Construction of Mutant Glucose Oxidases with Increased Dye-Mediated Dehydrogenase Activity

Abstract
:1. Introduction
2. Results and Discussion
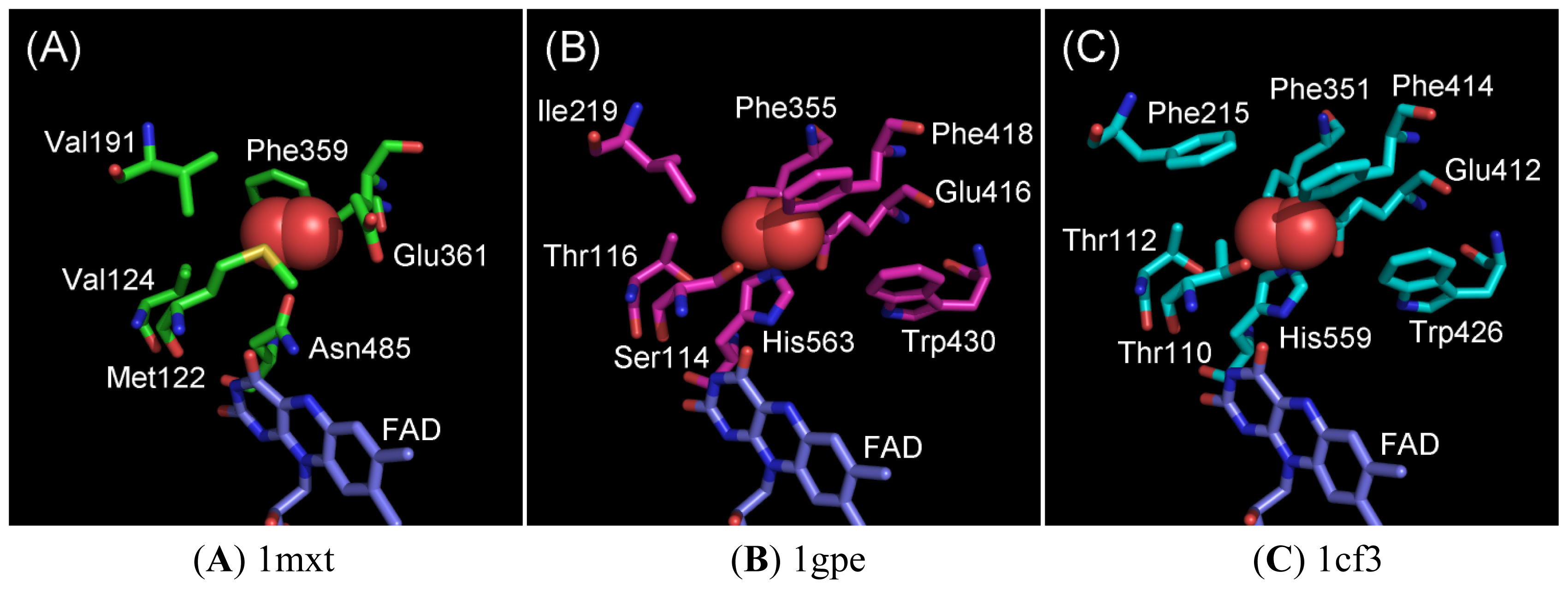
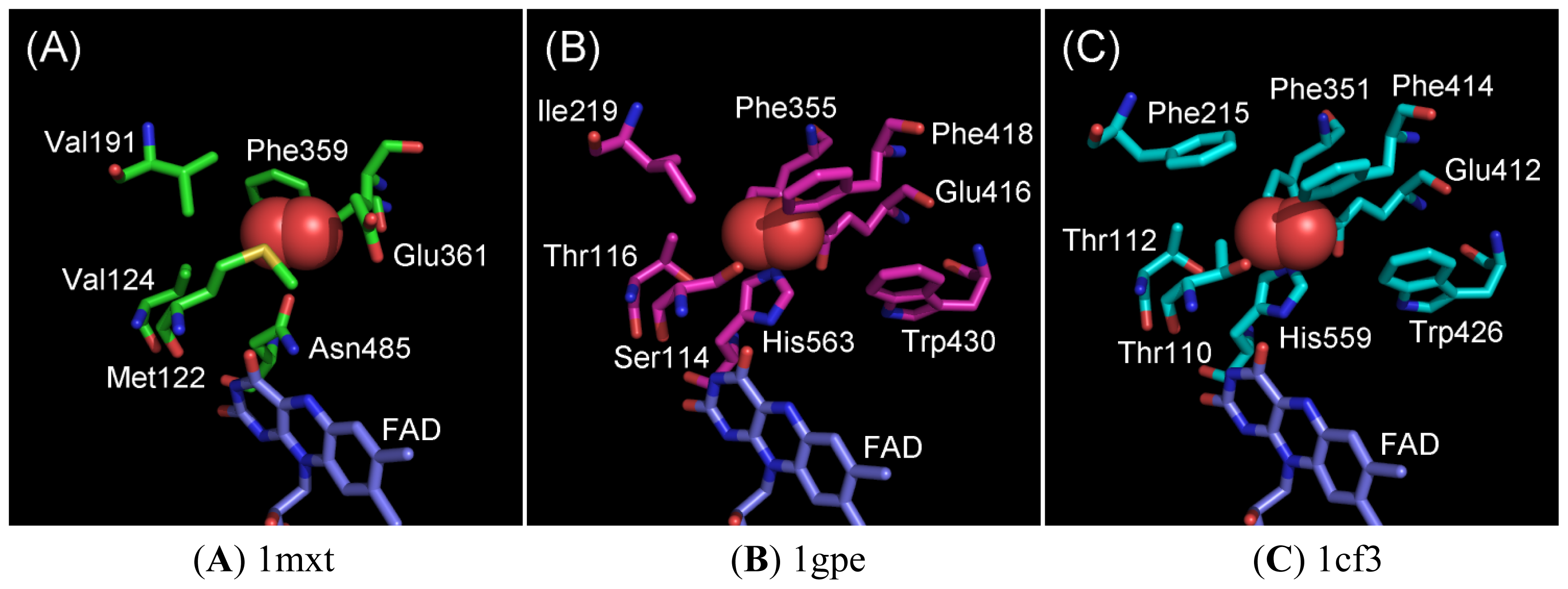
2.1. Oxygen-Interacting Structural Models for GOxs
2.2. Ala Substitutions within the Putative Functional Residues Responsible for Oxidative Half Reaction with Oxygen for 1gpe and 1cf3
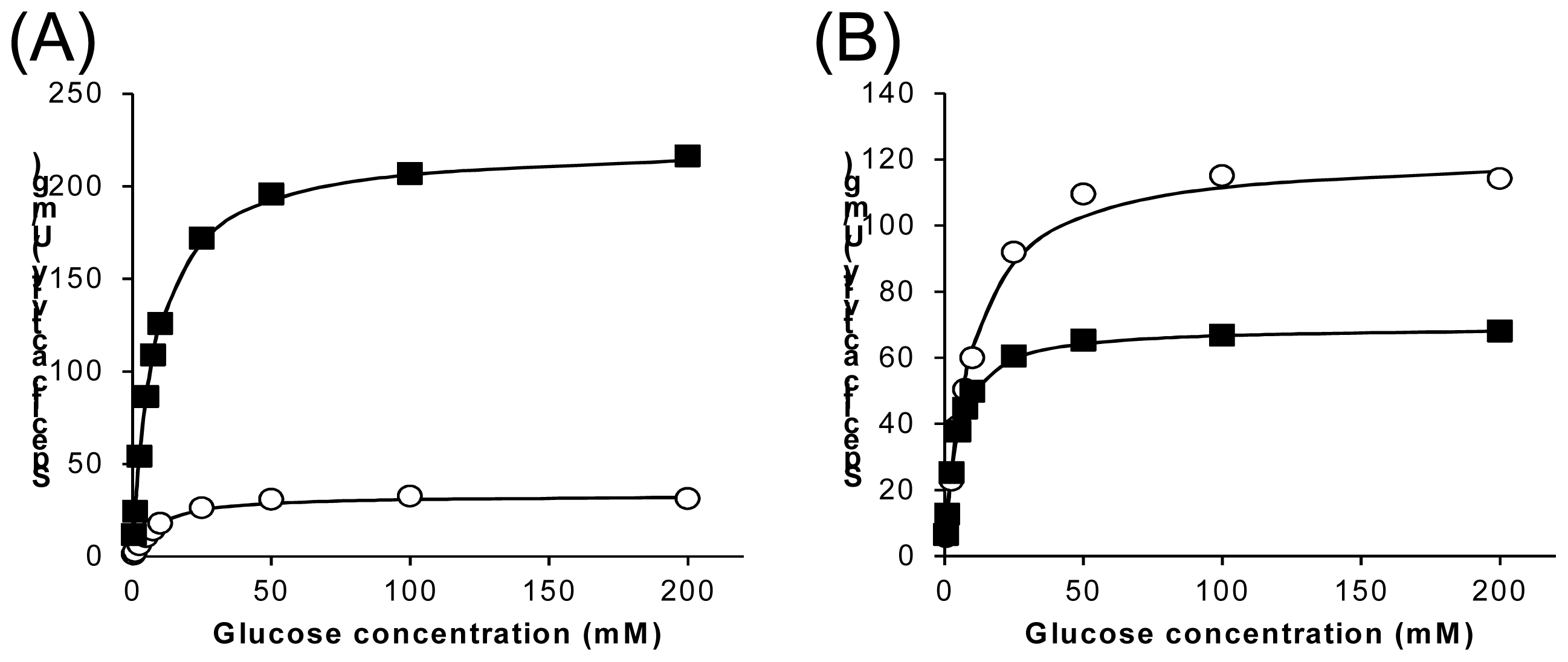
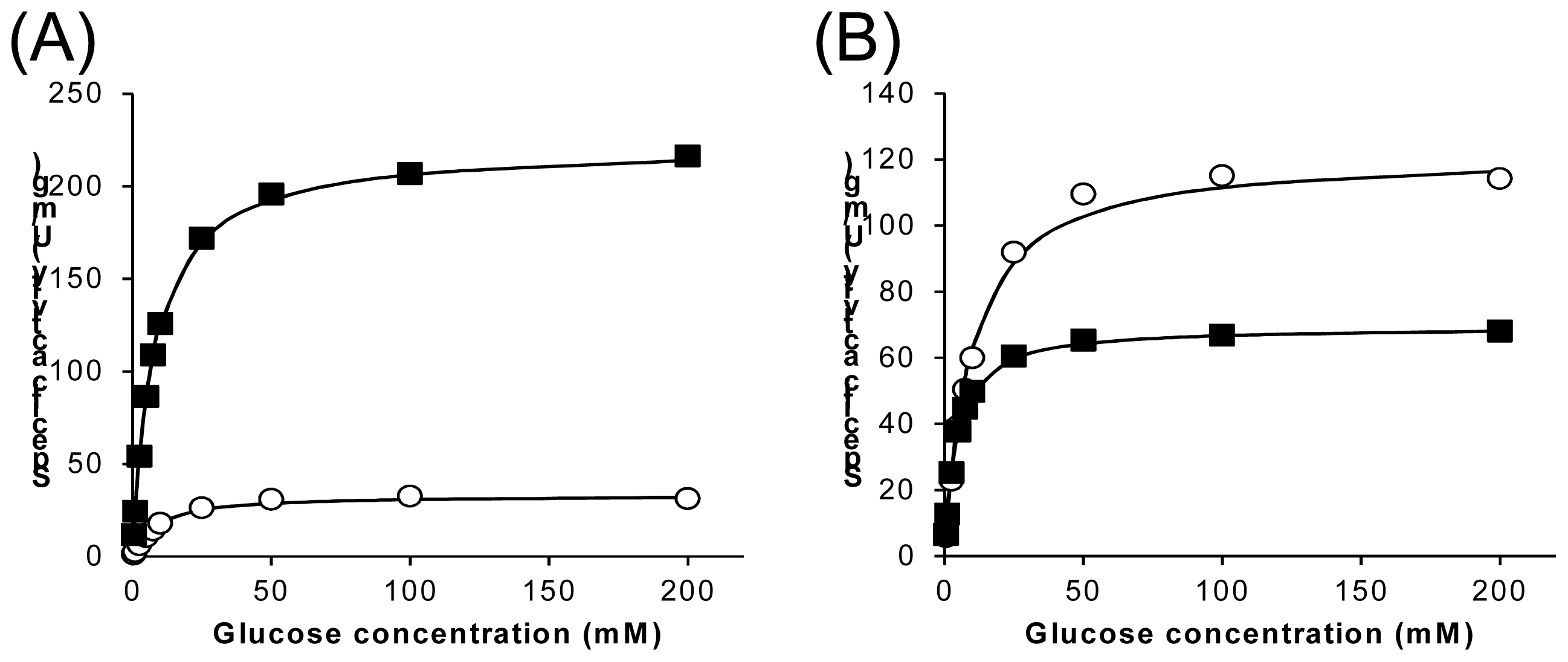
2.3. Characterization of Wild-Type 1gpe and Ser114Ala Mutant
3. Experimental Section
3.1. Bacterial strains and Plasmids
3.2. Site-Directed Mutagenesis
3.3. Enzyme Preparation
3.4. Enzyme Assay
4. Conclusions
- Conflict of InterestThe authors declare no conflict of interest.
References
- Pazur, J.H.; Kleppe, K.; Cepure, A. A glycoprotein structure for glucose oxidase from Aspergillus niger. Arch. Biochem. Biophys 1965, 111, 351–357. [Google Scholar]
- Kusai, K.; Sekuzu, I.; Hagihara, B.; Okunuki, K.; Yamauchi, S.; Nakai, M. Crystallization of glucose oxidase from Penicillium amagasakiense. Biochim. Biophys. Acta 1960, 40, 555–557. [Google Scholar]
- Frederick, K.R.; Tung, J.; Emerick, R.S.; Masiarz, F.R.; Chamberlain, S.H.; Vasavada, A.; Rosenberg, S.; Chakraborty, S.; Schopfer, L.M.; Massey, V. Glucose oxidase from Aspergillus niger: Cloning, gene sequence, secretion from Saccharomyces cerevisiae and kinetic analysis of a yeast-derived enzyme. J. Biol. Chem 1990, 265, 3793–3802. [Google Scholar]
- Hodgkins, M.; Mead, D.; Ballance, D.J.; Goodey, A.; Sudbery, P. Expression of the glucose oxidase gene from Aspergillus niger in Hansenula polymorpha and its use as a reporter gene to isolate regulatory mutations. Yeast 1993, 9, 625–635. [Google Scholar]
- Witt, S.; Singh, M.; Kalisz, H.M. Structural and kinetic properties of nonglycosylated recombinant Penicillium amagasakiense glucose oxidase expressed in Escherichia coli. Appl. Environ. Microbiol 1998, 64, 1405–1411. [Google Scholar]
- Updike, S.; Hicks, G. The enzyme electrode. Nature 1967, 214, 986–988. [Google Scholar]
- Ferri, S.; Kojima, K.; Sode, K. Review of glucose oxidases and glucose dehydrogenases: A bird’s eye view of glucose sensing enzymes. J. Diabetes Sci. Technol 2011, 5, 1068–1076. [Google Scholar]
- Kim, S.; Nibe, E.; Ferri, S.; Tsugawa, W.; Sode, K. Engineering of dye-mediated dehydrogenase property of fructosyl amino acid oxidases by site-directed mutagenesis studies of its putative proton relay system. Biotechnol. Lett 2010, 32, 1123–1129. [Google Scholar]
- Kim, S.; Nibe, E.; Tsugawa, W.; Kojima, K.; Ferri, S.; Sode, K. Construction of engineered fructosyl peptidyl oxidase for enzyme sensor applications under normal atmospheric conditions. Biotechnol. Lett 2012, 34, 491–497. [Google Scholar]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem 2004, 25, 1605–1612. [Google Scholar]
- Lario, P.I.; Sampson, N.; Vrielink, A. Sub-atomic resolution crystal structure of cholesterol oxidase: What atomic resolution crystallography reveals about enzyme mechanism and the role of the FAD cofactor in redox activity. J. Mol. Biol 2003, 326, 1635–1650. [Google Scholar]
- Kalisz, H.M.; Hecht, H.J.; Schomburg, D.; Schmid, R.D. Effects of carbohydrate depletion on the structure, stability and activity of glucose oxidase from Aspergillus niger. Biochim. Biophys. Acta 1991, 1080, 138–142. [Google Scholar]
- Zhu, Z.; Momeu, C.; Zakhartsev, M.; Schwaneberg, U. Making glucose oxidase fit for biofuel cell applications by directed protein evolution. Biosens. Bioelectron 2006, 21, 2046–2051. [Google Scholar]
- Zhu, Z.; Wang, M.; Gautam, A.; Nazor, J.; Momeu, C.; Prodanovic, R.; Schwaneberg, U. Directed evolution of glucose oxidase from Aspergillus niger for ferrocenemethanol-mediated electron transfer. Biotechnol. J 2007, 2, 241–248. [Google Scholar]
- Prodanovic, R.; Ostafe, R.; Scacioc, A.; Schwaneberg, U. Ultrahigh throughput screening system for directed glucose oxidase evolution in yeast cells. Comb. Chem. High Throughput Screening 2011, 14, 55–60. [Google Scholar]
- Roth, J.P.; Klinman, J.P. Catalysis of electron transfer during activation of O2 by the flavoprotein glucose oxidase. Proc. Natl. Acad. Sci. USA 2003, 100, 62–67. [Google Scholar]
- Holland, J.T.; Harper, J.C.; Dolan, P.L.; Manginell, M.M.; Arango, D.C.; Rawlings, J.A.; Apblett, C.A.; Brozik, S.M. Rational redesign of glucose oxidase for improved catalytic function and stability. PLoS One 2012, 7, e37924. [Google Scholar]
- Witt, S.; Wohlfahrt, G.; Schomburg, D.; Hecht, H.J.; Kalisz, H.M. Conserved arginine-516 of Penicillium amagasakiense glucose oxidase is essential for the efficient binding of β-d-glucose. Biochem. J 2000, 347, 553–559. [Google Scholar]

{kind=link}
{kind=link}
| 1gpe | 1cf3 | ||||||
|---|---|---|---|---|---|---|---|
| Ox (U/mg) | Dh (U/mg) | Dh/Ox (%) | Ox (U/mg) | Dh (U/mg) | Dh/Ox (%) | ||
| Wild-type | 36 (100%) | 6.5 (100%) | 18.2 (100%) | Wild-type | 31 (100%) | 8.3 (100%) | 27.1 (100%) |
| Ser114Ala | 11 (31.2%) | 25 (385%) | 224 (1233%) | Thr110Ala | 9.3 (30.4%) | 17 (203%) | 181 (668%) |
| Thr116Ala | n.d. | n.d. | - | Thr112Ala | 1.2 (3.9%) | 1.5 (17.5%) | 121 (445%) |
| Ile219Ala | n.d. | n.d. | - | ||||
| Phe355Ala | 1.1 × 10−2 (0.03%) | 1.6 × 10−2 (0.24%) | 145 (800%) | Phe351Ala | 3.3 × 10−2 (0.11%) | 3.6 × 10−2 (0.43%) | 108 (339%) |
| Glu416Ala | n.d. | n.d. | - | ||||
| Phe418Ala | 0.21 (0.58%) | 0.27 (4.1%) | 127 (702%) | Phe414Ala | n.d. | 7.6 × 10−2 (0.11%) | - |
| Trp430Ala | 0.31 (0.87%) | 0.79 (12.1%) | 252 (1387%) | Trp426Ala | 4.4 (14.3%) | 7.3 (87.3%) | 165 (610%) |
| His563Ala | n.d. | n.d. | - | ||||
| Km | Vmax | Vmax/Km | |||||
|---|---|---|---|---|---|---|---|
| Ox (mM) | Dh (mM) | Ox (U/mg) | Dh (U/mg) | Dh/Ox | Ox (U/mg·mM) | Dh (U/mg·mM) | |
| wild-type | 8.18 | 9.29 | 227 | 33.3 | 14.6% | 27.8 | 3.59 |
| Ser114Ala | 4.19 | 9.41 | 69.4 | 122 | 176% | 16.6 | 13.0 |
© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Horaguchi, Y.; Saito, S.; Kojima, K.; Tsugawa, W.; Ferri, S.; Sode, K. Construction of Mutant Glucose Oxidases with Increased Dye-Mediated Dehydrogenase Activity. Int. J. Mol. Sci. 2012, 13, 14149-14157. https://doi.org/10.3390/ijms131114149
Horaguchi Y, Saito S, Kojima K, Tsugawa W, Ferri S, Sode K. Construction of Mutant Glucose Oxidases with Increased Dye-Mediated Dehydrogenase Activity. International Journal of Molecular Sciences. 2012; 13(11):14149-14157. https://doi.org/10.3390/ijms131114149
Chicago/Turabian StyleHoraguchi, Yohei, Shoko Saito, Katsuhiro Kojima, Wakako Tsugawa, Stefano Ferri, and Koji Sode. 2012. "Construction of Mutant Glucose Oxidases with Increased Dye-Mediated Dehydrogenase Activity" International Journal of Molecular Sciences 13, no. 11: 14149-14157. https://doi.org/10.3390/ijms131114149