Modulation of Adipogenic Conditions for Prospective Use of hADSCs in Adipose Tissue Engineering





Abstract
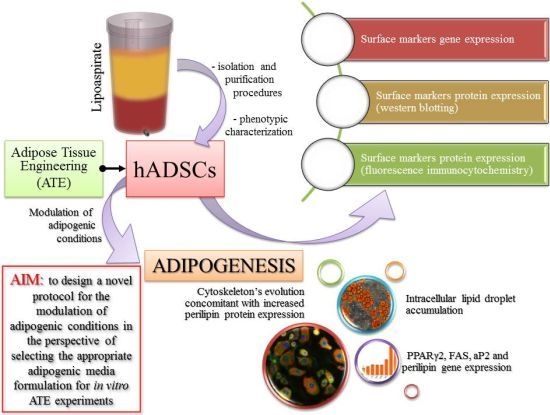
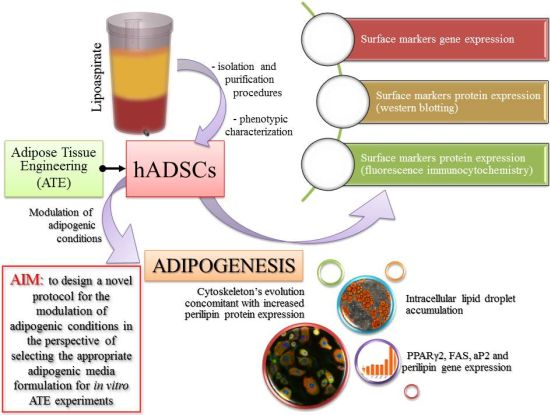
:
1. Introduction
2. Results and Discussion
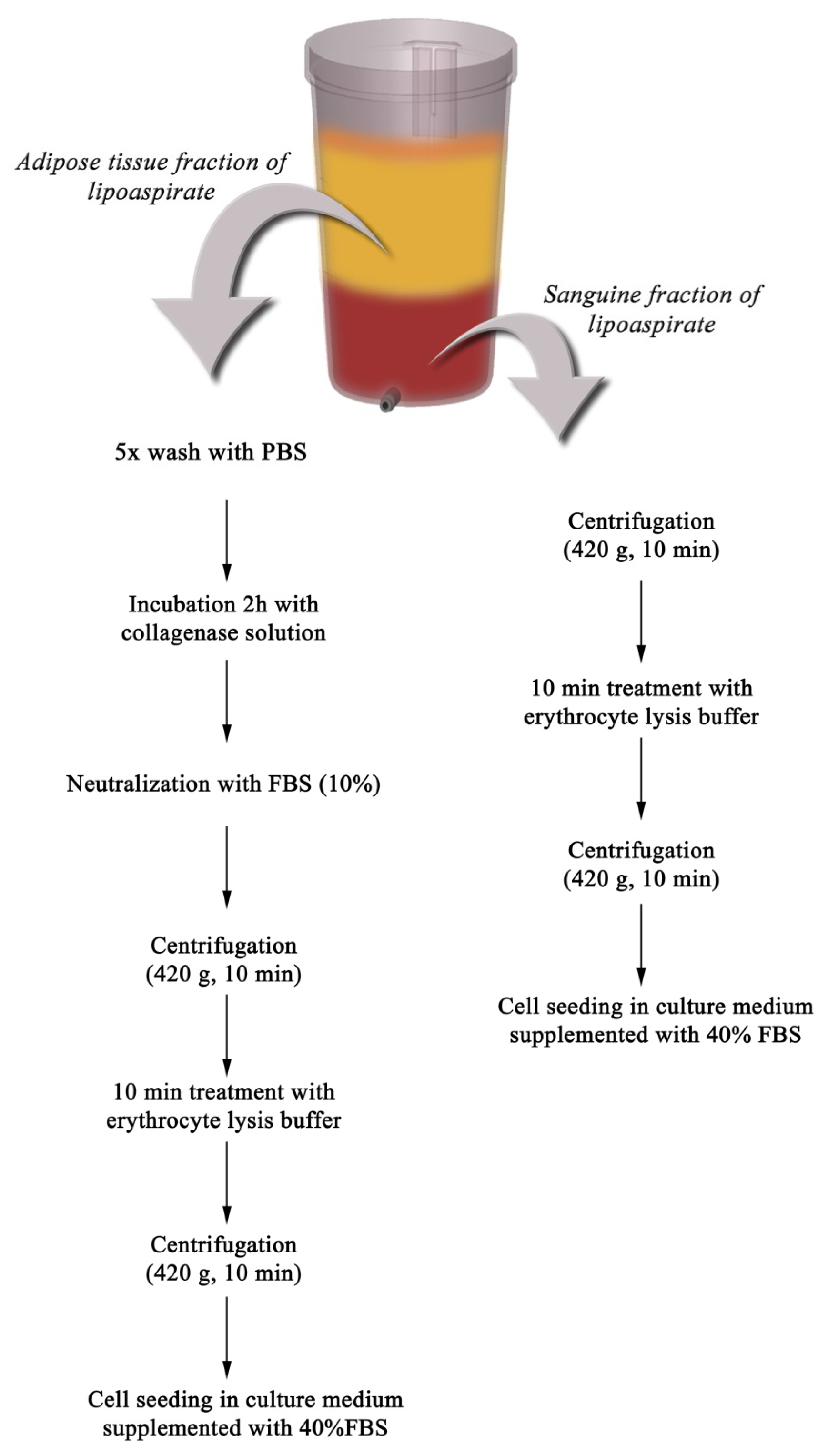
2.1. Primary Culture of PLAs-Derived Cells
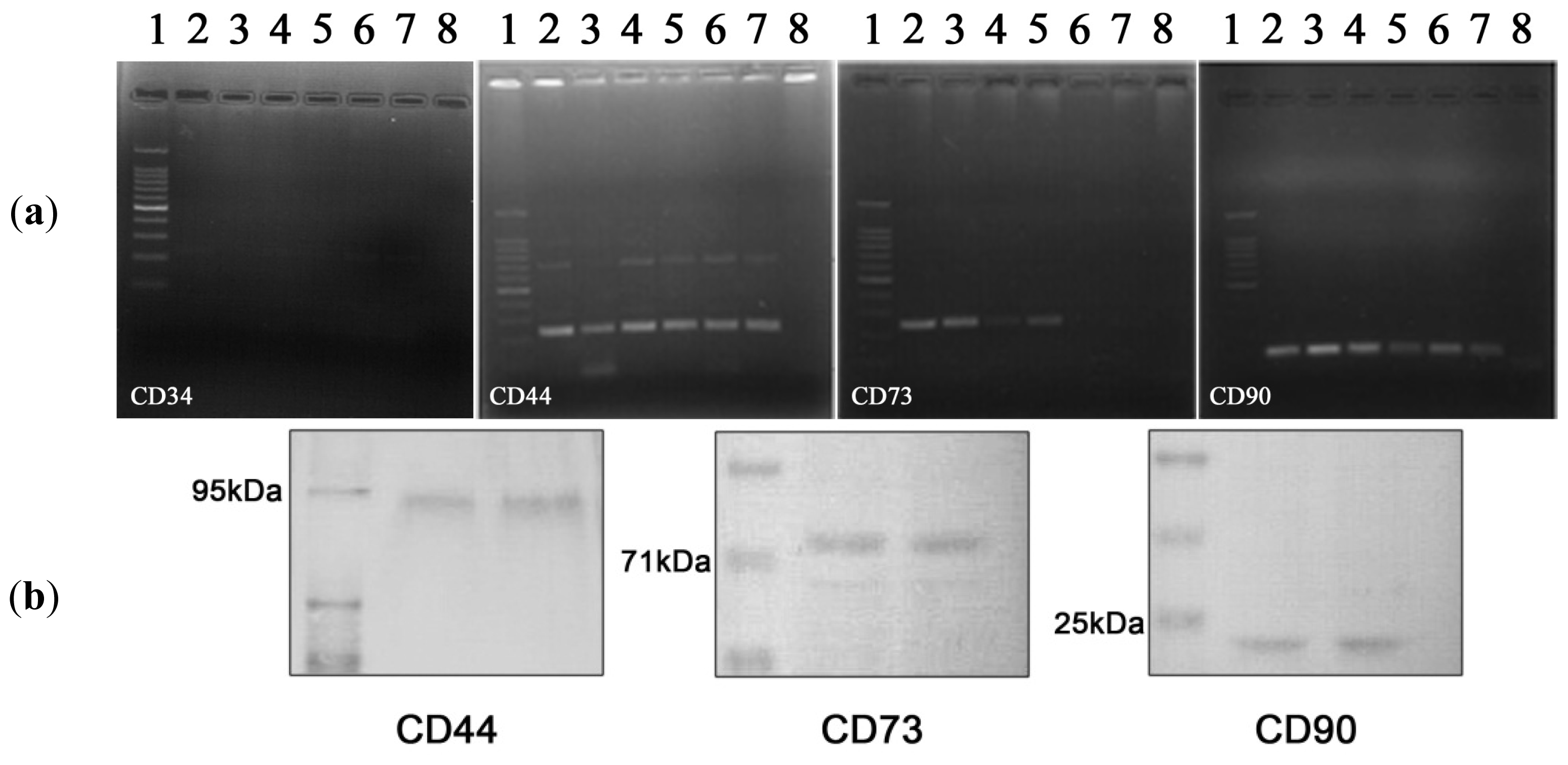
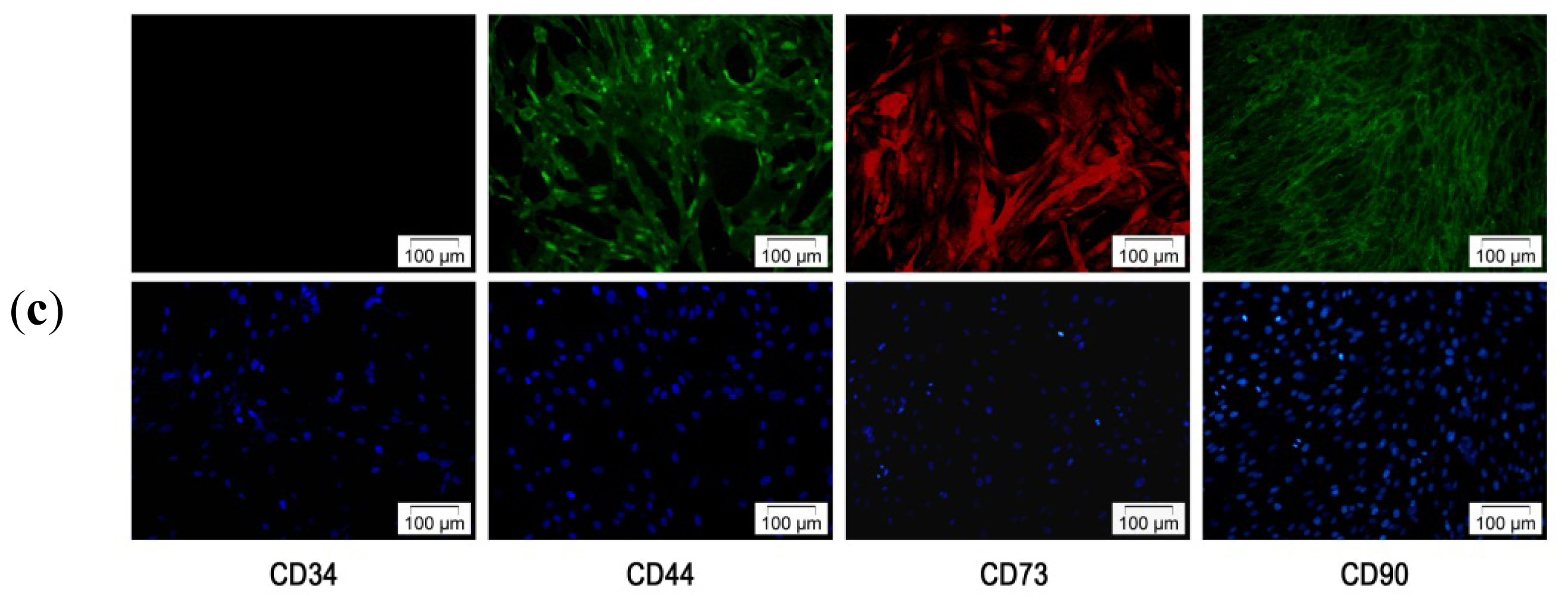
2.2. Characterization of hADSC
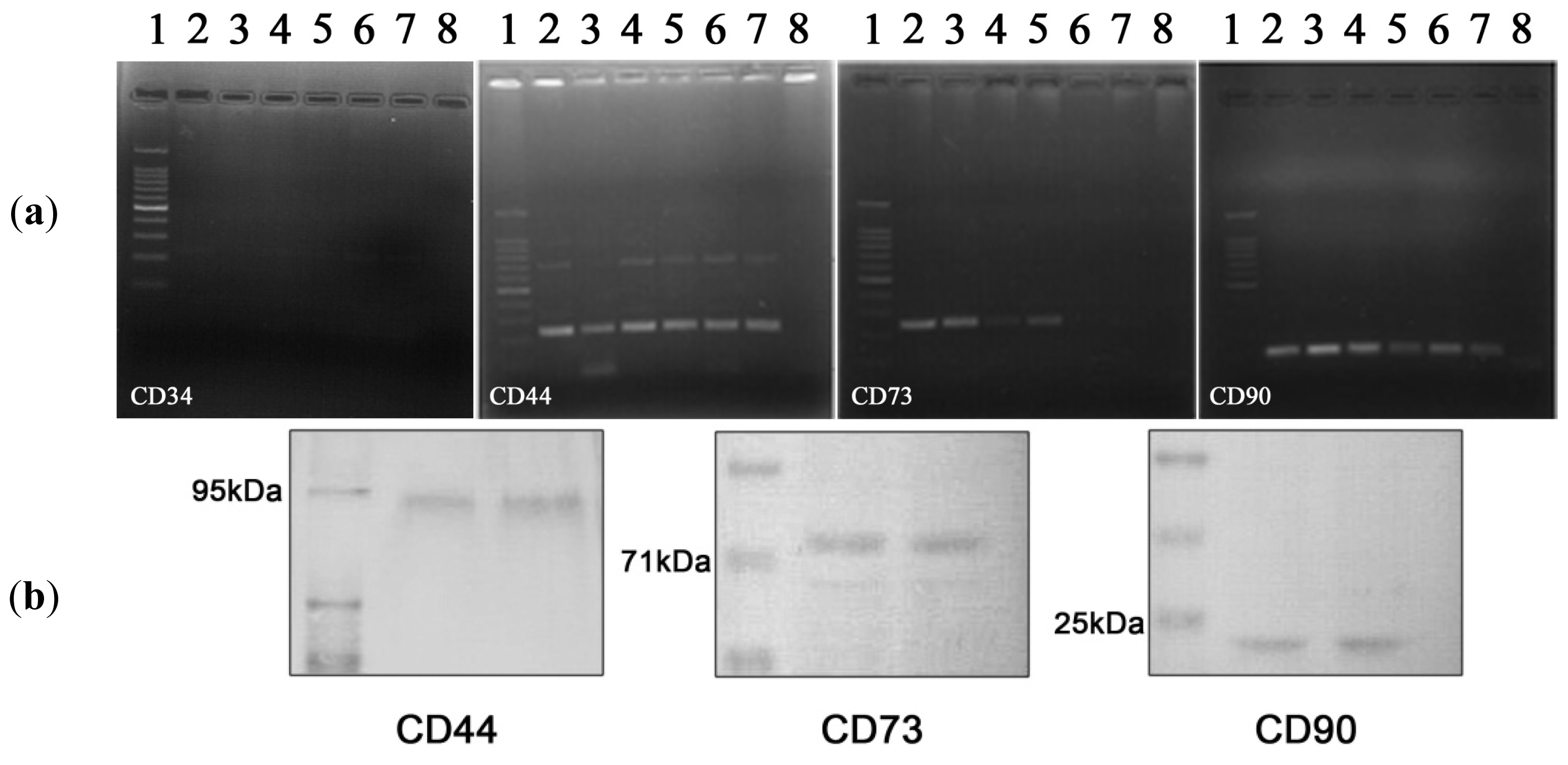
2.2.1. hADSCs Cell Surface Antigen Expression
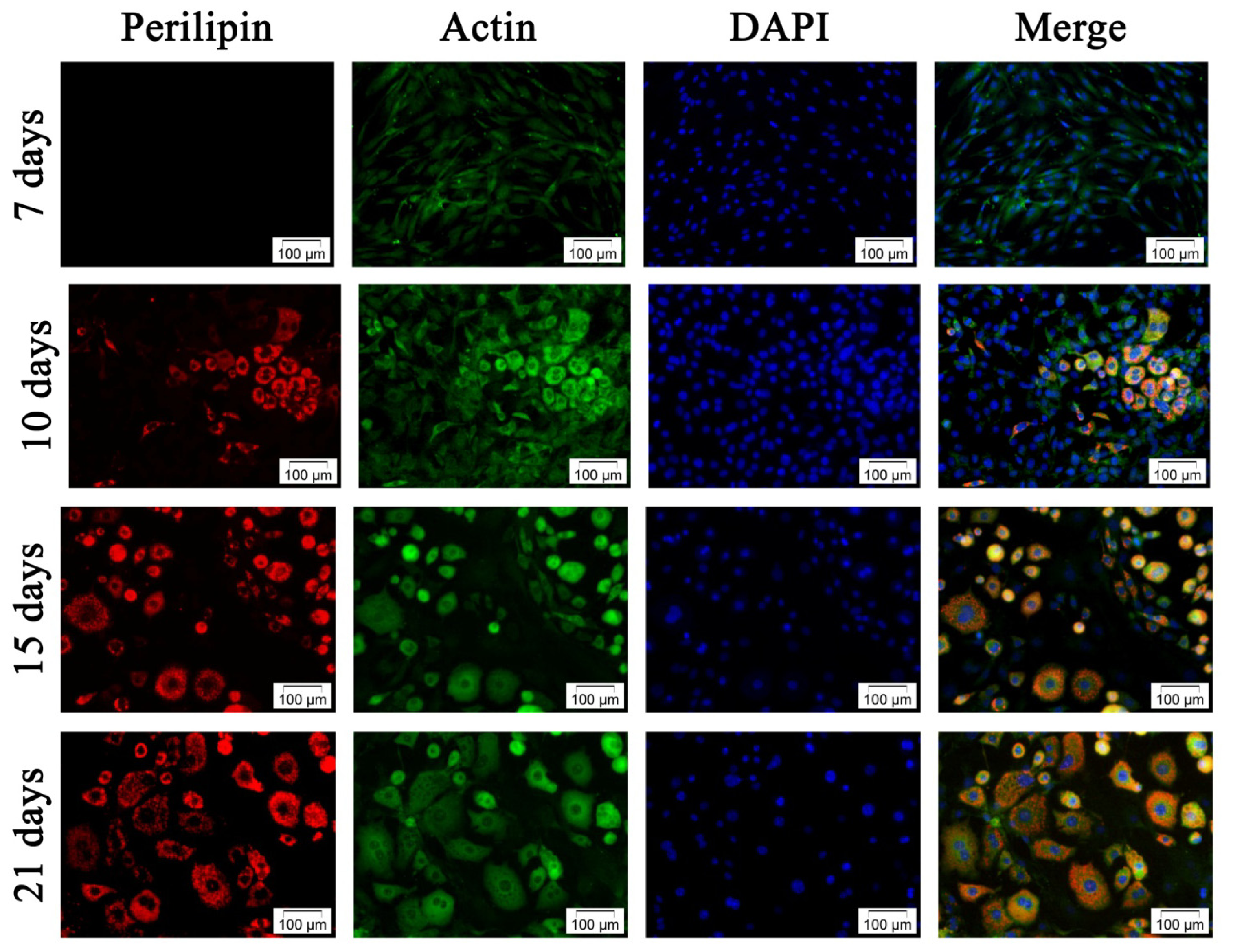
2.2.2. Adipogenic Differentiation of hADSC
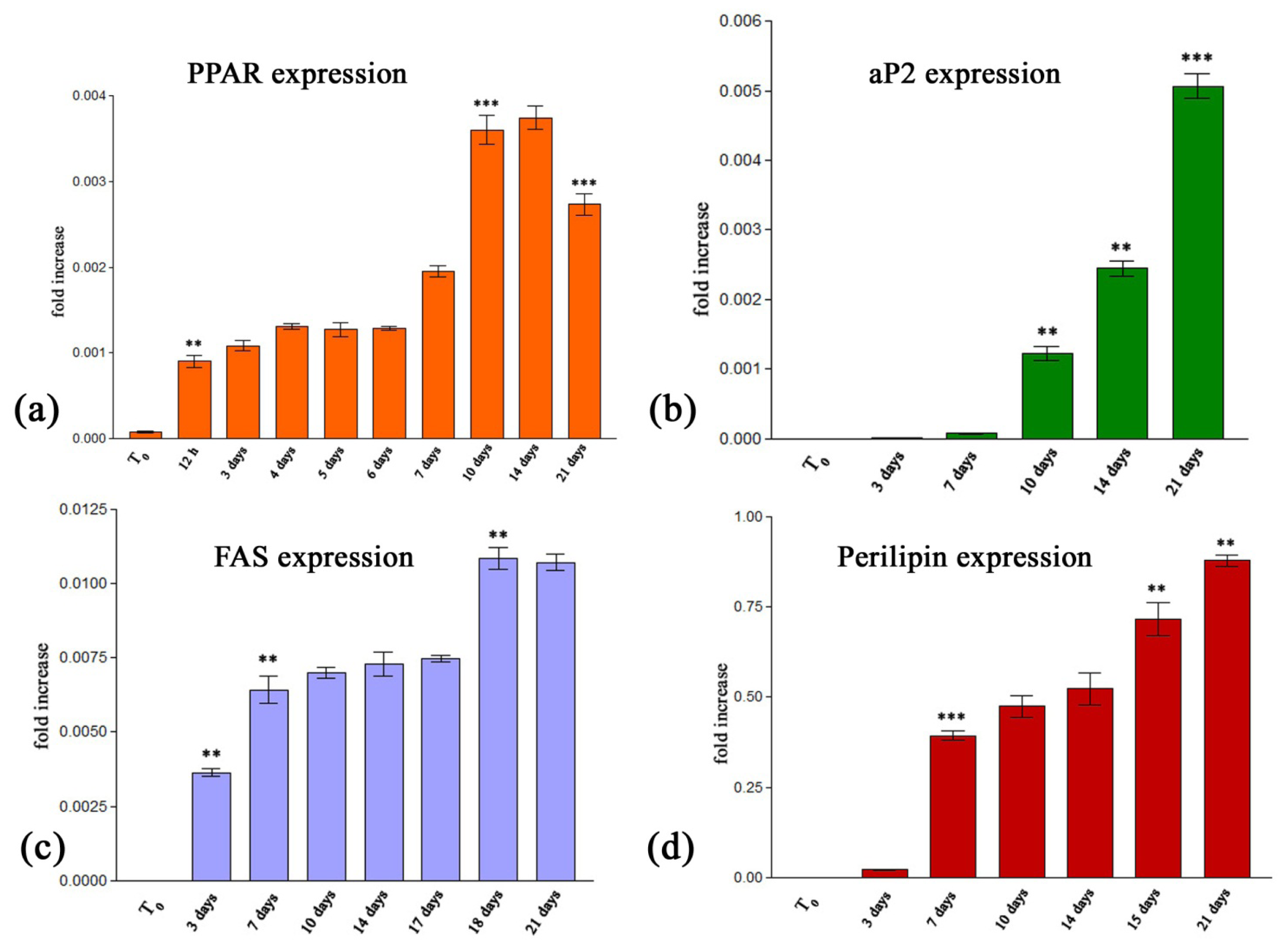
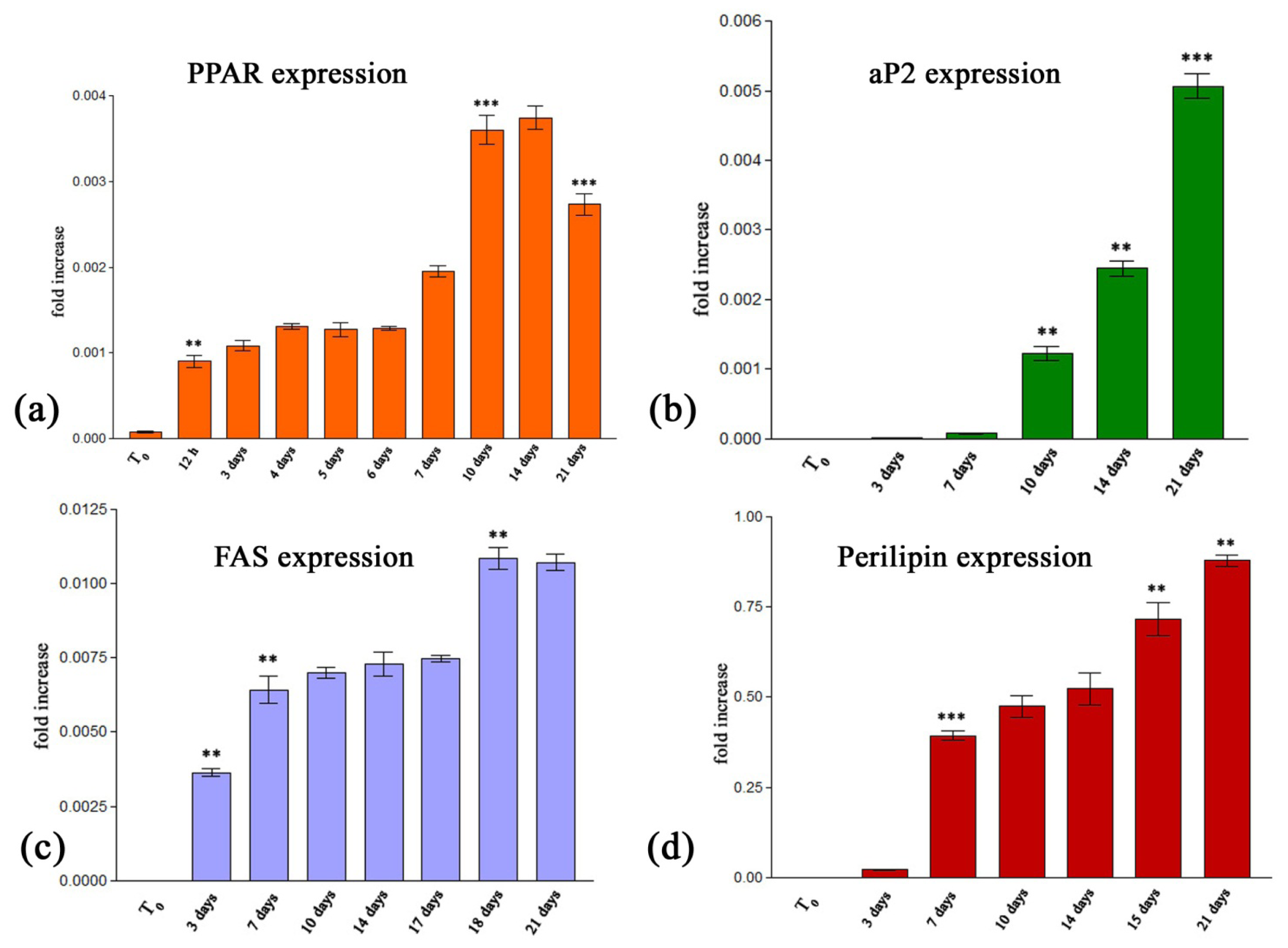
2.2.2.1. Quantitative RealTime RT-PCR Assay
3. Experimental Section
3.1. Isolation and Culture of Primary Cells
3.2. Subcultivation of the Cells
3.3. Characterization of Human Adipose Derived Stem Cells
3.3.1. Phenotypic Characterization
3.3.1.1. q-PCR
3.3.1.2. Western Blotting
3.3.1.3. Microscopic Immunophenotyping of hADSCs
3.3.2. Assessment of Adipogenic Differentiation
3.3.2.1. Staining of Lipid Droplets with Oil Red O
3.3.2.2. Fluorescent Double Staining of Perilipin and Actin Cytoskeleton
3.3.2.3. Quantitative RealTime RT-PCR
3.4. Statistical Analysis
4. Conclusions
Acknowledgments
References
- Trayhurn, P.; Beattie, J.H. Physiological role of adipose tissue: White adipose tissue as an endocrine and secretory organ. Proc. Nutr. Soc 2001, 60, 329–339. [Google Scholar]
- Frühbeck, G.; Ambrosi, J.G. The adipocyte: A model for integration of endocrine and metabolic signaling in energy metabolism regulation. Am. J. Physiol. Endocrinol. Metable 2001, 280, E827–E847. [Google Scholar]
- Helder, M.N.; Knippenberg, M.; Klein-Nulend, J.; Wuisman, P.I.J.M. Stem cells from adipose tissue allow challenging new concepts for regenerative medicine. Tissue Eng 2007, 13, 1799–1808. [Google Scholar]
- Gronthos, S.; Franklin, D.M.; Leddy, H.A.; Robey, P.G.; Storms, R.W.; Gimble, J.M. Surface protein characterization of human adipose tissue-derived stromal cells. J. Cell. Physiol 2001, 189, 54–63. [Google Scholar]
- Zuk, P.A.; Zhu, M.; Mizuno, H.; Huang, J.; Futrell, J.W.; Katz, A.J.; Benhaim, P.; Lorenz, H.P.; Hedrick, M.H. Multilineage cells from human adipose tissue: implications for cell-based therapies. Tissue Eng 2001, 7, 211–228. [Google Scholar]
- Cao, Y.; Sun, Z.; Liao, L.; Meng, Y.; Han, Q.; Zhao, R.C. Human adipose tissue-derived stem cells differentiate into endothelial cells in vitro and improve postnatal neovascularization in vivo. Biochem. Biophys. Res. Commun 2005, 332, 370–379. [Google Scholar]
- Planat-Benard, V.; Silvestre, J.S.; Cousin, B.; Andre, M.; Nibbelink, M.; Tamarat, R.; Clergue, M.; Manneville, C.; Saillan-Barreau, C.; Duriez, M.; et al. Plasticity of human adipose lineage cells toward endothelial cells: Physiological and therapeutic perspectives. Circulation 2004, 109, 656–663. [Google Scholar]
- Caplan, A.I. Adult mesenchymal stem cells for tissue engineering versus regenerative medicine. J. Cell. Physiol 2007, 213, 341–347. [Google Scholar]
- Gimble, J.M.; Katz, A.J.; Bunnell, B.A. Adipose-derived stem cells for regenerative medicine. Circ. Res 2007, 100, 1249–1260. [Google Scholar]
- Ebrahimian, T.G.; Pouzoulet, F.; Squiban, C.; Buard, V.; André, M.; Cousin, B.; Gourmelon, P.; Benderitter, M.; Casteilla, L.; Tamarat, R. Cell therapy based on adipose tissue-derived stromal cells promotes physiological and pathological wound healing. Arterioscler. Thromb. Vasc. Biol 2009, 29, 503–510. [Google Scholar]
- Blanton, M.W.; Hadad, I.; Johnstone, B.H.; Mund, J.A.; Rogers, P.I.; Eppley, B.L.; March, K.L. Adipose stromal cells and platelet-rich plasma therapies synergistically increase revascularization during wound healing. Plast. Reconstr. Surg 2009, 123, 56S–64S. [Google Scholar]
- Kim, W.S.; Park, B.S.; Sung, J.H.; Yang, J.M.; Park, S.B.; Kwak, S.J.; Park, J.S. Wound healing effect of adipose-derived stem cells: A critical role of secretory factors on human dermal fibroblasts. J. Dermatol. Sci 2007, 48, 15–24. [Google Scholar]
- Rehman, J.; Traktuev, D.; Li, J.; Merfeld-Clauss, S.; Temm-Grove, C.J.; Bovenkerk, J.E.; Pell, C.L.; Johnstone, B.H.; Considine, R.V.; March, K.L. Secretion of angiogenic and antiapoptotic factors by human adipose stromal cells. Circulation 2004, 109, 1292–1298. [Google Scholar]
- Badillo, A.T.; Redden, R.A.; Zhang, L.; Doolin, E.J.; Liechty, K.W. Treatment of diabetic wounds with fetal murine mesenchymal stromal cells enhances wound closure. Cell Tissue Res 2007, 329, 301–311. [Google Scholar]
- Rodbell, M. Metabolism of isolated fat cells. II. The similar effects of phospholipase c (clostridium perfringens alpha toxin) and of insulin on glucose and amino acid metabolism. J. Biol. Chem 1966, 241, 130–139. [Google Scholar]
- Rodbell, M.; Jones, A.B. Metabolism of isolated fat cells. 3. The similar inhibitory action of phospholipase c (clostridium perfringens alpha toxin) and of insulin on lipolysis stimulated by lipolytic hormones and theophylline. J. Biol. Chem 1966, 241, 140–142. [Google Scholar]
- Bjorntorp, P.; Karlsson, M.; Pertoft, H.; Pettersson, P.; Sjostrom, L.; Smith, U. Isolation and characterization of cells from rat adipose tissue developing into adipocytes. J. Lipid Res 1978, 19, 316–324. [Google Scholar]
- Van, R.L.; Bayliss, C.E.; Roncari, D.A. Cytological and enzymological characterization of adult human adipocyte precursors in culture. J. Clin. Invest 1976, 58, 699–704. [Google Scholar]
- Dominici, M.; Le Blanc, K.; Mueller, I.; Slaper-Cortenbach, I.; Marini, F.; Krause, D.; Deans, R.; Keating, A.; Prockop, D.; Horwitz, E. Minimal criteria for defining multipotent mesenchymal stromal cells. The Inetrnational Society for Cellular Therapy position statement. Cytotherapy 2006, 8, 315–317. [Google Scholar]
- Zuk, P.A.; Zhu, M.; Ashjian, P.; De Ugarte, D.A.; Huang, J.I.; Mizuno, H.; Alfonso, Z.C.; Fraser, J.K.; Benhaim, P.; Hedrick, M.H. Human adipose tissue is a source of multipotent stem cells. Mol. Biol. Cell 2002, 13, 4279–4295. [Google Scholar]
- Lowe, C.E.; O’Rahilly, S.; Rochford, J.J. Adipogenesis at a glance. J. Cell. Sci 2011, 124, 2681–2686. [Google Scholar]
- Rosen, E.D.; Spiegelman, B.M. Molecular regulation of adipogenesis. Annu. Rev. Cell Dev. Biol 2002, 16, 145–171. [Google Scholar]
- Spiegelman, B.M. PPARγ: Adipogenic regulator and thiazolidinedione receptor. Diabetes 1998, 4, 507–514. [Google Scholar]
- Fajas, L.; Fruchart, J.C.; Auwerx, J. Transcriptional control of adipogenesis. Curr. Opin. Cell Biol 1998, 10, 165–173. [Google Scholar]
- Rosen, E.D.; Spiegelman, B.M. PPARgamma: A nuclear regulator of metabolism, differentiation, and cell growth. J. Biol. Chem 2001, 276, 37731–37734. [Google Scholar]
- Moustaïd, N.; Sul, H.S. Regulation of expression of the fatty acid synthase gene in 3T3-Ll cells by differentiation and triiodothyronine. J. Biol. Chem 1991, 266, 18550–18554. [Google Scholar]
- Brasaemle, D.L.; Levin, D.M.; Adler-Wailes, D.C.; Londos, C. The lipolytic stimulation of 3T3-L1 adipocytes promotes the translocation of hormone-sensitive lipase to the surfaces of lipid storage droplets. Biochim. Biophys. Acta 2000, 1483, 251–262. [Google Scholar]
- Galateanu, B.; Dimonie, D.; Vasile, E.; Nae, S.; Cimpean, A.; Costache, M. Layer-shaped alginate hydrogels enhance the biological performance of human adipose-derived stem cells. BMC Biotechnol 2012, 12, 35. [Google Scholar]
- Stacey, D.H.; Hanson, S.E.; Lahvis, G.; Gutowski, K.A.; Masters, K.S. In vitro adipogenic differentiation of preadipocytes varies with differentiation stimulus, culture dimensionality, and scaffold composition. Tissue Eng 2009, 15, 3389–3399. [Google Scholar]
- Yoo, K.H.; Jang, I.K.; Lee, M.W.; Kim, H.E.; Yang, M.S.; Eom, Y.; Lee, J.E.; Kim, Y.J.; Yang, S.K.; Jung, H.L.; et al. Comparison of immunomodulatory properties of mesenchymal stem cells derived from adult human tissues. Cell Immunol. 2009, 259, 150–156. [Google Scholar]
- Hausman, G.J.; Dodson, M.V.; Ajuwon, K.; Azain, M.; Barnes, K.M.; Guan, L.L.; Jiang, Z.; Poulos, S.P.; Sainz, R.D.; Smith, S.; et al. The biology and regulation of preadipocytes and adipocytes in meat animals. J. Anim. Sci 2009, 87, 1218–1246. [Google Scholar]
- Oishi, Y.; Manabe, I.; Tobe, K.; Tsushima, K.; Shindo, T.; Fujiu, K.; Nishimura, G.; Maemura, K.; Yamauchi, T.; Kubota, N.; et al. Kruppel-like transcription factor KLF5 is a key regulator of adipocyte differentiation. Cell Metab 2005, 1, 27–39. [Google Scholar]
- Rahman, A.; Suresh, M.D.; Kumar, G.; Lee, S.H.; Hwang, H.S.; Kim, H.A.; Yun, J.W. Proteome analysis for 3T3-L1 adipocyte differentiation. J. Microbiol. Biotechnol 2008, 18, 1895–1902. [Google Scholar]
- Verstraeten, V.L.R.M.; Renes, J.; Ramaekers, F.C.S.; Kamps, M.; Kuijpers, H.J.; Verheyen, F.; Wabitsch, M.; Steijlen, P.M.; van Steensel, M.A.M.; Broers, J.L.V. Reorganization of the nuclear lamina and cytoskeleton in adipogenesis. Histochem. Cell Biol 2011, 135, 251–261. [Google Scholar]
- Kanzaki, M.; Watson, R.T.; Hou, J.C.; Stamnes, M.; Saltiel, A.R.; Pessin, J.E. Small GTP-binding protein TC10 differentially regulates two distinct populations of filamentous actin in 3T3L1 adipocytes. Mol. Biol. Cell 2002, 13, 2334–2346. [Google Scholar]
- Saladin, R.; Fajas, L.; Dana, S.; Halvorsen, Y.D.; Auwerx, J.; Briggs, M. Differential regulation of peroxisome proliferator activator receptor gamma 1 (PPAR gamma 1) and PPAR gamma 2 messenger RNA expression in the early stages of adipogenesis. Cell Growth Differ 1999, 10, 43–48. [Google Scholar]
- Zhang, L.; Paddon, C.; Lewis, M.D.; Grennan-Jones, F.; Gsa, M.L. Signalling suppresses PPARg2 generation and inhibits 3T3L1 adipogenesis. J. Endocrinol 2009, 202, 207–215. [Google Scholar]
- Tan, N.S.; Shaw, N.S.; Vinckenbosch, N.; Liu, P.; Yasmin, R.; Desvergne, B.; Wahli, W.; Noy, N. Selective Cooperation between Fatty Acid Binding Proteins and Peroxisome Proliferator-Activated Receptors in Regulating Transcription. Mol. Cell Biol 2002, 22, 5114–5127. [Google Scholar]
- Lawrence, J.W.; Kroll, D.J.; Eacho, P.I. Ligand-dependent interaction of hepatic fatty acid-binding protein with the nucleus. J. Lipid Res 2000, 41, 1390–1401. [Google Scholar]
- Clarke, C.D. Regulation of fatty acid synthase gene expression: An approach for reducing fataccumulation. J. Anim. Sci 1993, 71, 1957–1965. [Google Scholar]
- Prusty, D.; Park, B.H.; Davis, K.E.; Farmer, S.R. Activation of MEK/ERK Signaling Promotes Adipogenesis by Enhancing Peroxisome Proliferator-activated Receptor γ (PPARγ) and C/EBPα Gene Expression during the Differentiation of 3T3-L1 Preadipocytes. J. Biol. Chem 2002, 277, 46226–46232. [Google Scholar]
- Fernyhough, M.E.; Vierck, J.L.; Hausman, G.J.; Mir, P.S.; Okine, E.K.; Dodson, M.V. Primary adipocyte culture: Adipocyte purification methods may lead to a new understanding of adipose tissue growth and development. Cytotechnology 2005, 46, 163–172. [Google Scholar]
- Madonna, R.; Geng, Y.J.; De Caterina, R. Adipose tissue-derived stem cells. Characterization and potential for cardiovascular repair. Arterioscler. Thromb. Vasc. Biol 2009, 29, 1723–1729. [Google Scholar]
- Francis, M.P.; Sachs, P.C.; Elmore, L.W.; Holt, S.E. Isolating adipose-derived mesenchymal stem cells from lipoaspirate blood and saline fraction. Landers 2009, 6, 11–14. [Google Scholar]
- Bustin, S.A.; Benes, V.; Nolan, T.; Pfaffl, M.W. Quantitative real-time RT-PCR—A perspective. J. Mol. Endocrinol 2005, 34, 597–601. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Medium | Composition | |
|---|---|---|
| MD0 | Standard adipogenic differentiation medium (commercial formula code 811D-250), Cell Applications, San Diego, CA, USA | |
| MD1 | CM+ | 0.5 mM IBMX |
| 1 μM DEX | ||
| 1 μg/mL Troglitazone | ||
| 1 μM Insulin | ||
| 10 μg/mL Biotin | ||
| 200 μM Indomethacine | ||
| 0.1 mM Hydrocortisone | ||
| 0.2 mM 3,3′,5-Triiodothyronine | ||
| 10 μg/mL Transferrin | ||
| MD2 | CM+ | 10 μg/mL Biotin |
| 200 μM Indomethacine | ||
| 0.1 mM Hydrocortisone | ||
| 0.2 mM 3,3′,5-Triiodothyronine | ||
| 10 μg/mL Transferrin | ||
| MD3 | CM+ | 0.5 mM IBMX |
| 1 μM DEX | ||
| 1 μg/mL Troglitazone | ||
| 1 μM Insulin | ||
| MD4 | CM+ | 1 μM Insulin |
| 0.2 mM 3,3′,5-Triiodothyronine | ||
| P0 | P1 | P2 | P3 | |
|---|---|---|---|---|
| Day 1–3 | MD0 | MD1 | MD1 | MD3 |
| Day 4–21 | MD0 | MD1 | MD2 | MD4 |
| Target | Nucleotidic sequence | Fragment length |
|---|---|---|
| CD44 F | 5′-CATTCAAATCCGGAAGTGCT-3′ | 208 bp |
| CD44 R | 5′-GTTGCCAAACCACTGTTCCT-3′ | |
| CD73 | F 5′-CGCAACAATGGCACAATTAC-3′ | 224 bp |
| CD73 | R 5′-CTCGACACTTGGTGCAAGA-3′ | |
| CD90 | F 5′-TGAAGGTCCTCTACTTATCCGC-3′ | 111 bp |
| CD90 | R 5′-GCACTGTGACGTTCTGGGA-3′ | |
| CD34 | F 5′-CCACAGGAGAAAGGCTGGGCG-3′ | 157 bp |
| CD34 | R 5′-GCCTTGCCCCACCTAGCCGA-3′ |
| Target | Nucleotidic sequence | Fragment length |
|---|---|---|
| PPARγ2 F | 5′-TTACACAATGCTGGCCTCCTT-3′ | 99 bp |
| PPARγ2 | R 5′-AGGCTTTCGCAGGCTCTTTAG-3′ | |
| aP2 F | 5′-ATGGGATGGAAAATCAACCA-3′ | 104 bp |
| aP2 R | 5′-GTGGAAGTGACGCCTTTCAT-3′ | |
| Perilipin F | 5′-ATGCTTCCAGAAGACCTACA-3′ | 224 bp |
| Perilipin R | 5′-CAGCTCAGAAGCAATCTTTT-3′ | |
| FAS F | 5′-GCTGGAAGTCACCTATGAAG-3′ | 205 bp |
| FAS R | 5′-TGAAGTCGAAGAAGAAGGAG-3′ | |
| GAPDH F | 5′-AAGGTCGGAGTCAACGGATT-3′ | 224 bp |
| GAPDH R | 5′-CTCCTGGAAGATGGTGATGG-3′ |
© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Galateanu, B.; Dinescu, S.; Cimpean, A.; Dinischiotu, A.; Costache, M. Modulation of Adipogenic Conditions for Prospective Use of hADSCs in Adipose Tissue Engineering. Int. J. Mol. Sci. 2012, 13, 15881-15900. https://doi.org/10.3390/ijms131215881
Galateanu B, Dinescu S, Cimpean A, Dinischiotu A, Costache M. Modulation of Adipogenic Conditions for Prospective Use of hADSCs in Adipose Tissue Engineering. International Journal of Molecular Sciences. 2012; 13(12):15881-15900. https://doi.org/10.3390/ijms131215881
Chicago/Turabian StyleGalateanu, Bianca, Sorina Dinescu, Anisoara Cimpean, Anca Dinischiotu, and Marieta Costache. 2012. "Modulation of Adipogenic Conditions for Prospective Use of hADSCs in Adipose Tissue Engineering" International Journal of Molecular Sciences 13, no. 12: 15881-15900. https://doi.org/10.3390/ijms131215881
APA StyleGalateanu, B., Dinescu, S., Cimpean, A., Dinischiotu, A., & Costache, M. (2012). Modulation of Adipogenic Conditions for Prospective Use of hADSCs in Adipose Tissue Engineering. International Journal of Molecular Sciences, 13(12), 15881-15900. https://doi.org/10.3390/ijms131215881