Computational Identification and Modeling of Crosstalk between Phosphorylation, O-β-glycosylation and Methylation of FoxO3 and Implications for Cancer Therapeutics

Abstract
:
1. Introduction
2. Materials and Methods
2.1. Analysis Data
2.2. Prediction of Phosphorylation, Kinases Activity, and Solvent Accessibility of Human FoxO3
2.3. Prediction of O-β-GlcNAc Modifications and Yin Yang Sites in Human FoxO3
2.4. Prediction of Methylation in Human FoxO3
2.5. Selection Parameters for Posttranslational Modifications Residues
3. Results
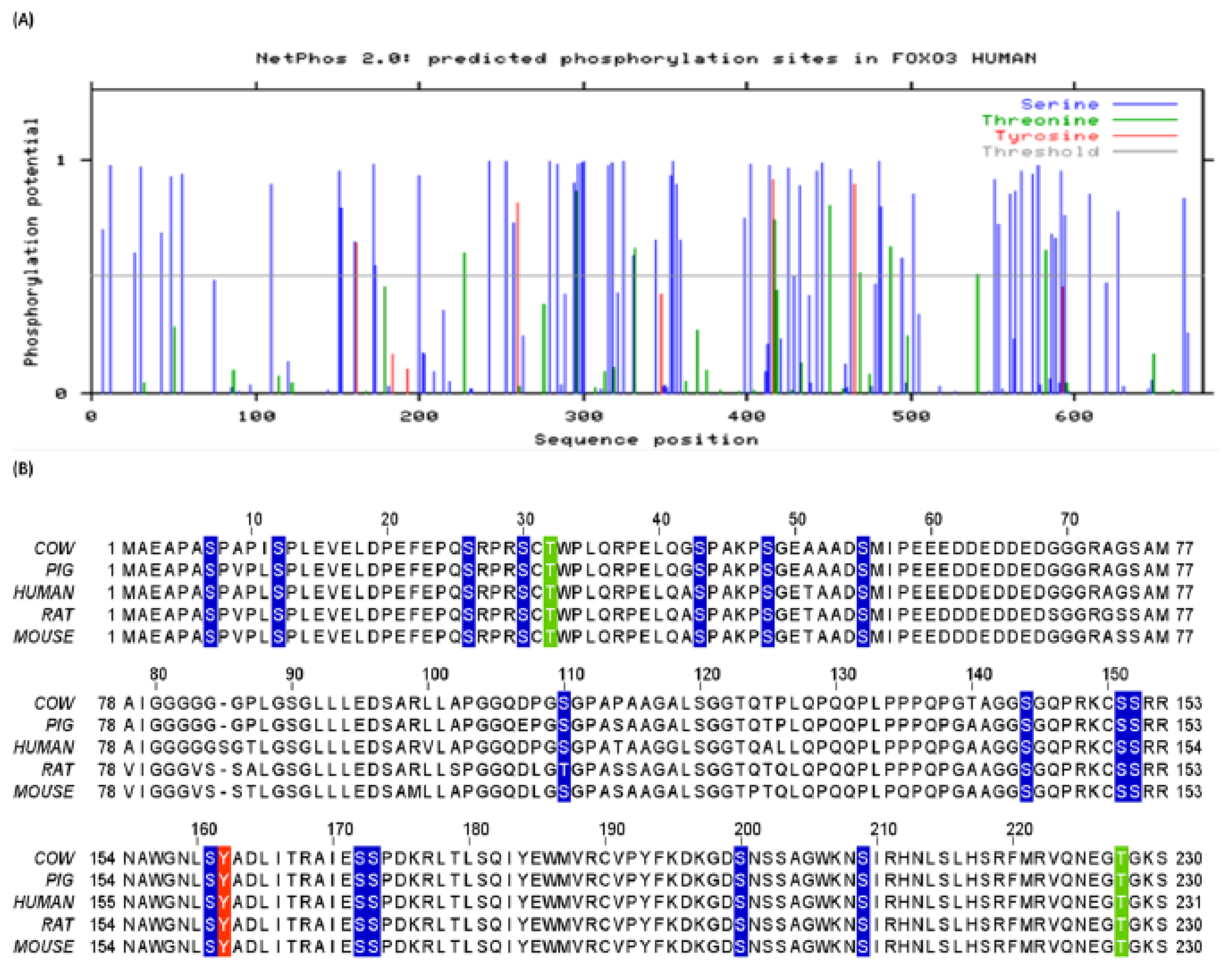
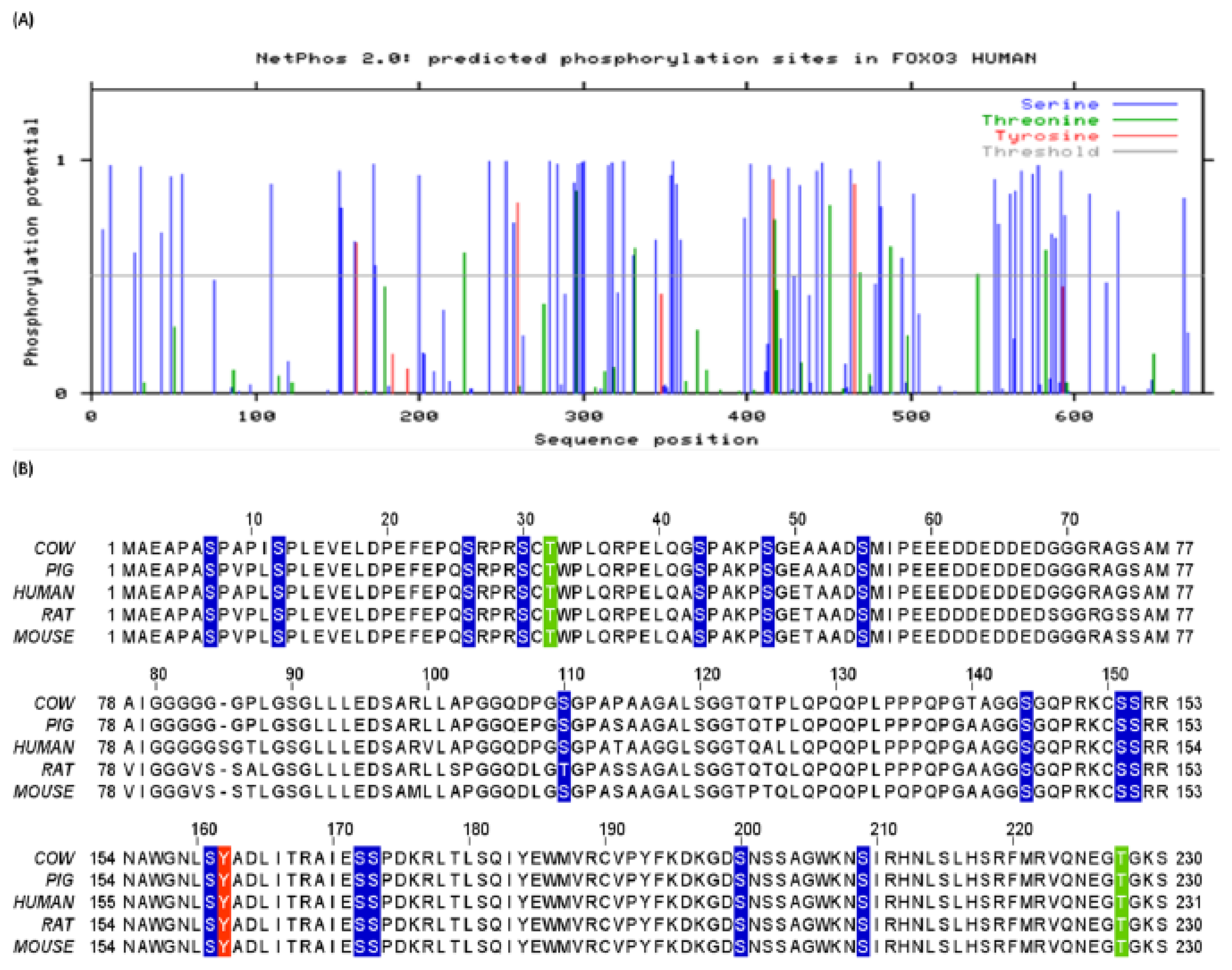
3.1. Human FoxO3 Possess Multiple Phosphorylation Sites
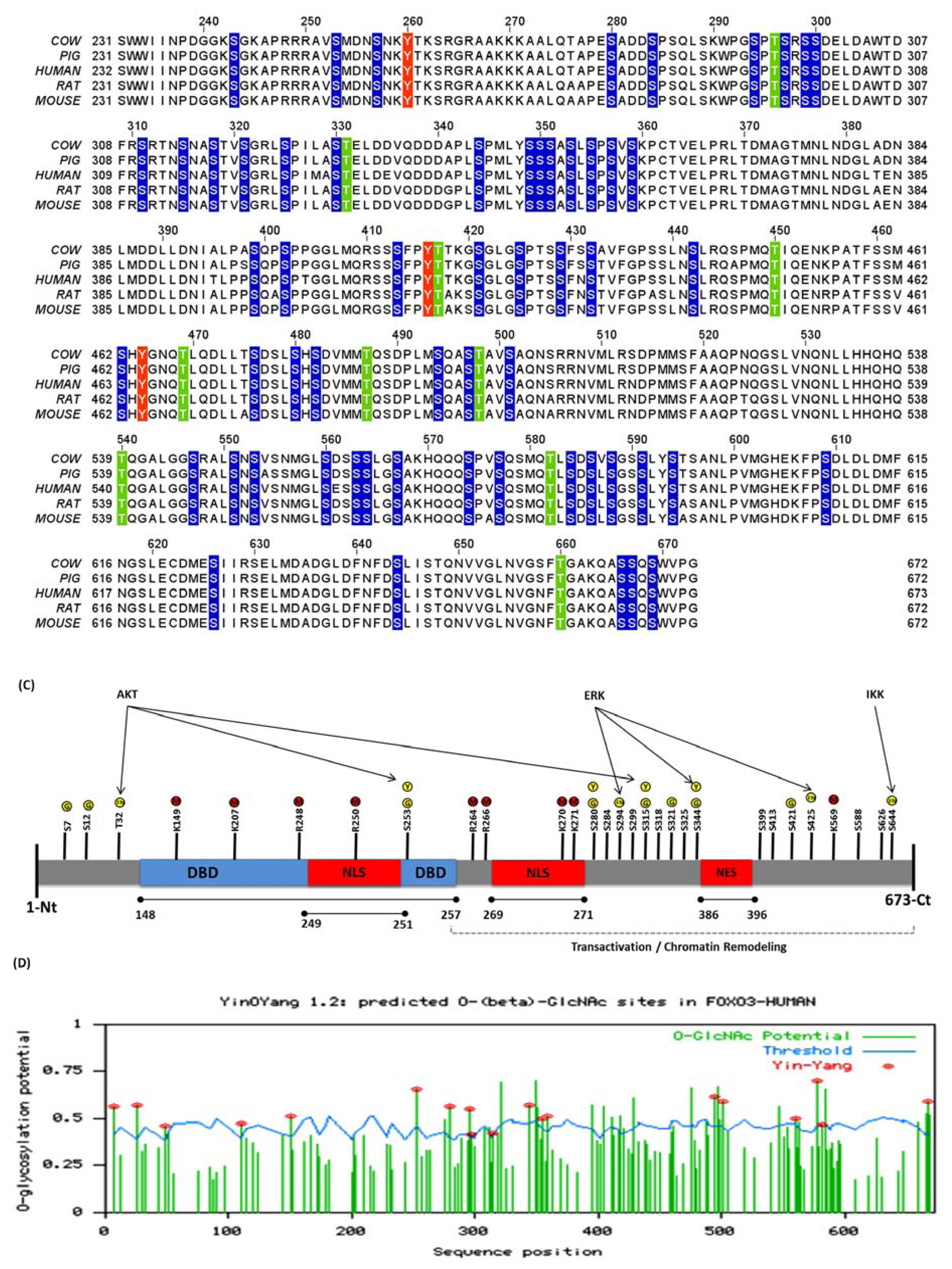
3.2. Human FoxO3 Is a Target of Multiple Oncogenic Kinases
3.3. Cdk5 Associated Phosphorylation of Human FoxO3
3.4. Phosphorylation of Human FoxO3 at Tyrosine Residues
3.5. O-β-GlcNAc Modifications and Yin Yang Sites in Human FoxO3
3.6. Identification of False-Negative Yin Yang Sites in Human FoxO3
3.7. Methylation Potential of Human FoxO3
4. Discussion
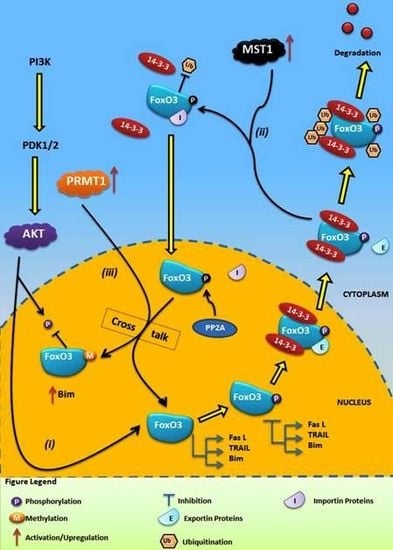
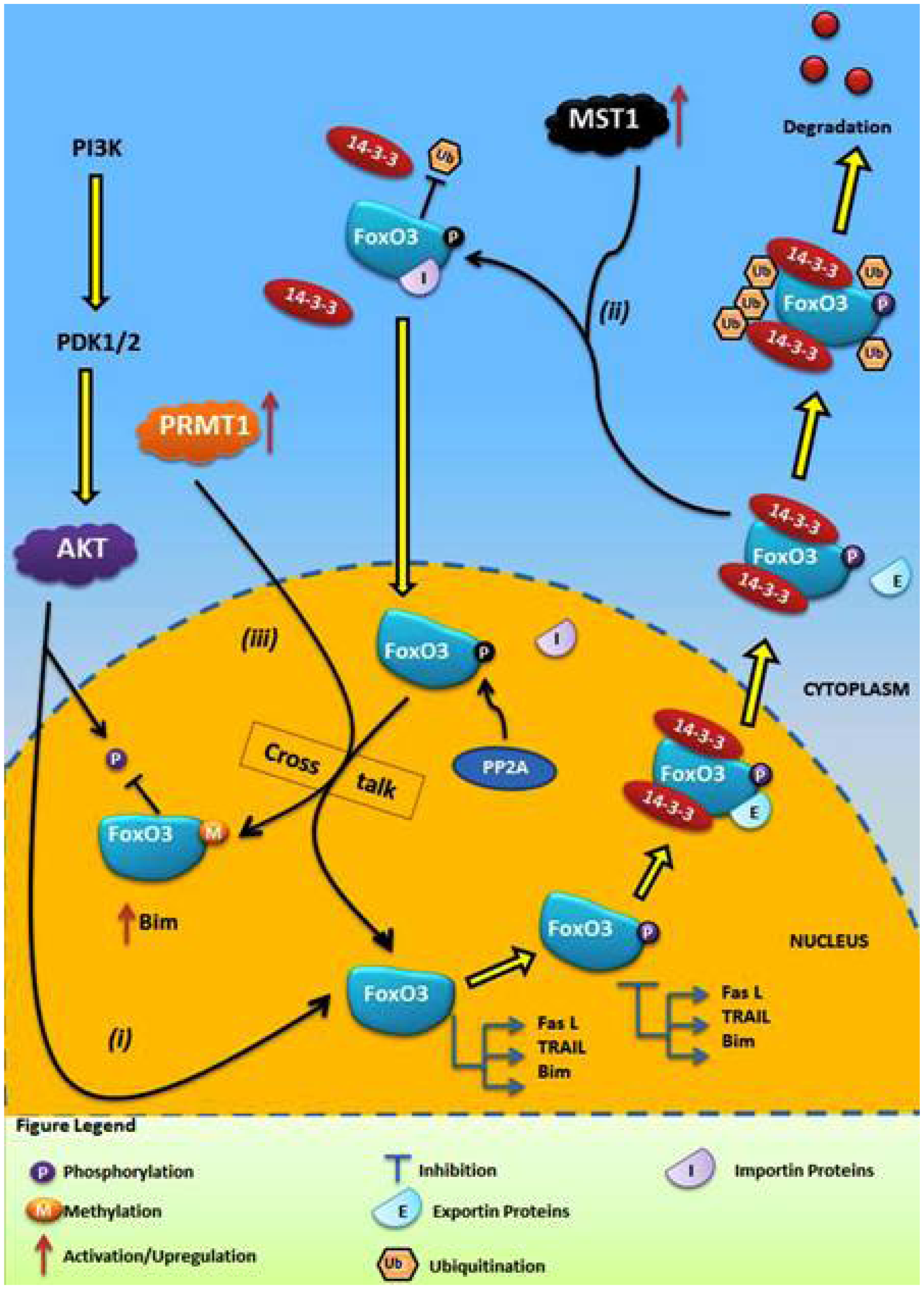
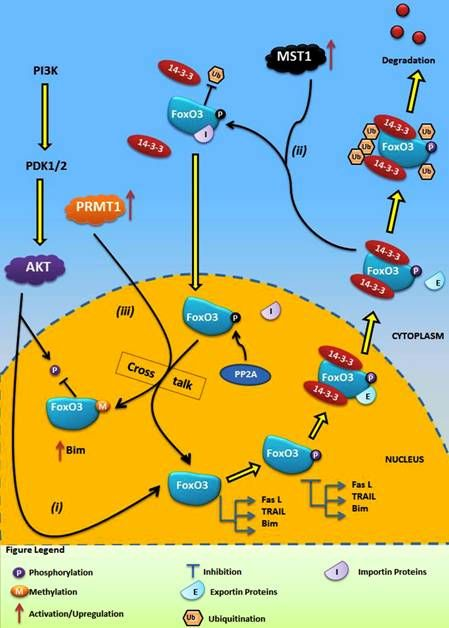
4.1. Crosstalk Between Phosphorylation and O-β-Glycosylation in Human FoxO3 Can Inhibit AKT, ERK, and IKK Pathways
4.2. Crosstalk Between Phosphorylation and Methylation in Human FoxO3
5. Conclusions
Acknowledgments
- Competing InterestsThe authors declare that they have no competing interests.
References
- Maiese, K.; Chong, Z.Z.; Shang, Y.C.; Hou, J. A “FOXO” in sight: Targeting Foxo proteins from conception to cancer. Med. Res. Rev 2009, 29, 395–418. [Google Scholar]
- Greer, E.L.; Brunet, A. FOXO transcription factors at the interface between longevity and tumor suppression. Oncogene 2005, 24, 7410–7425. [Google Scholar]
- Barr, F.G. Gene fusions involving PAX and FOX family members in alveolar rhabdomyosarcoma. Oncogene 2001, 20, 5736–5746. [Google Scholar]
- Hu, M.C.; Lee, D.F.; Xia, W.; Golfman, L.S.; Ou-Yang, F.; Yang, J.Y.; Zou, Y.; Bao, S.; Hanada, N.; Saso, H.; et al. IkappaB kinase promotes tumorigenesis through inhibition of forkhead FOXO3a. Cell 2004, 117, 225–237. [Google Scholar]
- Yang, J.Y.; Zong, C.S.; Xia, W.; Yamaguchi, H.; Ding, Q.; Xie, X.; Lang, J.Y.; Lai, C.C.; Chang, C.J.; Huang, W.C.; et al. ERK promotes tumorigenesis by inhibiting FOXO3a via MDM2-mediated degradation. Nat. Cell Biol 2008, 10, 138–148. [Google Scholar]
- Kornblau, S.M.; Singh, N.; Qiu, Y.; Chen, W.; Zhang, N.; Coombes, K.R. Highly phosphorylated FOXO3A is an adverse prognostic factor in acute myeloid leukemia. Clin. Cancer Res 2010, 16, 1865–1874. [Google Scholar]
- Furukawa-Hibi, Y.; Yoshida-Araki, K.; Ohta, T.; Ikeda, K.; Motoyama, N. FOXO forkhead transcription factors induce G(2)-M checkpoint in response to oxidative stress. J. Biol. Chem 2002, 277, 26729–26732. [Google Scholar]
- Kikuchi, S.; Nagai, T.; Kunitama, M.; Kirito, K.; Ozawa, K.; Komatsu, N. Active FKHRL1 overcomes imatinib resistance in chronic myelogenous leukemia-derived cell lines via the production of tumor necrosis factor-related apoptosis-inducing ligand. Cancer Sci 2007, 98, 1949–1958. [Google Scholar]
- Jacobsen, E.A.; Ananieva, O.; Brown, M.L.; Chang, Y. Growth, differentiation, and malignant transformation of pre-B cells mediated by inducible activation of v-Abl oncogene. J. Immunol 2006, 176, 6831–6838. [Google Scholar]
- Zhao, Y.; Wang, Y.; Zhu, W.G. Applications of post-translational modifications of FoxO family proteins in biological functions. J. Mol. Cell Biol 2011, 3, 276–282. [Google Scholar]
- Smith, G.R.; Shanley, D.P. Modelling the response of FOXO transcription factors to multiple post-translational modifications made by ageing-related signalling pathways. PLoS One 2010, 5, e11092. [Google Scholar]
- Jagani, Z.; Singh, A.; Khosravi-Far, R. FoxO tumor suppressors and BCR-ABL-induced leukemia: a matter of evasion of apoptosis. Biochim. Biophys. Acta 2008, 1785, 63–84. [Google Scholar]
- Myatt, S.S.; Lam, E.W. The emerging roles of forkhead box (Fox) proteins in cancer. Nat. Rev. Cancer 2007, 7, 847–859. [Google Scholar]
- van der Horst, A.; Burgering, B.M. Stressing the role of FoxO proteins in lifespan and disease. Nat. Rev. Mol. Cell Biol 2007, 8, 440–450. [Google Scholar]
- Strahl, B.D.; Allis, C.D. The language of covalent histone modifications. Nature 2000, 403, 41–45. [Google Scholar]
- Berger, S.L. The complex language of chromatin regulation during transcription. Nature 2007, 447, 407–412. [Google Scholar]
- Hart, G.W.; Greis, K.D.; Dong, L.Y.; Blomberg, M.A.; Chou, T.Y.; Jiang, M.S.; Roquemore, E.P.; Snow, D.M.; Kreppel, L.K.; Cole, R.N.; et al. O-linked N-acetylglucosamine: The “yin-yang” of Ser/Thr phosphorylation? Nuclear and cytoplasmic glycosylation. Adv. Exp. Med. Biol 1995, 376, 115–123. [Google Scholar]
- Kamemura, K.; Hart, G.W. Dynamic interplay between O-glycosylation and O-phosphorylation of nucleocytoplasmic proteins: A new paradigm for metabolic control of signal transduction and transcription. Prog. Nucleic Acid Res. Mol. Biol 2003, 73, 107–136. [Google Scholar]
- Zeidan, Q.; Hart, G.W. The intersections between O-GlcNAcylation and phosphorylation: Implications for multiple signaling pathways.
- Butt, A.M.; Khan, I.B.; Hussain, M.; Idress, M.; Lu, J.; Tong, Y. Role of post translational modifications and novel crosstalk between phosphorylation and O-beta-GlcNAc modifications in human claudin-1, −3 and −4. Mol. Biol. Rep 2012, 39, 1359–1369. [Google Scholar]
- Estève, P.-O.; Chang, Y.; Samaranayake, M.; Upadhyay, A.K.; Horton, J.R.; Feehery, G.R.; Cheng, X.; Pradhan, S. A methylation and phosphorylation switch between an adjacent lysine and serine determines human DNMT1 stability. Nat. Struct. Mol. Biol 2011, 18, 42–48. [Google Scholar]
- Ambler, R.P.; Rees, M.W. Epsilon-N-Methyl-lysine in bacterial flagellar protein. Nature 1959, 184, 56–57. [Google Scholar]
- Bedford, M.T.; Richard, S. Arginine methylation an emerging regulator of protein function. Mol. Cell 2005, 18, 263–272. [Google Scholar]
- Lee, D.Y.; Teyssier, C.; Strahl, B.D.; Stallcup, M.R. Role of protein methylation in regulation of transcription. Endocr. Rev 2005, 26, 147–170. [Google Scholar]
- Bannister, A.J.; Kouzarides, T. Reversing histone methylation. Nature 2005, 436, 1103–1106. [Google Scholar]
- Gupta, P.; Ho, P.C.; Huq, M.D.; Khan, A.A.; Tsai, N.P.; Wei, L.N. PKCepsilon stimulated arginine methylation of RIP140 for its nuclear-cytoplasmic export in adipocyte differentiation. PLoS One 2008, 3, e2658. [Google Scholar]
- Chen, W.; Daines, M.O.; Hershey, G.K. Methylation of STAT6 modulates STAT6 phosphorylation, nuclear translocation, and DNA-binding activity. J. Immunol 2004, 172, 6744–6750. [Google Scholar]
- Boeckmann, B.; Bairoch, A.; Apweiler, R.; Blatter, M.C.; Estreicher, A.; Gasteiger, E.; Martin, M.J.; Michoud, K.; O’Donovan, C.; Phan, I.; et al. The SWISS-PROT protein knowledgebase and its supplement TrEMBL in 2003. Nucleic Acids Res 2003, 31, 365–370. [Google Scholar]
- Altschul, S.F.; Madden, T.L.; Schaffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res 1997, 25, 3389–3402. [Google Scholar]
- Larkin, M.A.; Blackshields, G.; Brown, N.P.; Chenna, R.; McGettigan, P.A.; McWilliam, H.; Valentin, F.; Wallace, I.M.; Wilm, A.; Lopez, R.; et al. Clustal W and Clustal X version 2.0. Bioinformatics 2007, 23, 2947–2948. [Google Scholar]
- Blom, N.; Gammeltoft, S.; Brunak, S. Sequence and structure-based prediction of eukaryotic protein phosphorylation sites. J. Mol. Biol 1999, 294, 1351–1362. [Google Scholar]
- Obenauer, J.C.; Cantley, L.C.; Yaffe, M.B. Scansite 2.0: Proteome-wide prediction of cell signaling interactions using short sequence motifs. Nucleic Acids Res 2003, 31, 3635–3641. [Google Scholar]
- Gupta, R.; Brunak, S. Prediction of glycosylation across the human proteome and the correlation to protein function. Pac. Symp. Biocomput 2002, 310–322. [Google Scholar]
- Chen, H.; Xue, Y.; Huang, N.; Yao, X.; Sun, Z. MeMo: A web tool for prediction of protein methylation modifications. Nucleic Acids Res 2006, 34, W249–W253. [Google Scholar]
- Shao, J.; Xu, D.; Tsai, S.N.; Wang, Y.; Ngai, S.M. Computational identification of protein methylation sites through bi-profile Bayes feature extraction. PLoS One 2009, 4, e4920. [Google Scholar]
- Shien, D.M.; Lee, T.Y.; Chang, W.C.; Hsu, J.B.; Horng, J.T.; Hsu, P.C.; Wang, T.Y.; Huang, H.D. Incorporating structural characteristics for identification of protein methylation sites. J. Comput. Chem 2009, 30, 1532–1543. [Google Scholar]
- Dinkel, H.; Chica, C.; Via, A.; Gould, C.M.; Jensen, L.J.; Gibson, T.J.; Diella, F. Phospho.ELM: A database of phosphorylation sites—update 2011. Nucleic Acids Res 2011, 39, D261–D267. [Google Scholar]
- Mayya, V.; Lundgren, D.H.; Hwang, S.I.; Rezaul, K.; Wu, L.; Eng, J.K.; Rodionov, V.; Han, D.K. Quantitative phosphoproteomic analysis of T cell receptor signaling reveals system-wide modulation of protein-protein interactions. Sci. Signal 2009, 2, ra46. [Google Scholar]
- Lehtinen, M.K.; Yuan, Z.; Boag, P.R.; Yang, Y.; Villen, J.; Becker, E.B.; DiBacco, S.; de la Iglesia, N.; Gygi, S.; Blackwell, T.K.; Bonni, A. A conserved MST-FOXO signaling pathway mediates oxidative-stress responses and extends life span. Cell 2006, 125, 987–1001. [Google Scholar]
- Brunet, A.; Park, J.; Tran, H.; Hu, L.S.; Hemmings, B.A.; Greenberg, M.E. Protein kinase SGK mediates survival signals by phosphorylating the forkhead transcription factor FKHRL1 (FOXO3a). Mol. Cell. Biol 2001, 21, 952–965. [Google Scholar]
- Dephoure, N.; Zhou, C.; Villen, J.; Beausoleil, S.A.; Bakalarski, C.E.; Elledge, S.J.; Gygi, S.P. A quantitative atlas of mitotic phosphorylation. Proc. Natl. Acad. Sci. USA 2008, 105, 10762–10767. [Google Scholar]
- Rena, G.; Woods, Y.L.; Prescott, A.R.; Peggie, M.; Unterman, T.G.; Williams, M.R.; Cohen, P. Two novel phosphorylation sites on FKHR that are critical for its nuclear exclusion. EMBO J 2002, 21, 2263–2271. [Google Scholar]
- Kelly, P.A.; Rahmani, Z. DYRK1A enhances the mitogen-activated protein kinase cascade in PC12 cells by forming a complex with Ras, B-Raf, and MEK1. Mol. Biol. Cell 2005, 16, 3562–3573. [Google Scholar]
- Greer, E.L.; Oskoui, P.R.; Banko, M.R.; Maniar, J.M.; Gygi, M.P.; Gygi, S.P.; Brunet, A. The energy sensor AMP-activated protein kinase directly regulates the mammalian FOXO3 transcription factor. J. Biol. Chem 2007, 282, 30107–30119. [Google Scholar]
- Cantin, G.T.; Yi, W.; Lu, B.; Park, S.K.; Xu, T.; Lee, J.D.; Yates, J.R., III. Combining protein-based IMAC, peptide-based IMAC, and MudPIT for efficient phosphoproteomic analysis. J. Proteome Res. 2008, 7, 1346–1351. [Google Scholar]
- Brunet, A.; Bonni, A.; Zigmond, M.J.; Lin, M.Z.; Juo, P.; Hu, L.S.; Anderson, M.J.; Arden, K.C.; Blenis, J.; Greenberg, M.E. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell 1999, 96, 857–868. [Google Scholar]
- Strock, C.J.; Park, J.I.; Nakakura, E.K.; Bova, G.S.; Isaacs, J.T.; Ball, D.W.; Nelkin, B.D. Cyclin-dependent kinase 5 activity controls cell motility and metastatic potential of prostate cancer cells. Cancer Res 2006, 66, 7509–7515. [Google Scholar]
- Feldmann, G.; Mishra, A.; Hong, S.M.; Bisht, S.; Strock, C.J.; Ball, D.W.; Goggins, M.; Maitra, A.; Nelkin, B.D. Inhibiting the cyclin-dependent kinase CDK5 blocks pancreatic cancer formation and progression through the suppression of Ras-Ral signaling. Cancer Res 2010, 70, 4460–4469. [Google Scholar]
- Shukla, S.; Shukla, M.; Maclennan, G.T.; Fu, P.; Gupta, S. Deregulation of FOXO3A during prostate cancer progression. Int. J. Oncol 2009, 34, 1613–1620. [Google Scholar]
- Roy, S.K.; Srivastava, R.K.; Shankar, S. Inhibition of PI3K/AKT and MAPK/ERK pathways causes activation of FOXO transcription factor, leading to cell cycle arrest and apoptosis in pancreatic cancer. J. Mol. Signal 2010, 5, 10. [Google Scholar]
- Mareel, M.; Leroy, A. Clinical, cellular, and molecular aspects of cancer invasion. Physiol. Rev 2003, 83, 337–376. [Google Scholar]
- Steeg, P.S. Metastasis suppressors alter the signal transduction of cancer cells. Nat. Rev. Cancer 2003, 3, 55–63. [Google Scholar]
- Baselga, J. Targeting tyrosine kinases in cancer: The second wave. Science 2006, 312, 1175–1178. [Google Scholar]
- Yamagata, K.; Daitoku, H.; Takahashi, Y.; Namiki, K.; Hisatake, K.; Kako, K.; Mukai, H.; Kasuya, Y.; Fukamizu, A. Arginine methylation of FOXO transcription factors inhibits their phosphorylation by Akt. Mol. Cell 2008, 32, 221–231. [Google Scholar]
- Housley, M.P.; Rodgers, J.T.; Udeshi, N.D.; Kelly, T.J.; Shabanowitz, J.; Hunt, D.F.; Puigserver, P.; Hart, G.W. O-GlcNAc regulates FoxO activation in response to glucose. J. Biol. Chem 2008, 283, 16283–16292. [Google Scholar]
- Ho, S.R.; Wang, K.; Whisenhunt, T.R.; Huang, P.; Zhu, X.; Kudlow, J.E.; Paterson, A.J. O-GlcNAcylation enhances FOXO4 transcriptional regulation in response to stress. FEBS Lett 2010, 584, 49–54. [Google Scholar]
- Yang, J.Y.; Hung, M.C. A new fork for clinical application: targeting forkhead transcription factors in cancer. Clin. Cancer Res 2009, 15, 752–757. [Google Scholar]
- Fu, Z.; Tindall, D.J. FOXOs, cancer and regulation of apoptosis. Oncogene 2008, 27, 2312–2319. [Google Scholar]
- Vogelstein, B.; Kinzler, K.W. Cancer genes and the pathways they control. Nat. Med 2004, 10, 789–799. [Google Scholar]
- Real, P.J.; Benito, A.; Cuevas, J.; Berciano, M.T.; de Juan, A.; Coffer, P.; Gomez-Roman, J.; Lafarga, M.; Lopez-Vega, J.M.; Fernandez-Luna, J.L. Blockade of epidermal growth factor receptors chemosensitizes breast cancer cells through up-regulation of Bnip3L. Cancer Res 2005, 65, 8151–8157. [Google Scholar]
- Yao, H.J.; Ju, R.J.; Wang, X.X.; Zhang, Y.; Li, R.J.; Yu, Y.; Zhang, L.; Lu, W.L. The antitumor efficacy of functional paclitaxel nanomicelles in treating resistant breast cancers by oral delivery. Biomaterials 2011, 32, 3285–3302. [Google Scholar]
- Yan, L.; Lavin, V.A.; Moser, L.R.; Cui, Q.; Kanies, C.; Yang, E. PP2A regulates the pro-apoptotic activity of FOXO1. J. Biol. Chem 2008, 283, 7411–7420. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Residues | Phosphorylation | Scansite | O-GlcNAc | Yin Yang | |||||
|---|---|---|---|---|---|---|---|---|---|
| Name | Pos | CS | EV | CD | Kinases | SA | EV | CD | CD |
| Serine | 7 | * | [38] | Y | - | - | - | Y | Y |
| 12 | * | [38] | Y | ERK1; AKT | 0.8 | - | - | - | |
| 26 | * | - | Y | GSK3 | 4.8 | - | Y | Y | |
| 30 | * | - | Y | - | - | - | - | - | |
| 43 | * | - | Y | Cdk5 | 1.7 | - | - | - | |
| 48 | * | - | Y | - | - | - | Y | Y | |
| 55 | * | - | Y | - | - | - | - | - | |
| 110 | * | - | Y | - | - | - | Y | Y | |
| 144 | * | - | Y | - | - | - | Y | - | |
| 151 | * | - | Y | PKC; PKA | 1.7 | - | Y | Y | |
| 152 | * | - | Y | - | - | - | - | - | |
| 161 | * | - | Y | - | - | - | - | - | |
| 172 | * | - | Y | - | - | - | - | - | |
| 173 | * | - | Y | Cdk5; Cdc2 | 3.6 | - | - | - | |
| 200 | * | - | Y | - | - | - | - | - | |
| 209 | * | [39] | Y | PKC | 1.9 | - | - | - | |
| 243 | * | - | Y | - | - | - | - | - | |
| 253 | * | [40] | Y | AKT E; PKA | 0.6 | - | Y | Y | |
| 257 | * | - | Y | - | - | - | - | - | |
| 280 | * | [41] | Y | CK2; GSK3 | 2.3 | - | Y | Y | |
| 284 | * | [41] | Y | ERK | 1.9 | - | - | - | |
| 294 | * | [5] | Y | Cdk5; Cdc2; ERK1 E, p38 MAPK | 1.4 | - | - | FN | |
| 297 | * | - | Y | - | - | - | Y | Y | |
| 299 | * | [41] | Y | - | - | - | - | - | |
| 300 | * | - | Y | CK2 | 2.0 | - | - | - | |
| 311 | * | - | - | - | - | - | Y | - | |
| 315 | * | [40] | Y | AKT E, Clk2 | 1.6 | - | Y | Y | |
| 318 | * | [42] | Y | CK1 E | 1.7 | - | - | - | |
| 321 | * | [42] | Y | CK1 E | 1.9 | - | Y | - | |
| 325 | * | [43] | Y | ERK1 | 0.5 | - | - | - | |
| 330 | * | - | Y | - | - | - | - | - | |
| 344 | * | [5] | Y | ERK1 E | 0.6 | - | Y | Y | |
| 349 | * | - | - | - | - | - | Y | - | |
| 350 | * | - | - | - | - | - | Y | - | |
| 351 | * | - | - | GSK3 | 0.6 | - | Y | - | |
| 353 | * | - | Y | CK1; PKC; PKC δ | 0.7 | - | - | - | |
| 355 | * | - | Y | ERK1; Cdk5 | 0.5 | - | Y | Y | |
| 357 | * | - | Y | - | - | - | - | - | |
| 359 | * | - | Y | - | - | - | Y | Y | |
| 399 | * | [44] | Y | ATMK; AMPK E | 2.6 | - | - | - | |
| 402 | * | - | Y | ERK1 | 1.9 | - | - | ||
| 411 | * | - | - | - | - | - | Y | - | |
| 413 | * | [44] | Y | AKT; PKC | 1.1 | - | - | - | |
| 421 | * | [45] | Y | CK1 | 0.5 | - | Y | - | |
| 425 | * | [5] | Y | Cdc2; Cdk5; GSK3; ERK1 E | 0.7 | - | - | FN | |
| 428 | * | - | - | - | - | - | Y | - | |
| 429 | * | - | Y | - | - | - | - | - | |
| 432 | * | - | Y | CK1 | 0.5 | - | - | - | |
| 442 | * | - | Y | - | - | - | - | - | |
| 446 | * | - | Y | - | - | - | - | - | |
| 463 | * | - | Y | CK1 | 0.9 | - | - | - | |
| 476 | * | - | - | - | - | - | Y | - | |
| 480 | * | - | Y | - | - | - | - | - | |
| 482 | * | - | Y | - | - | - | - | - | |
| 494 | * | - | Y | - | - | - | Y | Y | |
| 497 | * | - | - | PKC | 0.6 | - | Y | - | |
| 501 | * | - | Y | - | - | - | Y | Y | |
| 547 | * | - | - | - | - | - | Y | - | |
| 551 | * | - | Y | - | - | - | - | - | |
| 553 | * | - | Y | PKC | 1.2 | - | - | - | |
| 560 | * | - | Y | - | - | - | Y | Y | |
| 563 | * | - | - | - | - | - | Y | - | |
| 564 | * | - | Y | - | - | - | - | - | |
| 567 | * | - | Y | PKC | 0.7 | - | - | - | |
| 574 | * | - | Y | - | - | - | - | - | |
| 577 | * | - | Y | ATMK | 0.7 | - | Y | Y | |
| 584 | * | - | - | - | - | - | Y | - | |
| 586 | * | - | Y | - | - | - | - | - | |
| 588 | * | [44] | Y | AMPK E | 0.6 | - | - | - | |
| 591 | * | - | Y | PKC; CK1 | 0.7 | - | - | - | |
| 594 | * | - | Y | - | - | - | - | - | |
| 609 | * | - | Y | - | - | - | - | - | |
| 626 | * | [44] | Y | - | - | - | - | - | |
| 644 | * | [4] | Y | - | - | - | - | FN | |
| 666 | * | - | - | - | - | - | Y | - | |
| 667 | * | - | Y | - | - | - | Y | Y | |
| 669 | * | - | - | - | - | - | Y | - | |
| Threonine | 32 | * | [46] | Y | AKT E, PKA | 0.5 | - | - | FN |
| 228 | * | - | Y | - | - | - | - | - | |
| 276 | * | - | - | - | - | - | Y | - | |
| 296 | * | - | Y | PKC | 2.5 | - | Y | Y | |
| 331 | * | - | Y | CK2 | 1.0 | - | - | - | |
| 395 | - | - | - | - | - | - | Y | - | |
| 404 | - | - | - | - | - | - | Y | - | |
| 417 | * | - | Y | PKC | 2.3 | - | - | - | |
| 418 | - | - | - | - | - | - | Y | - | |
| 450 | * | - | Y | - | - | - | - | - | |
| 469 | * | - | Y | - | - | - | - | - | |
| 487 | * | - | Y | DNA PK | 1.2 | - | - | - | |
| 498 | * | - | - | - | - | - | Y | - | |
| 540 | * | - | Y | - | - | - | - | - | |
| 582 | * | - | Y | PKC; CK1 | 1.0 | - | Y | Y | |
| 660 | * | - | - | - | - | - | Y | - | |
| Tyrosine | 162 | * | - | Y | Lck kinase | 0.6 | - | - | - |
| 260 | * | - | Y | - | - | - | - | - | |
| 416 | * | - | Y | - | - | - | - | - | |
| 465 | * | - | Y | Grb2 SH2 | - | - | - | - | |
| Residues | Conservation Status | Flanking Sequence | Methylation Status | ||
|---|---|---|---|---|---|
| Name | Position | CD | EV | ||
| Arg | 248 | * | GKSGKAPRRR | Y | Based on sequence similarity with mouse FOXO1 methylation sites [54] |
| Arg | 250 | * | PRRRAVSMD | Y | Based on sequence similarity with mouse FOXO1 methylation sites [54] |
| Arg | 264 | * | NKYTKSRGRAAKK | Y | - |
| Arg | 266 | * | YTKSRGRAAKKKA | Y | - |
| Lys | 149 | * | GGSGQPRKCSSRR | Y | - |
| Lys | 207 | * | SNSSAGWKNSIRH | Y | - |
| Lys | 270 | * | SRGRAAKKKAALQ | Y | - |
| Lys | 271 | * | SRGRAAKKKAALQ | Y | - |
| Lys | 569 | * | SSSLGSAKHQQQS | Y | - |
© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Butt, A.M.; Feng, D.; Idrees, M.; Tong, Y.; Lu, J. Computational Identification and Modeling of Crosstalk between Phosphorylation, O-β-glycosylation and Methylation of FoxO3 and Implications for Cancer Therapeutics. Int. J. Mol. Sci. 2012, 13, 2918-2938. https://doi.org/10.3390/ijms13032918
Butt AM, Feng D, Idrees M, Tong Y, Lu J. Computational Identification and Modeling of Crosstalk between Phosphorylation, O-β-glycosylation and Methylation of FoxO3 and Implications for Cancer Therapeutics. International Journal of Molecular Sciences. 2012; 13(3):2918-2938. https://doi.org/10.3390/ijms13032918
Chicago/Turabian StyleButt, Azeem Mehmood, Dandan Feng, Muhammad Idrees, Yigang Tong, and Jun Lu. 2012. "Computational Identification and Modeling of Crosstalk between Phosphorylation, O-β-glycosylation and Methylation of FoxO3 and Implications for Cancer Therapeutics" International Journal of Molecular Sciences 13, no. 3: 2918-2938. https://doi.org/10.3390/ijms13032918