X-Ray Repair Cross Complementing Protein 1 in Base Excision Repair

Abstract
:1. X-Ray Repair Cross Complementing Protein 1 (XRCC1)
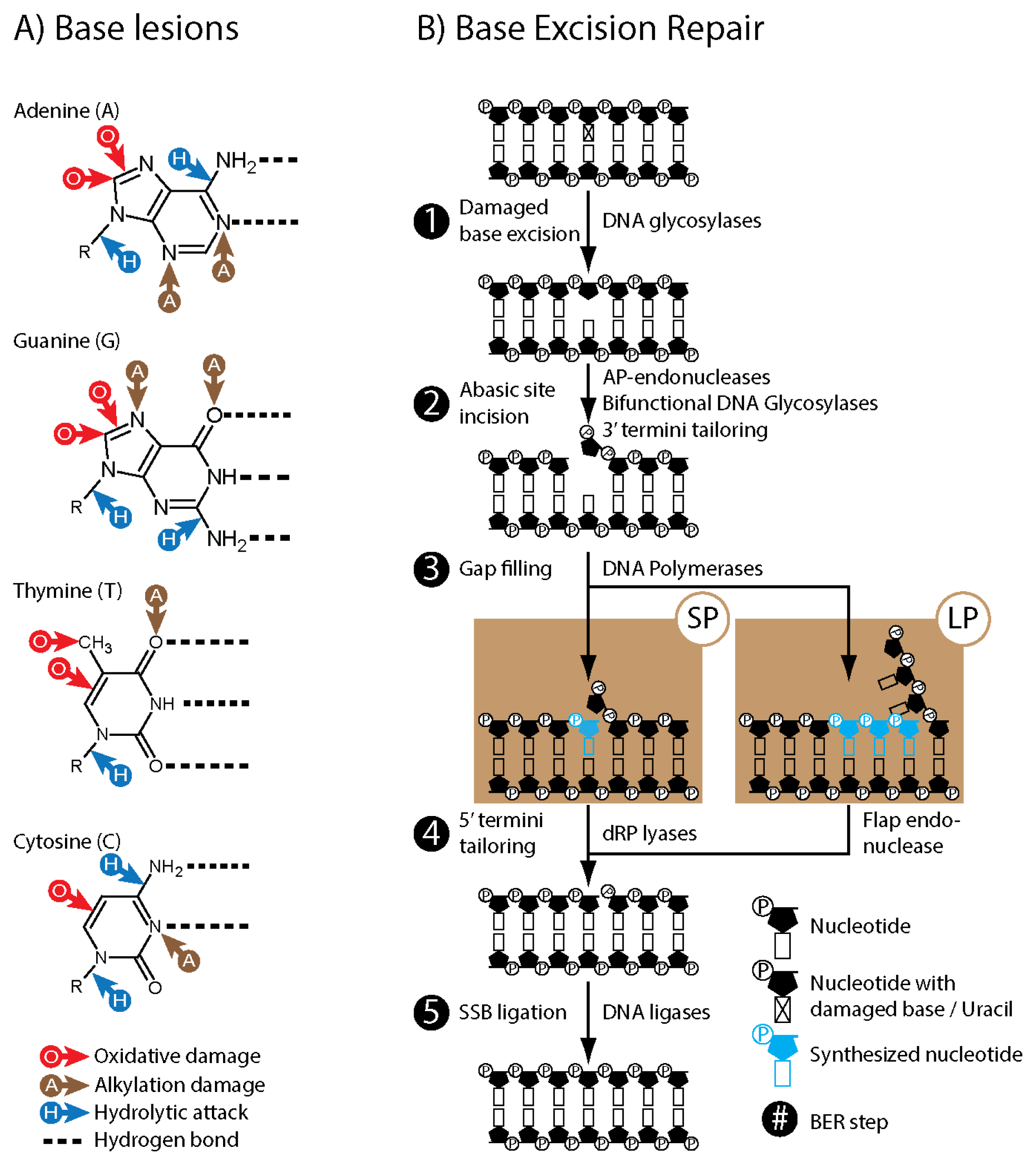
2. Base Excision Repair (BER)
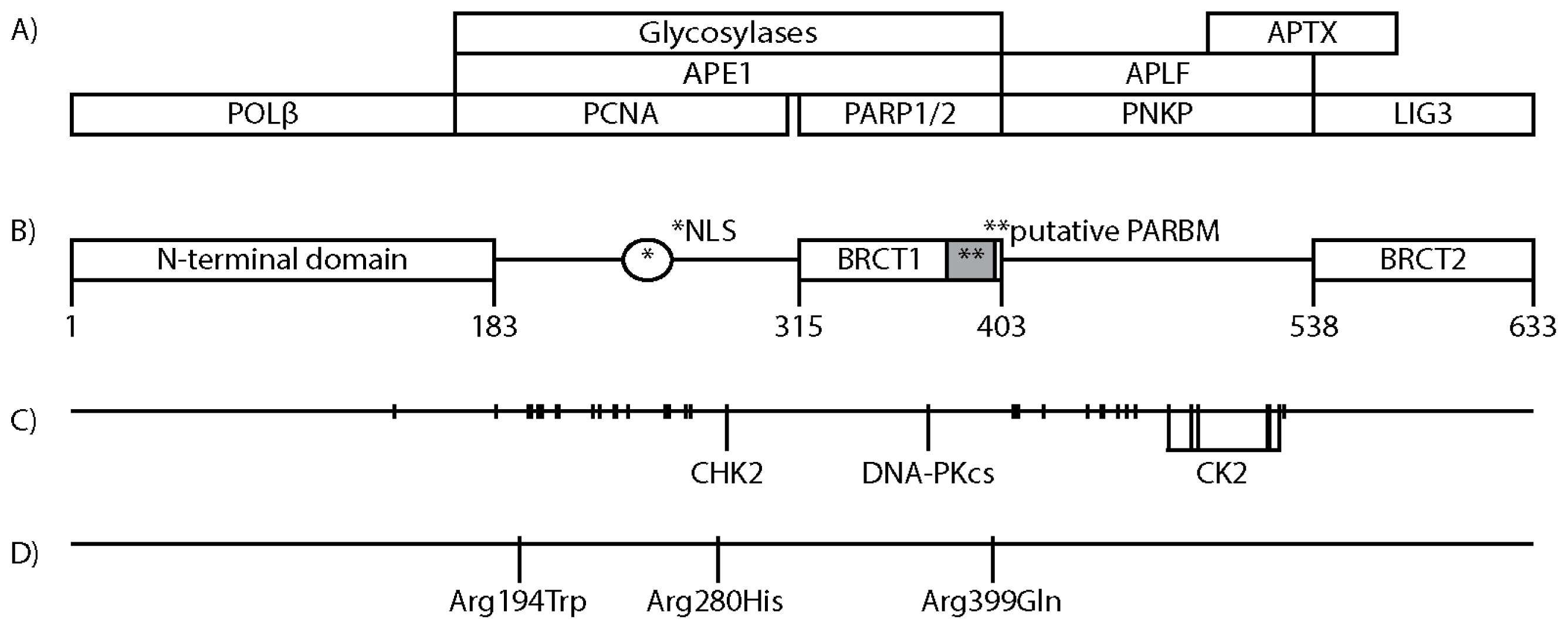
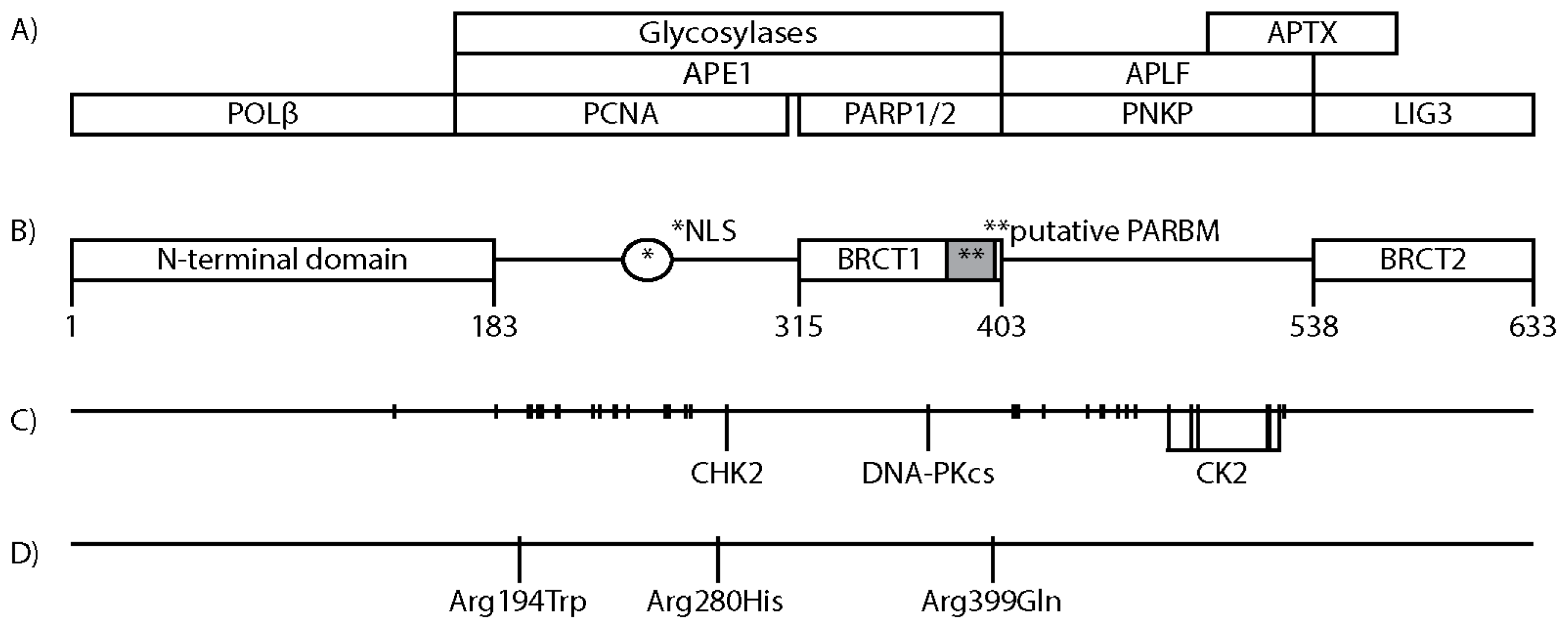
3. XRCC1 Structure and Protein Interacting Regions
4. Posttranslational Modifications of XRCC1
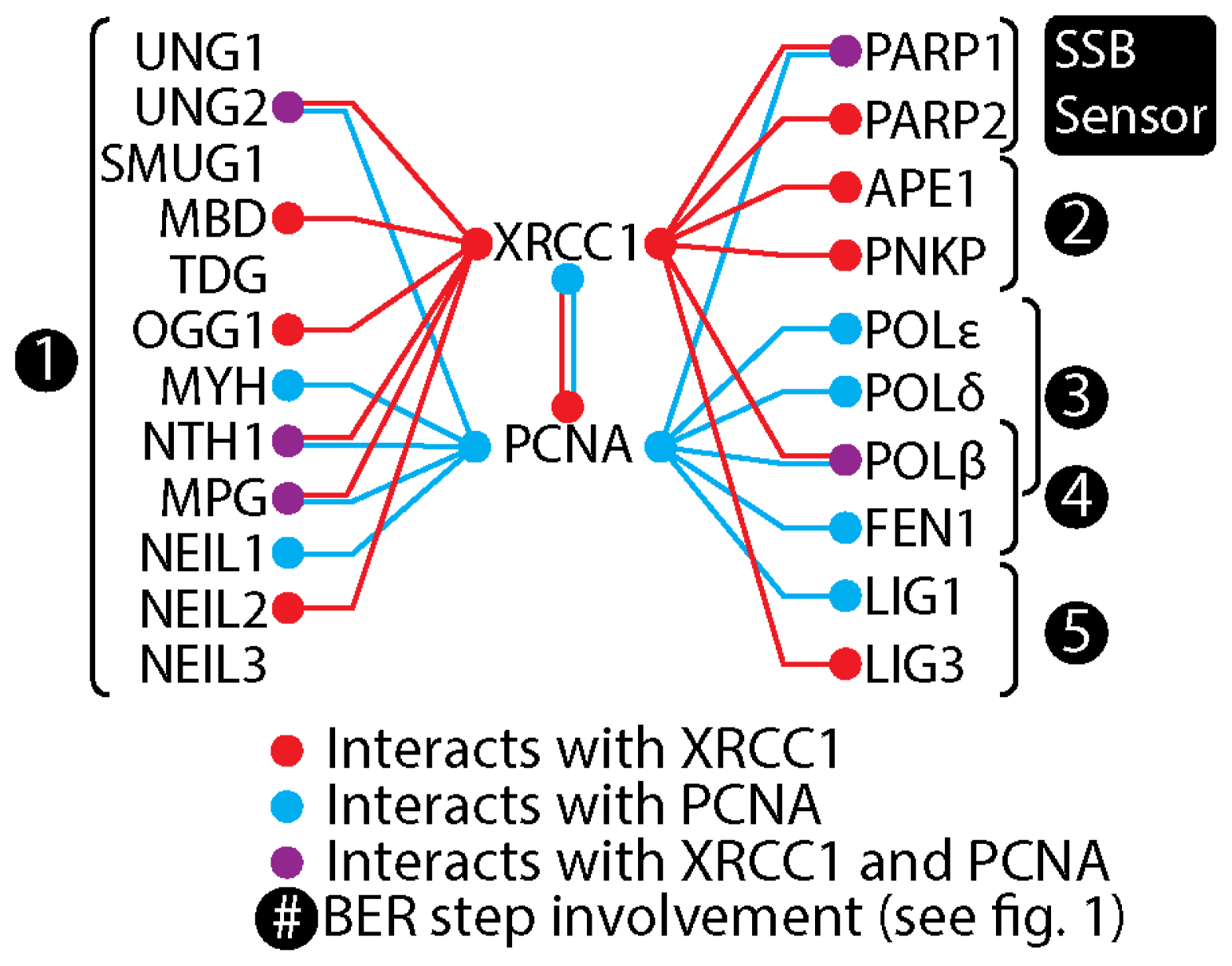
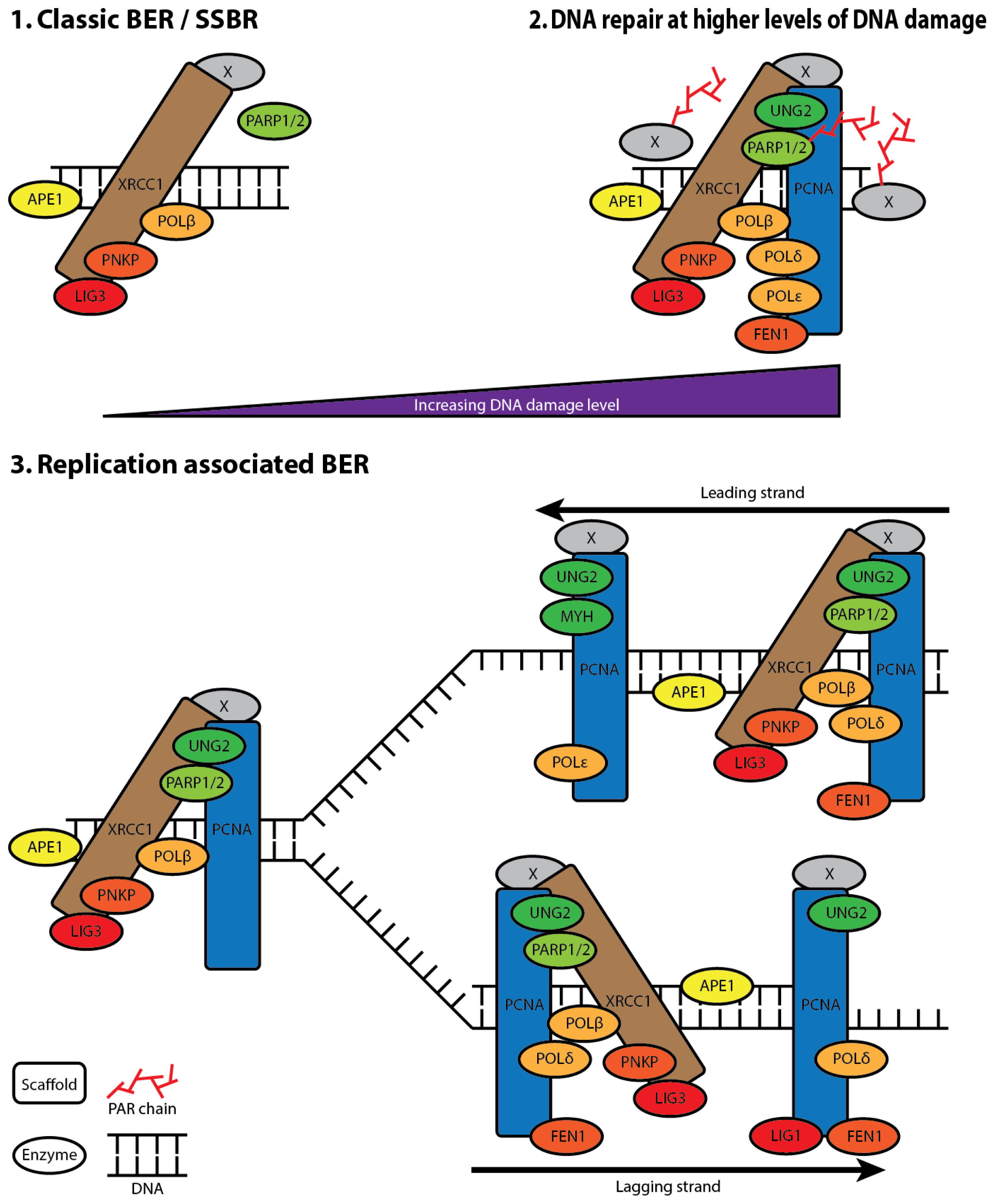
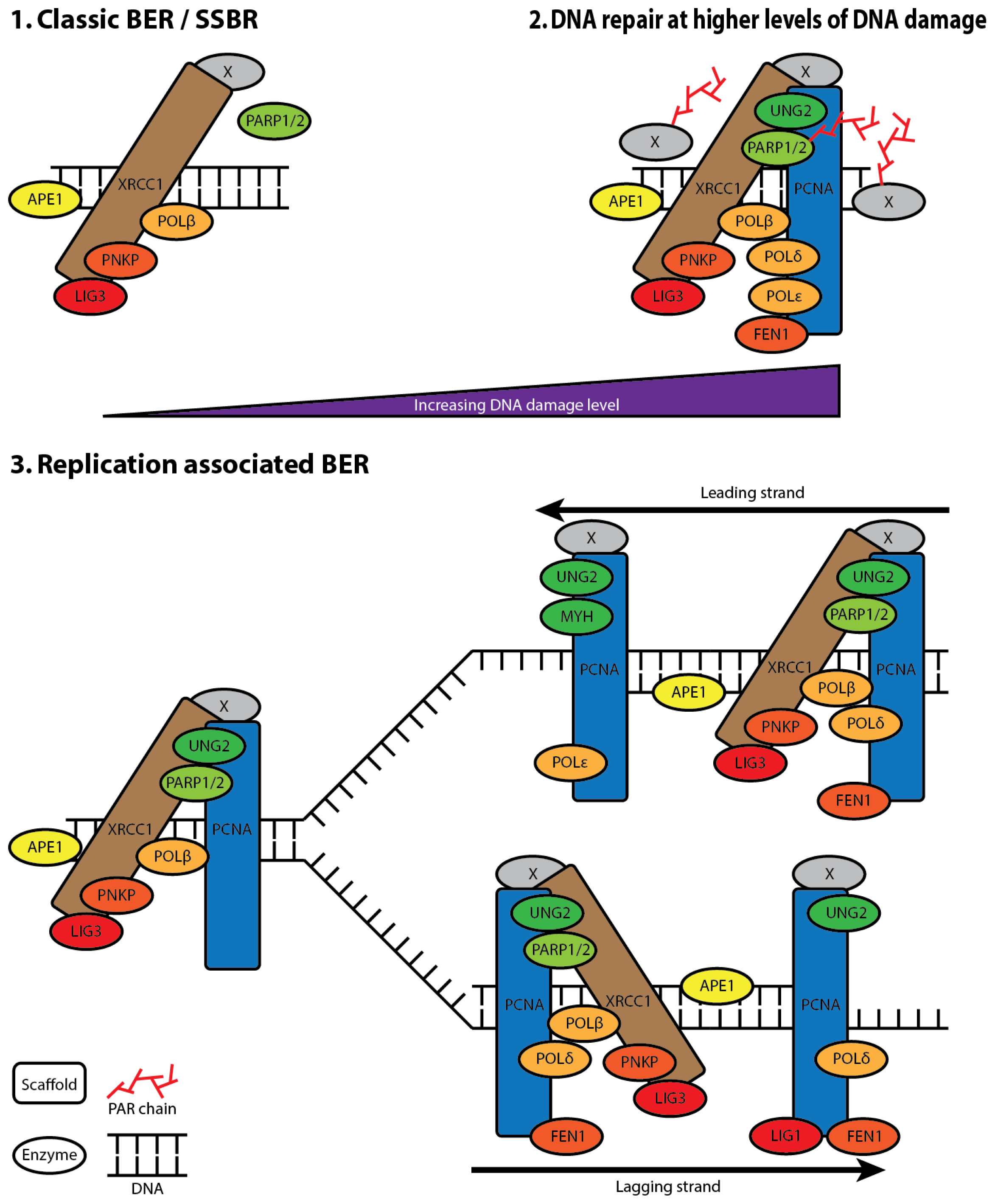
5. XRCC1 Multiprotein Complexes
6. The Composition of XRCC1 Associated Multiprotein Complexes
7. XRCC1 Recruitment to DNA Damage
8. XRCC1 Variants
9. XRCC1 and Disease
Acknowledgements
- Conflict of InterestThe authors declare no conflict of interest.
References
- Thompson, L.H.; Brookman, K.W.; Jones, N.J.; Allen, S.A.; Carrano, A.V. Molecular cloning of the human XRCC1 gene, which corrects defective DNA strand break repair and sister chromatid exchange. Mol. Cell Biol 1990, 10, 6160–6171. [Google Scholar]
- Tebbs, R.S.; Flannery, M.L.; Meneses, J.J.; Hartmann, A.; Tucker, J.D.; Thompson, L.H.; Cleaver, J.E.; Pedersen, R.A. Requirement for the Xrcc1 DNA base excision repair gene during early mouse development. Dev. Biol 1999, 208, 513–529. [Google Scholar]
- Xanthoudakis, S.; Smeyne, R.J.; Wallace, J.D.; Curran, T. The redox/DNA repair protein, Ref-1, is essential for early embryonic development in mice. Proc. Natl. Acad. Sci. USA 1996, 93, 8919–8923. [Google Scholar]
- Stucki, M.; Pascucci, B.; Parlanti, E.; Fortini, P.; Wilson, S.H.; Hubscher, U.; Dogliotti, E. Mammalian base excision repair by DNA polymerases delta and epsilon. Oncogene 1998, 17, 835–843. [Google Scholar]
- Puebla-Osorio, N.; Lacey, D.B.; Alt, F.W.; Zhu, C. Early embryonic lethality due to targeted inactivation of DNA ligase III. Mol. Cell Biol 2006, 26, 3935–3941. [Google Scholar]
- Audebert, M.; Salles, B.; Calsou, P. Involvement of poly(ADP-ribose) polymerase-1 and XRCC1/DNA ligase III in an alternative route for DNA double-strand breaks rejoining. J. Biol. Chem 2004, 279, 55117–55126. [Google Scholar]
- Moser, J.; Kool, H.; Giakzidis, I.; Caldecott, K.; Mullenders, L.H.; Fousteri, M.I. Sealing of chromosomal DNA nicks during nucleotide excision repair requires XRCC1 and DNA ligase III alpha in a cell-cycle-specific manner. Mol. Cell 2007, 27, 311–323. [Google Scholar]
- Lee, Y.; Katyal, S.; Li, Y.; El-Khamisy, S.F.; Russell, H.R.; Caldecott, K.W.; McKinnon, P.J. The genesis of cerebellar interneurons and the prevention of neural DNA damage require XRCC1. Nat. Neurosci 2009, 12, 973–980. [Google Scholar]
- Tebbs, R.S.; Thompson, L.H.; Cleaver, J.E. Rescue of Xrcc1 knockout mouse embryo lethality by transgene-complementation. DNA Repair (Amst.) 2003, 2, 1405–1417. [Google Scholar]
- McNeill, D.R.; Lin, P.C.; Miller, M.G.; Pistell, P.J.; de Souza-Pinto, N.C.; Fishbein, K.W.; Spencer, R.G.; Liu, Y.; Pettan-Brewer, C.; Ladiges, W.C.; et al. XRCC1 haploinsufficiency in mice has little effect on aging, but adversely modifies exposure-dependent susceptibility. Nucleic Acids Res 2011, 39, 7992–8004. [Google Scholar]
- Lindahl, T.; Barnes, D.E. Repair of endogenous DNA damage. Cold Spring Harb. Symp. Quant. Biol 2000, 65, 127–133. [Google Scholar]
- Araten, D.J.; Golde, D.W.; Zhang, R.H.; Thaler, H.T.; Gargiulo, L.; Notaro, R.; Luzzatto, L. A quantitative measurement of the human somatic mutation rate. Cancer Res 2005, 65, 8111–8117. [Google Scholar]
- Masson, M.; Niedergang, C.; Schreiber, V.; Muller, S.; Menissier-de Murcia, J.; de Murcia, G. XRCC1 is specifically associated with poly(ADP-ribose) polymerase and negatively regulates its activity following DNA damage. Mol. Cell Biol 1998, 18, 3563–3571. [Google Scholar]
- Fan, J.; Otterlei, M.; Wong, H.K.; Tomkinson, A.E.; Wilson, D.M., Jr. XRCC1 co-localizes and physically interacts with PCNA. Nucleic Acids Res. 2004, 32, 2193–2201. [Google Scholar]
- Akbari, M.; Solvang-Garten, K.; Hanssen-Bauer, A.; Lieske, N.V.; Pettersen, H.S.; Pettersen, G.K.; Wilson, D.M., Jr; Krokan, H.E.; Otterlei, M. Direct interaction between XRCC1 and UNG2 facilitates rapid repair of uracil in DNA by XRCC1 complexes. DNA Repair (Amst.) 2010, 9, 785–795. [Google Scholar]
- Hanssen-Bauer, A.; Solvang-Garten, K.; Sundheim, O.; Pena-Diaz, J.; Andersen, S.; Slupphaug, G.; Krokan, H.E.; Wilson, D.M., Jr; Akbari, M.; Otterlei, M. XRCC1 coordinates disparate responses and multiprotein repair complexes depending on the nature and context of the DNA damage. Environ. Mol. Mutagen. 2011, 52, 623–635. [Google Scholar]
- Kubota, Y.; Nash, R.A.; Klungland, A.; Schar, P.; Barnes, D.E.; Lindahl, T. Reconstitution of DNA base excision-repair with purified human proteins: interaction between DNA polymerase beta and the XRCC1 protein. EMBO J 1996, 15, 6662–6670. [Google Scholar]
- Caldecott, K.W.; Aoufouchi, S.; Johnson, P.; Shall, S. XRCC1 polypeptide interacts with DNA polymerase beta and possibly poly (ADP-ribose) polymerase, and DNA ligase III is a novel molecular “nick-sensor” in vitro. Nucleic Acids Res 1996, 24, 4387–4394. [Google Scholar]
- Marintchev, A.; Mullen, M.A.; Maciejewski, M.W.; Pan, B.; Gryk, M.R.; Mullen, G.P. Solution structure of the single-strand break repair protein XRCC1 N-terminal domain. Nat. Struct. Biol 1999, 6, 884–893. [Google Scholar]
- Mani, R.S.; Karimi-Busheri, F.; Fanta, M.; Caldecott, K.W.; Cass, C.E.; Weinfeld, M. Biophysical characterization of human XRCC1 and its binding to damaged and undamaged DNA. Biochemistry 2004, 43, 16505–16514. [Google Scholar]
- Cuneo, M.J.; London, R.E. Oxidation state of the XRCC1 N-terminal domain regulates DNA polymerase beta binding affinity. Proc. Natl. Acad. Sci. USA 2010, 107, 6805–6810. [Google Scholar]
- Parsons, J.L.; Tait, P.S.; Finch, D.; Dianova, I.; Allinson, S.L.; Dianov, G.L. CHIP-mediated degradation and DNA damage-dependent stabilization regulate base excision repair proteins. Mol. Cell 2008, 29, 477–487. [Google Scholar]
- Gilljam, K.M.; Feyzi, E.; Aas, P.A.; Sousa, M.M.; Muller, R.; Vagbo, C.B.; Catterall, T.C.; Liabakk, N.B.; Slupphaug, G.; Drablos, F.; et al. Identification of a novel, widespread, and functionally important PCNA-binding motif. J. Cell Biol 2009, 186, 645–654. [Google Scholar] [Green Version]
- Warbrick, E. The puzzle of PCNA’s many partners. Bioessays 2000, 22, 997–1006. [Google Scholar]
- Marsin, S.; Vidal, A.E.; Sossou, M.; Menissier-de Murcia, J.; Le Page, F.; Boiteux, S.; de Murcia, G.; Radicella, J.P. Role of XRCC1 in the coordination and stimulation of oxidative DNA damage repair initiated by the DNA glycosylase hOGG1. J. Biol. Chem 2003, 278, 44068–44074. [Google Scholar]
- Campalans, A.; Marsin, S.; Nakabeppu, Y.; O’Connor, T.R.; Boiteux, S.; Radicella, J.P. XRCC1 interactions with multiple DNA glycosylases: A model for its recruitment to base excision repair. DNA Repair (Amst.) 2005, 4, 826–835. [Google Scholar]
- Vidal, A.E.; Boiteux, S.; Hickson, I.D.; Radicella, J.P. XRCC1 coordinates the initial and late stages of DNA abasic site repair through protein-protein interactions. EMBO J 2001, 20, 6530–6539. [Google Scholar]
- Bork, P.; Hofmann, K.; Bucher, P.; Neuwald, A.F.; Altschul, S.F.; Koonin, E.V. A superfamily of conserved domains in DNA damage-responsive cell cycle checkpoint proteins. FASEB J 1997, 11, 68–76. [Google Scholar]
- Taylor, R.M.; Thistlethwaite, A.; Caldecott, K.W. Central role for the XRCC1 BRCT I domain in mammalian DNA single-strand break repair. Mol. Cell Biol 2002, 22, 2556–2563. [Google Scholar]
- Kubota, Y.; Horiuchi, S. Independent roles of XRCC1’s two BRCT motifs in recovery from methylation damage. DNA Repair (Amst.) 2003, 2, 407–415. [Google Scholar]
- Hanssen-Bauer, A.; Solvang-Garten, K.; Gilljam, K.M.; Torseth, K.; Wilson, D.M., Jr; Akbari, M.; Otterlei, M. The region of XRCC1 which harbours the three most common nonsynonymous polymorphic variants, is essential for the scaffolding function of XRCC1. DNA Repair (Amst.) 2012, 11, 357–366. [Google Scholar] [Green Version]
- Pleschke, J.M.; Kleczkowska, H.E.; Strohm, M.; Althaus, F.R. Poly(ADP-ribose) binds to specific domains in DNA damage checkpoint proteins. J. Biol. Chem 2000, 275, 40974–40980. [Google Scholar]
- Schreiber, V.; Ame, J.C.; Dolle, P.; Schultz, I.; Rinaldi, B.; Fraulob, V.; Menissier-de Murcia, J.; de Murcia, G. Poly(ADP-ribose) polymerase-2 (PARP-2) is required for efficient base excision DNA repair in association with PARP-1 and XRCC1. J. Biol. Chem 2002, 277, 23028–23036. [Google Scholar]
- Nash, R.A.; Caldecott, K.W.; Barnes, D.E.; Lindahl, T. XRCC1 protein interacts with one of two distinct forms of DNA ligase III. Biochemistry 1997, 36, 5207–5211. [Google Scholar]
- Taylor, R.M.; Wickstead, B.; Cronin, S.; Caldecott, K.W. Role of a BRCT domain in the interaction of DNA ligase III-alpha with the DNA repair protein XRCC1. Curr. Biol 1998, 8, 877–880. [Google Scholar]
- Caldecott, K.W.; Tucker, J.D.; Stanker, L.H.; Thompson, L.H. Characterization of the XRCC1-DNA ligase III complex in vitro and its absence from mutant hamster cells. Nucleic Acids Res 1995, 23, 4836–4843. [Google Scholar]
- Beernink, P.T.; Hwang, M.; Ramirez, M.; Murphy, M.B.; Doyle, S.A.; Thelen, M.P. Specificity of protein interactions mediated by BRCT domains of the XRCC1 DNA repair protein. J. Biol. Chem 2005, 280, 30206–30213. [Google Scholar]
- Levy, N.; Martz, A.; Bresson, A.; Spenlehauer, C.; de Murcia, G.; Menissier-de Murcia, J. XRCC1 is phosphorylated by DNA-dependent protein kinase in response to DNA damage. Nucleic Acids Res 2006, 34, 32–41. [Google Scholar]
- Cuneo, M.J.; Gabel, S.A.; Krahn, J.M.; Ricker, M.A.; London, R.E. The structural basis for partitioning of the XRCC1/DNA ligase III-alpha BRCT-mediated dimer complexes. Nucleic Acids Res. 2011. [Google Scholar] [CrossRef]
- Bekker-Jensen, S.; Fugger, K.; Danielsen, J.R.; Gromova, I.; Sehested, M.; Celis, J.; Bartek, J.; Lukas, J.; Mailand, N. Human Xip1 (C2orf13) is a novel regulator of cellular responses to DNA strand breaks. J. Biol. Chem 2007, 282, 19638–19643. [Google Scholar]
- Iles, N.; Rulten, S.; El-Khamisy, S.F.; Caldecott, K.W. APLF (C2orf13) is a novel human protein involved in the cellular response to chromosomal DNA strand breaks. Mol. Cell Biol 2007, 27, 3793–3803. [Google Scholar]
- Loizou, J.I.; El-Khamisy, S.F.; Zlatanou, A.; Moore, D.J.; Chan, D.W.; Qin, J.; Sarno, S.; Meggio, F.; Pinna, L.A.; Caldecott, K.W. The protein kinase CK2 facilitates repair of chromosomal DNA single-strand breaks. Cell 2004, 117, 17–28. [Google Scholar]
- Luo, H.; Chan, D.W.; Yang, T.; Rodriguez, M.; Chen, B.P.; Leng, M.; Mu, J.J.; Chen, D.; Songyang, Z.; Wang, Y.; Qin, J. A new XRCC1-containing complex and its role in cellular survival of methyl methanesulfonate treatment. Mol. Cell Biol 2004, 24, 8356–8365. [Google Scholar]
- Kanno, S.; Kuzuoka, H.; Sasao, S.; Hong, Z.; Lan, L.; Nakajima, S.; Yasui, A. A novel human AP endonuclease with conserved zinc-finger-like motifs involved in DNA strand break responses. EMBO J 2007, 26, 2094–2103. [Google Scholar]
- Date, H.; Igarashi, S.; Sano, Y.; Takahashi, T.; Takano, H.; Tsuji, S.; Nishizawa, M.; Onodera, O. The FHA domain of aprataxin interacts with the C-terminal region of XRCC1. Biochem. Biophys. Res. Commun 2004, 325, 1279–1285. [Google Scholar]
- Hofmann, K.; Bucher, P. The FHA domain: A putative nuclear signalling domain found in protein kinases and transcription factors. Trends Biochem. Sci 1995, 20, 347–349. [Google Scholar]
- Whitehouse, C.J.; Taylor, R.M.; Thistlethwaite, A.; Zhang, H.; Karimi-Busheri, F.; Lasko, D.D.; Weinfeld, M.; Caldecott, K.W. XRCC1 stimulates human polynucleotide kinase activity at damaged DNA termini and accelerates DNA single-strand break repair. Cell 2001, 104, 107–117. [Google Scholar]
- Almeida, K.H.; Sobol, R.W. A unified view of base excision repair: Lesion-dependent protein complexes regulated by post-translational modification. DNA Repair (Amst.) 2007, 6, 695–711. [Google Scholar]
- Chou, W.C.; Wang, H.C.; Wong, F.H.; Ding, S.L.; Wu, P.E.; Shieh, S.Y.; Shen, C.Y. Chk2-dependent phosphorylation of XRCC1 in the DNA damage response promotes base excision repair. EMBO J 2008, 27, 3140–3150. [Google Scholar]
- Hanif, I.M.; Shazib, M.A.; Ahmad, K.A.; Pervaiz, S. Casein Kinase II: An attractive target for anti-cancer drug design. Int. J. Biochem. Cell Biol 2010, 42, 1602–1605. [Google Scholar]
- Akbari, M.; Otterlei, M.; Pena-Diaz, J.; Aas, P.A.; Kavli, B.; Liabakk, N.B.; Hagen, L.; Imai, K.; Durandy, A.; Slupphaug, G.; Krokan, H.E. Repair of U/G and U/A in DNA by UNG2-associated repair complexes takes place predominantly by short-patch repair both in proliferating and growth-arrested cells. Nucleic Acids Res 2004, 32, 5486–5498. [Google Scholar]
- Heale, J.T.; Ball, A.R., Jr; Schmiesing, J.A.; Kim, J.S.; Kong, X.; Zhou, S.; Hudson, D.F.; Earnshaw, W.C.; Yokomori, K. Condensin I interacts with the PARP-1-XRCC1 complex and functions in DNA single-strand break repair. Mol. Cell 2006, 21, 837–848. [Google Scholar]
- Parlanti, E.; Locatelli, G.; Maga, G.; Dogliotti, E. Human base excision repair complex is physically associated to DNA replication and cell cycle regulatory proteins. Nucleic Acids Res 2007, 35, 1569–1577. [Google Scholar]
- Prasad, R.; Shock, D.D.; Beard, W.A.; Wilson, S.H. Substrate channeling in mammalian base excision repair pathways: Passing the baton. J. Biol. Chem 2010, 285, 40479–40488. [Google Scholar]
- Caldecott, K.W. Mammalian DNA single-strand break repair: An X-ra(y)ted affair. Bioessays 2001, 23, 447–455. [Google Scholar]
- Otterlei, M.; Warbrick, E.; Nagelhus, T.A.; Haug, T.; Slupphaug, G.; Akbari, M.; Aas, P.A.; Steinsbekk, K.; Bakke, O.; Krokan, H.E. Post-replicative base excision repair in replication foci. EMBO J 1999, 18, 3834–3844. [Google Scholar]
- Caldecott, K.W.; McKeown, C.K.; Tucker, J.D.; Ljungquist, S.; Thompson, L.H. An interaction between the mammalian DNA repair protein XRCC1 and DNA ligase III. Mol. Cell Biol 1994, 14, 68–76. [Google Scholar]
- Akbari, M.; Krokan, H.E. Base excision repair efficiency and mechanism in nuclear extracts are influenced by the ratio between volume of nuclear extraction buffer and nuclei-implications for comparative studies. Mutat. Res 2011, 736, 33–38. [Google Scholar]
- Eissenberg, J.C.; Ayyagari, R.; Gomes, X.V.; Burgers, P.M. Mutations in yeast proliferating cell nuclear antigen define distinct sites for interaction with DNA polymerase delta and DNA polymerase epsilon. Mol. Cell Biol 1997, 17, 6367–6378. [Google Scholar]
- Levin, D.S.; Bai, W.; Yao, N.; O’Donnell, M.; Tomkinson, A.E. An interaction between DNA ligase I and proliferating cell nuclear antigen: Implications for Okazaki fragment synthesis and joining. Proc. Natl. Acad. Sci. USA 1997, 94, 12863–12868. [Google Scholar]
- Zhang, P.; Mo, J.Y.; Perez, A.; Leon, A.; Liu, L.; Mazloum, N.; Xu, H.; Lee, M.Y. Direct interaction of proliferating cell nuclear antigen with the p125 catalytic subunit of mammalian DNA polymerase delta. J. Biol. Chem 1999, 274, 26647–26653. [Google Scholar]
- Parker, A.; Gu, Y.; Mahoney, W.; Lee, S.H.; Singh, K.K.; Lu, A.L. Human homolog of the MutY repair protein (hMYH) physically interacts with proteins involved in long patch DNA base excision repair. J. Biol. Chem 2001, 276, 5547–5555. [Google Scholar]
- Kedar, P.S.; Kim, S.J.; Robertson, A.; Hou, E.; Prasad, R.; Horton, J.K.; Wilson, S.H. Direct interaction between mammalian DNA polymerase beta and proliferating cell nuclear antigen. J. Biol. Chem 2002, 277, 31115–31123. [Google Scholar]
- Frouin, I.; Maga, G.; Denegri, M.; Riva, F.; Savio, M.; Spadari, S.; Prosperi, E.; Scovassi, A.I. Human proliferating cell nuclear antigen, poly(ADP-ribose) polymerase-1, and p21waf1/cip1. A dynamic exchange of partners. J. Biol. Chem 2003, 278, 39265–39268. [Google Scholar]
- Oyama, M.; Wakasugi, M.; Hama, T.; Hashidume, H.; Iwakami, Y.; Imai, R.; Hoshino, S.; Morioka, H.; Ishigaki, Y.; Nikaido, O.; Matsunaga, T. Human NTH1 physically interacts with p53 and proliferating cell nuclear antigen. Biochem. Biophys. Res. Commun 2004, 321, 183–191. [Google Scholar]
- Xia, L.; Zheng, L.; Lee, H.W.; Bates, S.E.; Federico, L.; Shen, B.; O’Connor, T.R. Human 3-methyladenine-DNA glycosylase: Effect of sequence context on excision, association with PCNA, and stimulation by AP endonuclease. J. Mol. Biol 2005, 346, 1259–1274. [Google Scholar]
- Li, X.; Li, J.; Harrington, J.; Lieber, M.R.; Burgers, P.M. Lagging strand DNA synthesis at the eukaryotic replication fork involves binding and stimulation of FEN-1 by proliferating cell nuclear antigen. J. Biol. Chem 1995, 270, 22109–22112. [Google Scholar]
- Loor, G.; Zhang, S.J.; Zhang, P.; Toomey, N.L.; Lee, M.Y. Identification of DNA replication and cell cycle proteins that interact with PCNA. Nucleic Acids Res 1997, 25, 5041–5046. [Google Scholar]
- Dou, H.; Theriot, C.A.; Das, A.; Hegde, M.L.; Matsumoto, Y.; Boldogh, I.; Hazra, T.K.; Bhakat, K.K.; Mitra, S. Interaction of the human DNA glycosylase NEIL1 with proliferating cell nuclear antigen. The potential for replication-associated repair of oxidized bases in mammalian genomes. J. Biol. Chem 2008, 283, 3130–3140. [Google Scholar]
- Bryant, H.E.; Petermann, E.; Schultz, N.; Jemth, A.S.; Loseva, O.; Issaeva, N.; Johansson, F.; Fernandez, S.; McGlynn, P.; Helleday, T. PARP is activated at stalled forks to mediate Mre11-dependent replication restart and recombination. EMBO J 2009, 28, 2601–2615. [Google Scholar]
- Wood, R.D.; Mitchell, M.; Lindahl, T. Human DNA repair genes, 2005. Mutat. Res 2005, 577, 275–283. [Google Scholar]
- Odell, I.D.; Wallace, S.S.; Pederson, D.S. Rules of engagement for base excision repair in chromatin. J. Cell Physiol 2012, 228, 258–266. [Google Scholar]
- Ame, J.C.; Rolli, V.; Schreiber, V.; Niedergang, C.; Apiou, F.; Decker, P.; Muller, S.; Hoger, T.; Menissier-de Murcia, J.; de Murcia, G. PARP-2, A novel mammalian DNA damage-dependent poly(ADP-ribose) polymerase. J. Biol. Chem 1999, 274, 17860–17868. [Google Scholar]
- Gradwohl, G.; Menissier de Murcia, J.M.; Molinete, M.; Simonin, F.; Koken, M.; Hoeijmakers, J.H.; de Murcia, G. The second zinc-finger domain of poly(ADP-ribose) polymerase determines specificity for single-stranded breaks in DNA. Proc. Natl. Acad. Sci. USA 1990, 87, 2990–2994. [Google Scholar]
- Okano, S.; Lan, L.; Caldecott, K.W.; Mori, T.; Yasui, A. Spatial and temporal cellular responses to single-strand breaks in human cells. Mol. Cell Biol 2003, 23, 3974–3981. [Google Scholar]
- D’Amours, D.; Desnoyers, S.; D’Silva, I.; Poirier, G.G. Poly(ADP-ribosyl)ation reactions in the regulation of nuclear functions. Biochem. J. 1999, 342 Pt 2, 249–268. [Google Scholar]
- Schreiber, V.; Dantzer, F.; Ame, J.C.; de Murcia, G. Poly(ADP-ribose): Novel functions for an old molecule. Nat. Rev. Mol. Cell Biol 2006, 7, 517–528. [Google Scholar]
- Gagne, J.P.; Isabelle, M.; Lo, K.S.; Bourassa, S.; Hendzel, M.J.; Dawson, V.L.; Dawson, T.M.; Poirier, G.G. Proteome-wide identification of poly(ADP-ribose) binding proteins and poly(ADP-ribose)-associated protein complexes. Nucleic Acids Res 2008, 36, 6959–6976. [Google Scholar] [Green Version]
- El-Khamisy, S.F.; Masutani, M.; Suzuki, H.; Caldecott, K.W. A requirement for PARP-1 for the assembly or stability of XRCC1 nuclear foci at sites of oxidative DNA damage. Nucleic Acids Res 2003, 31, 5526–5533. [Google Scholar]
- Mortusewicz, O.; Ame, J.C.; Schreiber, V.; Leonhardt, H. Feedback-regulated poly(ADP-ribosyl)ation by PARP-1 is required for rapid response to DNA damage in living cells. Nucleic Acids Res 2007, 35, 7665–7675. [Google Scholar]
- Godon, C.; Cordelieres, F.P.; Biard, D.; Giocanti, N.; Megnin-Chanet, F.; Hall, J.; Favaudon, V. PARP inhibition versus PARP-1 silencing: Different outcomes in terms of single-strand break repair and radiation susceptibility. Nucleic Acids Res 2008, 36, 4454–4464. [Google Scholar]
- Ginsberg, G.; Angle, K.; Guyton, K.; Sonawane, B. Polymorphism in the DNA repair enzyme XRCC1: Utility of current database and implications for human health risk assessment. Mutat. Res 2011, 727, 1–15. [Google Scholar]
- Berquist, B.R.; Singh, D.K.; Fan, J.; Kim, D.; Gillenwater, E.; Kulkarni, A.; Bohr, V.A.; Ackerman, E.J.; Tomkinson, A.E.; Wilson, D.M. Functional capacity of XRCC1 protein variants identified in DNA repair-deficient Chinese hamster ovary cell lines and the human population. Nucleic Acids Res 2010, 38, 5023–5035. [Google Scholar]
- Xue, H.; Ni, P.; Lin, B.; Xu, H.; Huang, G. X-ray repair cross-complementing group 1 (XRCC1) genetic polymorphisms and gastric cancer risk: A HuGE review and meta-analysis. Am. J. Epidemiol 2011, 173, 363–375. [Google Scholar]
- Huang, J.; Zhang, J.; Zhao, Y.; Liao, B.; Liu, J.; Li, L.; Liao, M.; Wang, L. The Arg194Trp polymorphism in the XRCC1 gene and cancer risk in Chinese Mainland population: A meta-analysis. Mol. Biol. Rep 2011, 38, 4565–4573. [Google Scholar]
- Gsur, A.; Bernhart, K.; Baierl, A.; Feik, E.; Fuhrlinger, G.; Hofer, P.; Leeb, G.; Mach, K.; Micksche, M. No association of XRCC1 polymorphisms Arg194Trp and Arg399Gln with colorectal cancer risk. Cancer Epidemiol 2011, 35, e38–e41. [Google Scholar]
- Chen, B.; Zhou, Y.; Yang, P.; Wu, X.T. Polymorphisms of XRCC1 and gastric cancer susceptibility: A meta-analysis. Mol. Biol. Rep 2011, 39, 1305–1313. [Google Scholar]
- Li, H.; Ha, T.C.; Tai, B.C. XRCC1 gene polymorphisms and breast cancer risk in different populations: A meta-analysis. Breast 2009, 18, 183–191. [Google Scholar]
- Jiang, J.; Liang, X.; Zhou, X.; Huang, R.; Chu, Z.; Zhan, Q.; Lin, H. DNA repair gene X-ray repair cross complementing group 1 Arg194Trp polymorphism on the risk of lung cancer: A meta-analysis on 22 studies. J. Thorac. Oncol 2010, 5, 1741–1747. [Google Scholar]
- Wu, K.; Su, D.; Lin, K.; Luo, J.; Au, W.W. XRCC1 Arg399Gln gene polymorphism and breast cancer risk: A meta-analysis based on case-control studies. Asian Pac. J. Cancer Prev 2011, 12, 2237–2243. [Google Scholar]
- El-Khamisy, S.F.; Saifi, G.M.; Weinfeld, M.; Johansson, F.; Helleday, T.; Lupski, J.R.; Caldecott, K.W. Defective DNA single-strand break repair in spinocerebellar ataxia with axonal neuropathy-1. Nature 2005, 434, 108–113. [Google Scholar]
- Plo, I.; Liao, Z.Y.; Barcelo, J.M.; Kohlhagen, G.; Caldecott, K.W.; Weinfeld, M.; Pommier, Y. Association of XRCC1 and tyrosyl DNA phosphodiesterase (Tdp1) for the repair of topoisomerase I-mediated DNA lesions. DNA Repair (Amst.) 2003, 2, 1087–1100. [Google Scholar]
- Date, H.; Onodera, O.; Tanaka, H.; Iwabuchi, K.; Uekawa, K.; Igarashi, S.; Koike, R.; Hiroi, T.; Yuasa, T.; Awaya, Y.; et al. Early-onset ataxia with ocular motor apraxia and hypoalbuminemia is caused by mutations in a new HIT superfamily gene. Nat. Genet 2001, 29, 184–188. [Google Scholar]
- Moreira, M.C.; Barbot, C.; Tachi, N.; Kozuka, N.; Uchida, E.; Gibson, T.; Mendonca, P.; Costa, M.; Barros, J.; Yanagisawa, T.; et al. The gene mutated in ataxia-ocular apraxia 1 encodes the new HIT/Zn-finger protein aprataxin. Nat. Genet 2001, 29, 189–193. [Google Scholar]
- Wei, X.; Chen, D.; Lv, T. A functional polymorphism in XRCC1 is associated with glioma risk: Evidence from a meta-analysis. Mol. Biol. Rep. 2012. [Google Scholar] [CrossRef]
- Gencer, M.; Dasdemir, S.; Cakmakoglu, B.; Cetinkaya, Y.; Varlibas, F.; Tireli, H.; Kucukali, C.I.; Ozkok, E.; Aydin, M. DNA repair genes in Parkinson’s disease. Genet. Test Mol. Biomarkers 2012, 16, 504–507. [Google Scholar]
- Coppede, F.; Migheli, F.; Lo Gerfo, A.; Fabbrizi, M.R.; Carlesi, C.; Mancuso, M.; Corti, S.; Mezzina, N.; del Bo, R.; Comi, G.P.; et al. Association study between XRCC1 gene polymorphisms and sporadic amyotrophic lateral sclerosis. Amyotroph. Lateral Scler 2010, 11, 122–124. [Google Scholar]
- Yosunkaya, E.; Kucukyuruk, B.; Onaran, I.; Gurel, C.B.; Uzan, M.; Kanigur-Sultuybek, G. Glioma risk associates with polymorphisms of DNA repair genes, XRCC1 and PARP1. Br. J. Neurosurg 2010, 24, 561–565. [Google Scholar]
- Rajaraman, P.; Hutchinson, A.; Wichner, S.; Black, P.M.; Fine, H.A.; Loeffler, J.S.; Selker, R.G.; Shapiro, W.R.; Rothman, N.; Linet, M.S.; Inskip, P.D. DNA repair gene polymorphisms and risk of adult meningioma, glioma, and acoustic neuroma. Neuro-oncology 2010, 12, 37–48. [Google Scholar]
- Audebert, M.; Salles, B.; Weinfeld, M.; Calsou, P. Involvement of polynucleotide kinase in a poly(ADP-ribose) polymerase-1-dependent DNA double-strand breaks rejoining pathway. J. Mol. Biol 2006, 356, 257–265. [Google Scholar]
- Han, J.; Hankinson, S.E.; Colditz, G.A.; Hunter, D.J. Genetic variation in XRCC1, sun exposure, and risk of skin cancer. Br. J. Cancer 2004, 91, 1604–1609. [Google Scholar]
- Santonocito, C.; Scapaticci, M.; Penitente, R.; Paradisi, A.; Capizzi, R.; Lanza-Silveri, S.; Ficarra, S.; Landi, F.; Zuppi, C.; Capoluongo, E. Polymorphisms in base excision DNA repair genes and association with melanoma risk in a pilot study on Central-South Italian population. Clin. Chim. Acta 2012, 413, 1519–1524. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Xrcc1 (ENST00000262887) | Africa | America | Asia | Europe | |||||
|---|---|---|---|---|---|---|---|---|---|
| aa substitution | SNP ID | Hom. | Het. | Hom. | Het. | Hom. | Het. | Hom. | Het. |
| Arg194Trp | rs1799782 | N.D. | 14.6% | 0.6% | 19.9% | 8.4% | 40.9% | 1.3% | 9.0% |
| Arg280His | rs25489 | N.D. | 2.8% | 0.6% | 12.2% | 1.0% | 16.8% | 0.5% | 11.6% |
| Arg399Gln | rs25487 | 0.8% | 22.0% | 9.4% | 43.1% | 6.6% | 36.7% | 11.9% | 45.4% |
© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Hanssen-Bauer, A.; Solvang-Garten, K.; Akbari, M.; Otterlei, M. X-Ray Repair Cross Complementing Protein 1 in Base Excision Repair. Int. J. Mol. Sci. 2012, 13, 17210-17229. https://doi.org/10.3390/ijms131217210
Hanssen-Bauer A, Solvang-Garten K, Akbari M, Otterlei M. X-Ray Repair Cross Complementing Protein 1 in Base Excision Repair. International Journal of Molecular Sciences. 2012; 13(12):17210-17229. https://doi.org/10.3390/ijms131217210
Chicago/Turabian StyleHanssen-Bauer, Audun, Karin Solvang-Garten, Mansour Akbari, and Marit Otterlei. 2012. "X-Ray Repair Cross Complementing Protein 1 in Base Excision Repair" International Journal of Molecular Sciences 13, no. 12: 17210-17229. https://doi.org/10.3390/ijms131217210
APA StyleHanssen-Bauer, A., Solvang-Garten, K., Akbari, M., & Otterlei, M. (2012). X-Ray Repair Cross Complementing Protein 1 in Base Excision Repair. International Journal of Molecular Sciences, 13(12), 17210-17229. https://doi.org/10.3390/ijms131217210