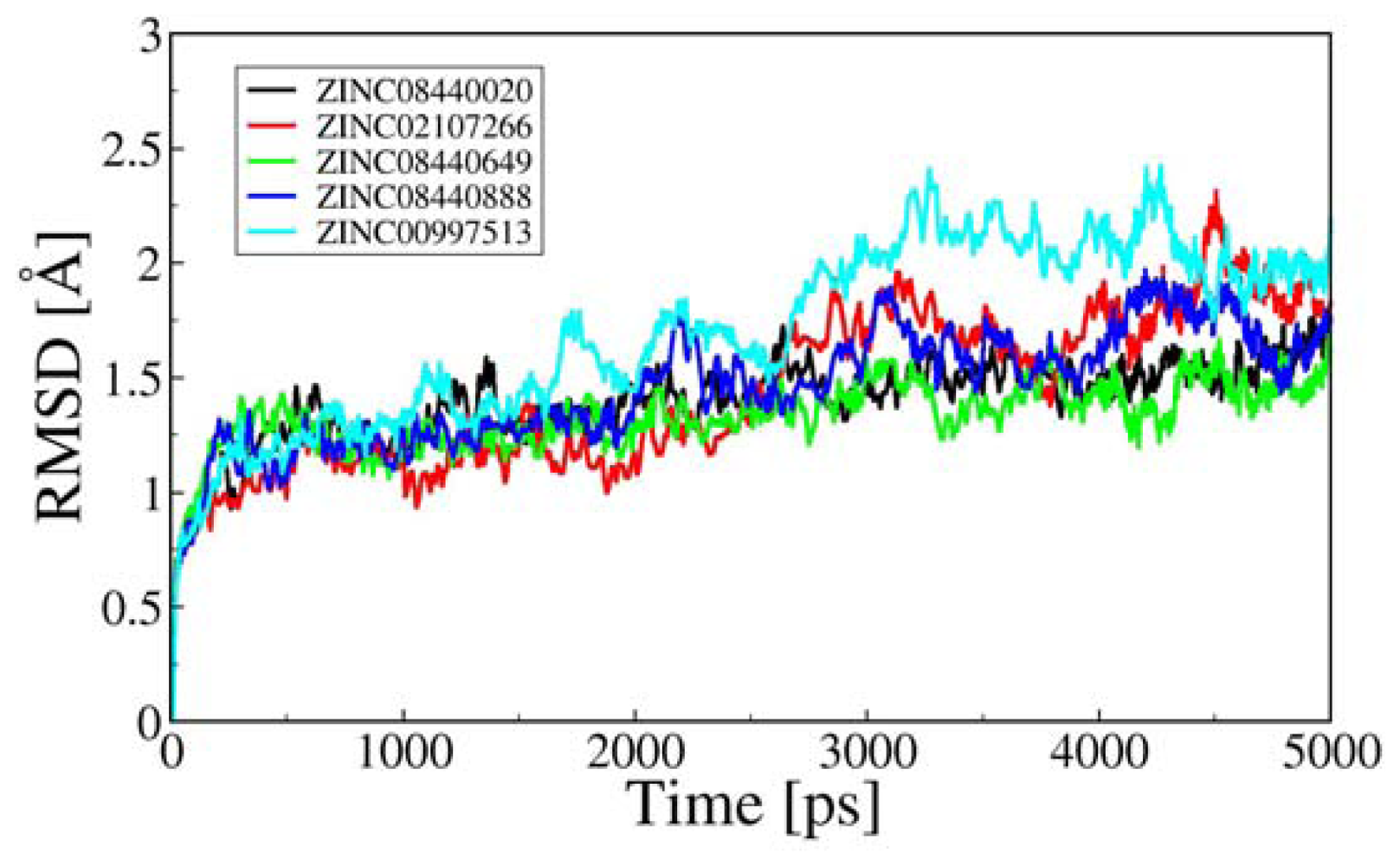
2.3. Molecular Dynamics Simulation
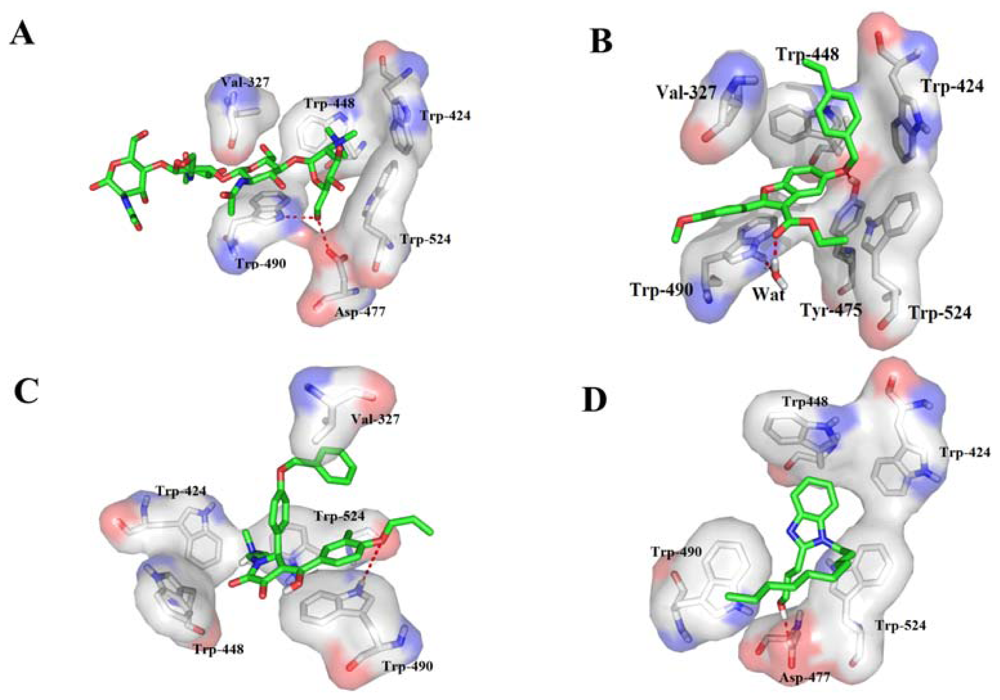
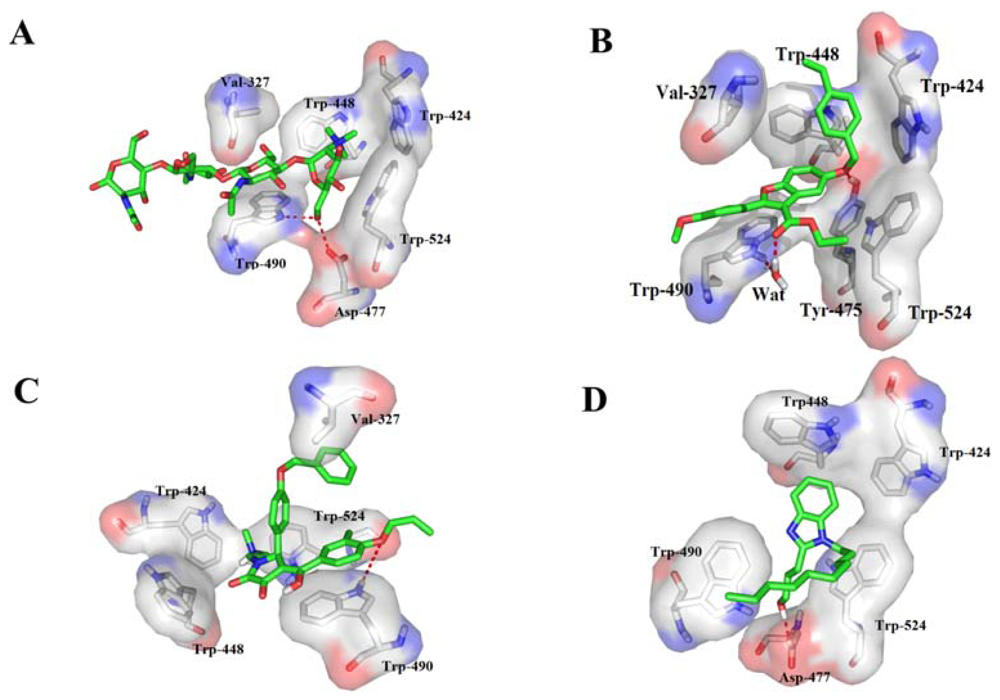
2.4. Binding Mode
2.5. Binding Free Energy Calculation
3. Materials and Methods
3.1. Lead-like Selection
3.2. Docking with Surflex-Dock
3.3. Docking with AUTODOCK
3.4. Molecular Dynamics (MD) Simulations
3.5. MM/PBSA Calculations
4. Conclusions
Acknowledgements
References
- Cohen, E. Chitin synthesis and degradation as targets for pesticide action. Arch. Insect Biochem. Physiol 1993, 22, 245–261. [Google Scholar]
- Kramer, K.J.; Koga, D. Insect chitin: Physical state, synthesis, degradation and metabolic regulation. Insect Biochem 1986, 16, 851–877. [Google Scholar]
- Jeuniaux, C. [111] Chitinases. Methods Enzymol. 1966, 8, 644–650. [Google Scholar]
- Keyhani, N.O.; Roseman, S. Physiological aspects of chitin catabolism in marine bacteria. Biochim. Biophys. Acta 1999, 1473, 108–122. [Google Scholar]
- Fukamizo, T.; Kramer, K.J. Mechanism of chitin hydrolysis by the binary chitinase system in insect moulting fluid. Insect Biochem 1985, 15, 141–145. [Google Scholar]
- Strasser, R.; Bondili, J.S.; Schoberer, J.; Svoboda, B.; Liebminger, E.; Glossl, J.; Altmann, F.; Steinkellner, H.; Mach, L. Enzymatic Properties and subcellular localization of arabidopsis beta-N-acetylhexosaminidases. Plant Physiol 2007, 145, 5–16. [Google Scholar]
- Yi, C.K. Increase in β-N-acetylglucosaminidase activity during germination of cotton seeds. Plant Physiol 1981, 67, 68–73. [Google Scholar]
- Watanabe, K. Biochemical studies on carbohydrates. J. Biochem 1936, 24, 297–303. [Google Scholar]
- Koga, D.; Isogai, A.; Sakuda, S.; Matsumoto, S.; Suzuki, A.; Kimura, S.; Ide, A. Specific inhibition of Bombyx mori chitinase by allosamidin. Agric. Biol. Chem 1987, 51, 471–476. [Google Scholar]
- Arai, N.; Shiomi, K.; Iwai, Y.; Omura, S. Argifin, a new chitinase inhibitor, produced by Gliocladium sp. FTD-0668. II. Isolation, physico-chemical properties, and structure elucidation. J. Antibiot 2000, 53, 609–614. [Google Scholar]
- Arai, N.; Shiomi, K.; Yamaguchi, Y.; Masuma, R.; Iwai, Y.; Turberg, A.; Kolbl, H.; Omura, S. Argadin, a new chitinase inhibitor, produced by Clonostachys sp. FO-7314. Chem. Pharmac. Bull 2000, 48, 1442–1446. [Google Scholar]
- Gouda, H.; Yanai, Y.; Sugawara, A.; Sunazuka, T.; Omura, S.; Hirono, S. Computational analysis of the binding affinities of the natural-product cyclopentapeptides argifin and argadin to chitinase B from Serratia marcescens. Bioorg. Med. Chem 2008, 16, 3565–3579. [Google Scholar]
- Yang, Q.; Liu, T.; Liu, F.; Qu, M.; Qian, X. A novel β-N-acetyl-d-hexosaminidase from the insect Ostrinia furnacalis (Guenée). FEBS J 2008, 275, 5690–5702. [Google Scholar]
- Liu, H.; Wang, X.; Wang, J.; Wang, J.; Li, Y.; Yang, L.; Li, G. Structural determinants of CX-4945 derivatives as protein kinase CK2 inhibitors: A computational study. Int. J. Mol. Sci 2011, 12, 7004–7021. [Google Scholar]
- Wang, X.; Yang, W.; Xu, X.; Zhang, H.; Li, Y.; Wang, Y. Studies of benzothiadiazine derivatives as hepatitis C virus NS5B polymerase inhibitors using 3D-QSAR, molecular docking and molecular dynamics. Curr. Med. Chem 2010, 17, 2788–2803. [Google Scholar]
- Irwin, J.J.; Raushel, F.M.; Shoichet, B.K. Virtual screening against metalloenzymes for inhibitors and substrates. Biochemistry 2005, 44, 12316–12328. [Google Scholar]
- Hou, T.; Chen, K.; McLaughlin, W.A.; Lu, B.; Wang, W. Computational analysis and prediction of the binding motif and protein interacting partners of the Abl SH3 domain. PLoS Comput. Biol 2006, 2. [Google Scholar] [CrossRef]
- Liu, T.; Zhang, H.; Liu, F.; Wu, Q.; Shen, X.; Yang, Q. Structural determinants of an insect β-N-acetyl-d-hexosaminidase specialized as a chitinolytic enzyme. J. Biol. Chem 2011, 286, 4049–4058. [Google Scholar]
- Pranav Kumar, S.; Kulkarni, V.M. Insights into the selective inhibition of Candida albicans secreted aspartyl protease: A docking analysis study. Bioorg. Med. Chem 2002, 10, 1153–1170. [Google Scholar]
- Wang, Y.; Li, Y.; Ma, Z.; Yang, W.; Ai, C. Mechanism of microRNA-target interaction: Molecular dynamics simulations and thermodynamics analysis. PLoS Comput. Biol 2010, 6. [Google Scholar] [CrossRef]
- Xu, X.; Yang, W.; Wang, X.; Li, Y.; Wang, Y.; Ai, C. Dynamic communication between androgen and coactivator: Mutually induced conformational perturbations in androgen receptor ligand-binding domain. Proteins Struct. Funct. Bioinforma 2011, 79, 1154–1171. [Google Scholar]
- Irwin, J.J.; Shoichet, B.K. ZINC-a free database of commercially available compounds for virtual screening. J. Chem. Inf. Model 2005, 45, 177–182. [Google Scholar]
- Liu, B.; Yu, F.; Yao, J.; Liao, Q.; Fan, B. Screening rules of lead compounds of herbicide, fungicide and insecticide. Chin. J. Pestic. Sci 2007, 3, 220–228. [Google Scholar]
- Welch, W.; Ruppert, J.; Jain, A.N. Hammerhead: Fast, fully automated docking of flexible ligands to protein binding sites. Chem. Biol 1996, 3, 449–462. [Google Scholar]
- AutoDock Homepage. Available online: http://autodock.scripps.edu/ accessed on 12 July 2011.
- Medina-Franco, J.L.; López-Vallejo, F.; Kuck, D.; Lyko, F. Natural products as DNA methyltransferase inhibitors: A computer-aided discovery approach. Mol. Divers 2011, 15, 1–12. [Google Scholar]
- Gasteiger, J.; Marsili, M. Iterative partial equalization of orbital electronegativity—A rapid access to atomic charges. Tetrahedron 1980, 36, 3219–3228. [Google Scholar]
- Morris, G.M.; Goodsell, D.S.; Halliday, R.S.; Huey, R.; Hart, W.E.; Belew, R.K.; Olson, A.J. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J. Comput. Chem 1998, 19, 1639–1662. [Google Scholar]
- Case, D.A.; Darden, T.; Cheatham, T.E., III; Simmerling, C.; Wang, J.; Duke, R.E.; Luo, R.; Crowley, M.; Walker, R.; Zhang, W.; et al. AMBER 10; University of California: San Francisco, CA, USA, 2008. [Google Scholar]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem 2004, 25, 1157–1174. [Google Scholar]
- Lee, M.C.; Duan, Y. Distinguish protein decoys by using a scoring function based on a new AMBER force field, short molecular dynamics simulations, and the generalized born solvent model. Proteins Struct. Funct. Bioinforma 2004, 55, 620–634. [Google Scholar]
- Weber, W.; Hünenberger, P.H.; McCammon, J.A. Molecular dynamics simulations of a polyalanine octapeptide under Ewald boundary conditions: Influence of artificial periodicity on peptide conformation. J. Phys. Chem. B 2000, 104, 3668–3675. [Google Scholar]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh Ewald method. J. Chem. Phys 1995, 103, 8577–8593. [Google Scholar]
- Ryckaert, J.P.; Ciccotti, G.; Berendsen, H.J.C. Numerical integration of the cartesian equations of motion of a system with constraints: Molecular dynamics of n-alkanes. J. Comput. Phys 1977, 23, 327–341. [Google Scholar]
- Kollman, P.A.; Massova, I.; Reyes, C.; Kuhn, B.; Huo, S.; Chong, L.; Lee, M.; Lee, T.; Duan, Y.; Wang, W. Calculating structures and free energies of complex molecules: Combining molecular mechanics and continuum models. Acc. Chem. Res 2000, 33, 889–897. [Google Scholar]
- Honig, B.; Nicholls, A. Classical electrostatics in biology and chemistry. Science 1995, 268, 1144–1149. [Google Scholar]
- Kuhn, B.; Kollman, P.A. Binding of a diverse set of ligands to avidin and streptavidin: An accurate quantitative prediction of their relative affinities by a combination of molecular mechanics and continuum solvent models. J. Med. Chem 2000, 43, 3786–3791. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| NO. | Compound (ZINC ID) | MW a | xLogP a | HBA a | HBD a | PSA a | Autodock4 Docking energy b | Surflex score b |
|---|---|---|---|---|---|---|---|---|
| 1 | 08440649 | 535 | 5.07 | 8 | 0 | 102 | −8.65 | 9.06 |
| 2 | 08440888 | 527 | 6.28 | 4 | 1 | 120 | −9.78 | 10.86 |
| 3 | 02107266 | 428 | 7 | 5 | 0 | 63 | −10.2 | 9.03 |
| 4 | 08440020 | 316 | 5.68 | 1 | 1 | 50 | −8.66 | 10.31 |
| 5 | 00997513 | 479 | 4.39 | 4 | 2 | 105 | −8.52 | 8.96 |
| 6 | 02931894 | 376 | 2.7 | 1 | 0 | 119 | −10.42 | 8.6 |
| 7 | 08440404 | 500 | 0 | 1 | 2 | 110 | −10.02 | 9.68 |
| 8 | 08431884 | 359 | 7 | 2 | 2 | 93 | −9.55 | 11.6 |
| 9 | 08440901 | 534 | 5.9 | 5 | 1 | 134 | −9.35 | 8.83 |
| 10 | 08430957 | 420 | 6.2 | 0 | 2 | 86 | −9.12 | 10.15 |
| 11 | 08437938 | 478 | 4.3 | 0 | 3 | 86 | −9.02 | 10.9 |
| 12 | 01823357 | 342 | 5 | 0 | 3 | 96 | −8.7 | 10.1 |
| 13 | 08431492 | 468 | 3 | 0 | 2 | 60 | −8.7 | 9.9 |
| 14 | 08430170 | 522 | 7 | 7 | 2 | 97 | −8.69 | 9.86 |
| 15 | 08424627 | 491 | 3.1 | 0 | 2 | 200 | −8.65 | 10.3 |
| 16 | 02728968 | 454 | 5.9 | 2 | 1 | 107 | −8.65 | 9.3 |
| 17 | 02087859 | 535 | 7.3 | 0 | 0 | 156 | −8.6 | 9.7 |
| 18 | 08430696 | 493 | 6.76 | 4 | 2 | 99 | −8.54 | 11.03 |
| 19 | 08425698 | 410 | 5.3 | 2 | 2 | 158 | −8.53 | 9.29 |
| 20 | 02083480 | 521 | 6.7 | 2 | 1 | 149 | −8.3 | 8.9 |
| 21 | 08441543 | 533 | 6.2 | 0 | 0 | 126 | −8.23 | 9.9 |
| 22 | 00622979 | 455 | 7 | 0 | 3 | 194 | −8.2 | 10.3 |
| 23 | 08441161 | 479 | 5.3 | 1 | 0 | 134 | −8.2 | 8.7 |
| 24 | 08440023 | 330 | 6.2 | 2 | 1 | 51 | −8.19 | 9.76 |
| 25 | 00707058 | 507 | 6.2 | 2 | 1 | 144 | −8.15 | 8.9 |
| 26 | 08426335 | 517 | 5.85 | 1 | 3 | 167 | −8.02 | 10.83 |
| 27 | 08431548 | 487 | 6.4 | 4 | 2 | 87 | −8.02 | 10.68 |
| 28 | 02083456 | 573 | 7 | 0 | 1 | 186 | −7.9 | 8.3 |
| 29 | 08431487 | 468 | 3 | 0 | 2 | 76 | −7.85 | 9.8 |
| 30 | 08441514 | 555 | 5.9 | 0 | 1 | 136 | −7.73 | 9.3 |
| 31 | 09312959 | 511 | 7 | 1 | 2 | 168 | −7.14 | 11.4 |
| 32 | 08440589 | 573 | 4.9 | 0 | 3 | 175 | −7.05 | 9.8 |
| 33 | 08433358 | 527 | 6.2 | 1 | 1 | 120 | −7.01 | 11.4 |
| 34 | 00702745 | 512 | 5.5 | 0 | 0 | 114 | −7.01 | 10.8 |
| 35 | 08433372 | 553 | 4.9 | 0 | 3 | 167 | −6.97 | 10.5 |
| 36 | 00479883 | 328 | 2.4 | 2 | 2 | 100 | −6.84 | 8.5 |
| 37 | 02285257 | 403 | 3 | 0 | 1 | 155 | −6.78 | 10.3 |
| 38 | 08441298 | 543 | 5.3 | 0 | 0 | 154 | −6.69 | 10.7 |
| 39 | 08430222 | 527 | 2.7 | 0 | 0 | 149 | −6.37 | 9.7 |
| 40 | 00702687 | 523 | 5.8 | 0 | 0 | 106 | −6.27 | 9.7 |
| 41 | 00929956 | 464 | 2.6 | 0 | 2 | 134 | −6.12 | 9.2 |
| 42 | 08426332 | 461 | 3.7 | 0 | 3 | 127 | −5.8 | 9.9 |
| 43 | 08441428 | 467 | 3.3 | 0 | 0 | 254 | −5.53 | 8.8 |
| 44 | 02184739 | 374 | 2.8 | 0 | 0 | 96 | −4.98 | 8.9 |
| 45 | 03354661 | 308 | 1.83 | 4 | 3 | 137 | −6.01 | 8.09 |
| Contribution | ZINC08440649 | ZINC08440888 | ZINC02107266 | ZINC08440020 | ZINC00997513 | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| Mean | Std | Mean | Std | Mean | Std | Mean | Std | Mean | Std | |
| ΔEele | −22.31 | 7.86 | −42.52 | 8.21 | −5.99 | 3.44 | −28.59 | 3.77 | −20.28 | 5.51 |
| ΔGvdw | −40.84 | 6.08 | −27.99 | 2.95 | −41.12 | 2.61 | −32.56 | 2.33 | −31.81 | 4.30 |
| ΔGnp | −7.44 | 0.68 | −6.02 | 0.50 | −5.87 | 0.25 | −6.03 | 0.22 | −5.43 | 0.45 |
| ΔGPB | 30.44 | 6.62 | 32.31 | 4.54 | 17.37 | 2.23 | 26.67 | 2.10 | 10.49 | 2.08 |
| ΔGcavity | −7.44 | 0.68 | −6.02 | 0.50 | −5.87 | 0.25 | −6.03 | 0.22 | −5.43 | 0.45 |
| ΔGgas | −63.16 | 11.76 | −70.52 | 8.20 | −47.11 | 4.35 | −61.14 | 4.02 | −52.09 | 8.02 |
| ΔGsol | 23.00 | 6.07 | 26.29 | 4.45 | 11.50 | 2.16 | 20.63 | 2.02 | 5.06 | 1.77 |
| −TΔS | 19.62 | 3.2 | 15.86 | 6.3 | 16.89 | 6.9 | 21.17 | 6.4 | 27.80 | 6.5 |
| ΔGbind a | −40.17 | 6.29 | −44.23 | 4.54 | −35.61 | 3.57 | −40.51 | 3.53 | −47.03 | 6.61 |
| ΔGbind b | −20.55 | - | −28.37 | - | −16.92 | - | −19.34 | - | −19.23 | - |
© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Liu, J.; Liu, M.; Yao, Y.; Wang, J.; Li, Y.; Li, G.; Wang, Y. Identification of Novel Potential β-N-Acetyl-D-Hexosaminidase Inhibitors by Virtual Screening, Molecular Dynamics Simulation and MM-PBSA Calculations. Int. J. Mol. Sci. 2012, 13, 4545-4563. https://doi.org/10.3390/ijms13044545
Liu J, Liu M, Yao Y, Wang J, Li Y, Li G, Wang Y. Identification of Novel Potential β-N-Acetyl-D-Hexosaminidase Inhibitors by Virtual Screening, Molecular Dynamics Simulation and MM-PBSA Calculations. International Journal of Molecular Sciences. 2012; 13(4):4545-4563. https://doi.org/10.3390/ijms13044545
Chicago/Turabian StyleLiu, Jianling, Mengmeng Liu, Yao Yao, Jinan Wang, Yan Li, Guohui Li, and Yonghua Wang. 2012. "Identification of Novel Potential β-N-Acetyl-D-Hexosaminidase Inhibitors by Virtual Screening, Molecular Dynamics Simulation and MM-PBSA Calculations" International Journal of Molecular Sciences 13, no. 4: 4545-4563. https://doi.org/10.3390/ijms13044545