Molecular Pathogenesis of Neuromyelitis Optica

Abstract
:1. Introduction
2. Pathogenesis of NMO
2.1. Aquaporin 4
2.2. NMO IgG
2.3. Triggers for Autoimmunity
2.4. Is Anti-AQP4 Antibody Pathogenic?
2.5. Immune Mediated Astrocytopathy
2.6. Immunopathogenesis
2.7. Histopathology of NMO
3. Conclusions
Acknowledgements
References
- Allbutt, T. On the opthalmoscopic signs of spinal disease. Lancet 1870, 1, 76–78. [Google Scholar]
- Erb, W. About the concurrence of optic neuritis and subacute myelitis. Arch. Psychiatr. Nervenkr 1879, 1, 146–157. [Google Scholar]
- Jarius, S.; Wildemann, B. On the contribution of thomas clifford allbutt, F.R.S., to the early history of neuromyelitis optica. J. Neurol 2012. [Google Scholar] [CrossRef]
- Devic, E. Myélite subaiguë compliquée de névrite optique. Bull. Med. (Paris) 1894, 8, 1033–1034. [Google Scholar]
- Totsuka, S. Clinico-pathological studies on the two cases of neuromyelitis optica (Devic’s disease) with a chronic clinical course, with especial reference on its relationship to multiple sclerosis. Folia Psychiatr. Neurol. Jpn 1962, 64, 1149–1165. [Google Scholar]
- Wingerchuk, D.M.; Hogancamp, W.F.; O’Brien, P.C.; Weinshenker, B.G. The clinical course of neuromyelitis optica (Devic’s syndrome). Neurology 1999, 53, 1107–1114. [Google Scholar]
- Lennon, V.A.; Wingerchuk, D.M.; Kryzer, T.J.; Pittock, S.J.; Lucchinetti, C.F.; Fujihara, K.; Nakashima, I.; Weinshenker, B.G. A serum autoantibody marker of neuromyelitis optica: Distinction from multiple sclerosis. Lancet 2004, 364, 2106–2112. [Google Scholar]
- Kim, J.S.; Park, Y.H.; Kim, S.M.; Kim, S.H.; Park, K.S.; Sung, J.J.; Lee, K.W. A case of chronic progressive myelopathy. Mult. Scler 2010, 16, 1255–1257. [Google Scholar]
- Wingerchuk, D.M.; Pittock, S.J.; Lucchinetti, C.F.; Lennon, V.A.; Weinshenker, B.G. A secondary progressive clinical course is uncommon in neuromyelitis optica. Neurology 2007, 68, 603–605. [Google Scholar]
- Jarius, S.; Ruprecht, K.; Wildemann, B.; Kuempfel, T.; Ringelstein, M.; Geis, C.; Kleiter, I.; Kleinschnitz, C.; Berthele, A.; Brettschneider, J.; et al. Contrasting disease patterns in seropositive and seronegative neuromyelitis optica: A multicentre study of 175 patients. J. Neuroinflamm 2012, 9, 14. [Google Scholar] [Green Version]
- Jarius, S.; Paul, F.; Franciotta, D.; Ruprecht, K.; Ringelstein, M.; Bergamaschi, R.; Rommer, P.; Kleiter, I.; Stich, O.; Reuss, R.; et al. Cerebrospinal fluid findings in aquaporin-4 antibody positive neuromyelitis optica: Results from 211 lumbar punctures. J. Neurol. Sci 2011, 306, 82–90. [Google Scholar]
- Pittock, S.J.; Lennon, V.A.; Krecke, K.; Wingerchuk, D.M.; Lucchinetti, C.F.; Weinshenker, B.G. Brain abnormalities in neuromyelitis optica. Arch. Neurol 2006, 63, 390–396. [Google Scholar]
- Wingerchuk, D.M.; Lennon, V.A.; Pittock, S.J.; Lucchinetti, C.F.; Weinshenker, B.G. Revised diagnostic criteria for neuromyelitis optica. Neurology 2006, 66, 1485–1489. [Google Scholar]
- Wingerchuk, D.M.; Lennon, V.A.; Lucchinetti, C.F.; Pittock, S.J.; Weinshenker, B.G. The spectrum of neuromyelitis optica. Lancet Neurol 2007, 6, 805–815. [Google Scholar]
- Bizzoco, E.; Lolli, F.; Repice, A.M.; Hakiki, B.; Falcini, M.; Barilaro, A.; Taiuti, R.; Siracusa, G.; Amato, M.P.; Biagioli, T.; et al. Prevalence of neuromyelitis optica spectrum disorder and phenotype distribution. J. Neurol 2009, 256, 1891–1898. [Google Scholar]
- McKeon, A.; Lennon, V.A.; Lotze, T.; Tenenbaum, S.; Ness, J.M.; Rensel, M.; Kuntz, N.L.; Fryer, J.P.; Homburger, H.; Hunter, J.; et al. CNS aquaporin-4 autoimmunity in children. Neurology 2008, 71, 93–100. [Google Scholar]
- Takahashi, T.; Miyazawa, I.; Misu, T.; Takano, R.; Nakashima, I.; Fujihara, K.; Tobita, M.; Itoyama, Y. Intractable hiccup and nausea in neuromyelitis optica with anti-aquaporin-4 antibody: A herald of acute exacerbations. J. Neurol. Neurosurg. Psychiatry 2008, 79, 1075–1078. [Google Scholar]
- Pittock, S.J.; Weinshenker, B.G.; Lucchinetti, C.F.; Wingerchuk, D.M.; Corboy, J.R.; Lennon, V.A. Neuromyelitis optica brain lesions localized at sites of high aquaporin 4 expression. Arch. Neurol 2006, 63, 964–968. [Google Scholar]
- Makino, T.; Ito, S.; Mori, M.; Yonezu, T.; Ogawa, Y.; Kuwabara, S. Diffuse and heterogeneous T2-hyperintense lesions in the splenium are characteristic of neuromyelitis optica. Mult. Scler 2012. [Google Scholar] [CrossRef]
- Ikeda, K.; Ito, H.; Hidaka, T.; Takazawa, T.; Sekine, T.; Yoshii, Y.; Hirayama, T.; Kawabe, K.; Kano, O.; Iwasaki, Y. Repeated non-enhancing tumefactive lesions in a patient with a neuromyelitis optica spectrum disorder. Intern. Med 2011, 50, 1061–1064. [Google Scholar]
- Newey, C.R.; Bermel, R.A. Fulminant cerebral demyelination in neuromyelitis optica. Neurology 2011, 77, 193. [Google Scholar]
- Orton, S.M.; Herrera, B.M.; Yee, I.M.; Valdar, W.; Ramagopalan, S.V.; Sadovnick, A.D.; Ebers, G.C. Canadian Collaborative Study G. Sex ratio of multiple sclerosis in canada: A longitudinal study. Lancet Neurol 2006, 5, 932–936. [Google Scholar]
- Wingerchuk, D.M.; Weinshenker, B.G. The emerging relationship between neuromyelitis optica and systemic rheumatologic autoimmune disease. Mult. Scler 2012, 18, 5–10. [Google Scholar]
- Wynn, D.R.; Rodriguez, M.; O’Fallon, W.M.; Kurland, L.T. A reappraisal of the epidemiology of multiple sclerosis in Olmsted county, Minnesota. Neurology 1990, 40, 780–786. [Google Scholar]
- Broadley, S.A.; Deans, J.; Sawcer, S.J.; Clayton, D.; Compston, D.A. Autoimmune disease in first-degree relatives of patients with multiple sclerosis. A UK survey. Brain 2000, 123, 1102–1111. [Google Scholar]
- Spadaro, M.; Amendolea, M.A.; Mazzucconi, M.G.; Fantozzi, R.; Di Lello, R.; Zangari, P.; Masala, G. Autoimmunity in multiple sclerosis: Study of a wide spectrum of autoantibodies. Mult. Scler 1999, 5, 121–125. [Google Scholar]
- Henderson, R.D.; Bain, C.J.; Pender, M.P. The occurrence of autoimmune diseases in patients with multiple sclerosis and their families. J. Clin. Neurosci 2000, 7, 434–437. [Google Scholar]
- Kim, S.H.; Kim, W.; Li, X.F.; Jung, I.J.; Kim, H.J. Does interferon β treatment exacerbate neuromyelitis optica spectrum disorder? Mult. Scler 2012. [Google Scholar] [CrossRef]
- Shimizu, J.; Hatanaka, Y.; Hasegawa, M.; Iwata, A.; Sugimoto, I.; Date, H.; Goto, J.; Shimizu, T.; Takatsu, M.; Sakurai, Y.; et al. IFNbeta-1b may severely exacerbate Japanese optic-spinal MS in neuromyelitis optica spectrum. Neurology 2010, 75, 1423–1427. [Google Scholar]
- Palace, J.; Leite, M.I.; Nairne, A.; Vincent, A. Interferon beta treatment in neuromyelitis optica: Increase in relapses and aquaporin 4 antibody titers. Arch. Neurol 2010, 67, 1016–1017. [Google Scholar]
- Watanabe, S.; Nakashima, I.; Misu, T.; Miyazawa, I.; Shiga, Y.; Fujihara, K.; Itoyama, Y. Therapeutic efficacy of plasma exchange in NMO-IgG-positive patients with neuromyelitis optica. Mult. Scler 2007, 13, 128–132. [Google Scholar]
- Papeix, C.; Vidal, J.S.; de Seze, J.; Pierrot-Deseilligny, C.; Tourbah, A.; Stankoff, B.; Lebrun, C.; Moreau, T.; Vermersch, P.; Fontaine, B.; et al. Immunosuppressive therapy is more effective than interferon in neuromyelitis optica. Mult. Scler 2007, 13, 256–259. [Google Scholar]
- Jacob, A.; Weinshenker, B.G.; Violich, I.; McLinskey, N.; Krupp, L.; Fox, R.J.; Wingerchuk, D.M.; Boggild, M.; Constantinescu, C.S.; Miller, A.; et al. Treatment of neuromyelitis optica with rituximab: Retrospective analysis of 25 patients. Arch. Neurol 2008, 65, 1443–1448. [Google Scholar]
- Kim, S.H.; Kim, W.; Li, X.F.; Jung, I.J.; Kim, H.J. Repeated treatment with rituximab based on the assessment of peripheral circulating memory B cells in patients with relapsing neuromyelitis optica over 2 years. Arch. Neurol 2011, 68, 1412–1420. [Google Scholar]
- Saadoun, S.; Waters, P.; Bell, B.A.; Vincent, A.; Verkman, A.S.; Papadopoulos, M.C. Intra-cerebral injection of neuromyelitis optica immunoglobulin G and human complement produces neuromyelitis optica lesions in mice. Brain 2010, 133, 349–361. [Google Scholar]
- Parratt, J.D.; Prineas, J.W. Neuromyelitis optica: A demyelinating disease characterized by acute destruction and regeneration of perivascular astrocytes. Mult. Scler 2010, 16, 1156–1172. [Google Scholar]
- Misu, T.; Fujihara, K.; Kakita, A.; Konno, H.; Nakamura, M.; Watanabe, S.; Takahashi, T.; Nakashima, I.; Takahashi, H.; Itoyama, Y. Loss of aquaporin 4 in lesions of neuromyelitis optica: Distinction from multiple sclerosis. Brain 2007, 130, 1224–1234. [Google Scholar]
- Preston, G.M.; Carroll, T.P.; Guggino, W.B.; Agre, P. Appearance of water channels in xenopus oocytes expressing red cell CHIP28 protein. Science 1992, 256, 385–387. [Google Scholar]
- Verkman, A.S. More than just water channels: Unexpected cellular roles of aquaporins. J. Cell Sci 2005, 118, 3225–3232. [Google Scholar]
- Nielsen, S.; Nagelhus, E.A.; Amiry-Moghaddam, M.; Bourque, C.; Agre, P.; Ottersen, O.P. Specialized membrane domains for water transport in glial cells: High-resolution immunogold cytochemistry of aquaporin-4 in rat brain. J. Neurosci 1997, 17, 171–180. [Google Scholar]
- Frigeri, A.; Gropper, M.A.; Umenishi, F.; Kawashima, M.; Brown, D.; Verkman, A.S. Localization of MIWC and GLIP water channel homologs in neuromuscular, epithelial and glandular tissues. J. Cell Sci 1995, 108, 2993–3002. [Google Scholar]
- Li, J.; Patil, R.V.; Verkman, A.S. Mildly abnormal retinal function in transgenic mice without muller cell aquaporin-4 water channels. Invest. Ophth. Vis. Sci 2002, 43, 573–579. [Google Scholar]
- Venero, J.L.; Vizuete, M.L.; Ilundain, A.A.; Machado, A.; Echevarria, M.; Cano, J. Detailed localization of aquaporin-4 messenger RNA in the CNS: Preferential expression in periventricular organs. Neuroscience 1999, 94, 239–250. [Google Scholar]
- Agre, P.; King, L.S.; Yasui, M.; Guggino, W.B.; Ottersen, O.P.; Fujiyoshi, Y.; Engel, A.; Nielsen, S. Aquaporin water channels—From atomic structure to clinical medicine. J. Physiol 2002, 542, 3–16. [Google Scholar]
- Lu, M.Q.; Lee, M.D.; Smith, B.L.; Jung, J.S.; Agre, P.; Verdijk, M.A.J.; Merkx, G.; Rijs, J.P.L.; Deen, P.M.T. The human AQP4 gene: Definition of the locus encoding two water channel polypeptides in brain. Proc. Natl. Acad. Sci. USA 1996, 93, 10908–10912. [Google Scholar]
- Neely, J.D.; Christensen, B.M.; Nielsen, S.; Agre, P. Heterotetrameric composition of aquaporin-4 water channels. Biochemistry 1999, 38, 11156–11163. [Google Scholar]
- Furman, C.S.; Gorelick-Feldman, D.A.; Davidson, K.G.; Yasumura, T.; Neely, J.D.; Agre, P.; Rash, J.E. Aquaporin-4 square array assembly: Opposing actions of M1 and M23 isoforms. Proc. Natl. Acad. Sci. USA 2003, 100, 13609–13614. [Google Scholar]
- Strand, L.; Moe, S.E.; Solbu, T.T.; Vaadal, M.; Holen, T. Roles of aquaporin-4 isoforms and amino acids in square array assembly. Biochemistry 2009, 48, 5785–5793. [Google Scholar]
- Suzuki, H.; Nishikawa, K.; Hiroaki, Y.; Fujiyoshi, Y. Formation of aquaporin-4 arrays is inhibited by palmitoylation of N-terminal cysteine residues. BBA-Biomembranes 2008, 1778, 1181–1189. [Google Scholar]
- Hinson, S.R.; Roemer, S.F.; Lucchinetti, C.F.; Fryer, J.P.; Kryzer, T.J.; Chamberlain, J.L.; Howe, C.L.; Pittock, S.J.; Lennon, V.A. Aquaporin-4-binding autoantibodies in patients with neuromyelitis optica impair glutamate transport by down-regulating EAAT2. J. Exp. Med 2008, 205, 2473–2481. [Google Scholar]
- Marignier, R.; Nicolle, A.; Watrin, C.; Touret, M.; Cavagna, S.; Varrin-Doyer, M.; Cavillon, G.; Rogemond, V.; Confavreux, C.; Honnorat, J.; et al. Oligodendrocytes are damaged by neuromyelitis optica immunoglobulin G via astrocyte injury. Brain 2010, 133, 2578–2591. [Google Scholar]
- Ratelade, J.; Bennett, J.L.; Verkman, A.S. Evidence against cellular internalization in vivo of NMO-IgG, aquaporin-4, and excitatory amino acid transporter 2 in neuromyelitis optica. J. Biol. Chem 2011, 286, 45156–45164. [Google Scholar]
- Manley, G.T.; Fujimura, M.; Ma, T.; Noshita, N.; Filiz, F.; Bollen, A.W.; Chan, P.; Verkman, A.S. Aquaporin-4 deletion in mice reduces brain edema after acute water intoxication and ischemic stroke. Nat. Med 2000, 6, 159–163. [Google Scholar]
- Saadoun, S.; Papadopoulos, M.C.; Watanabe, H.; Yan, D.; Manley, G.T.; Verkman, A.S. Involvement of aquaporin-4 in astroglial cell migration and glial scar formation. J. Cell Sci 2005, 118, 5691–5698. [Google Scholar]
- Papadopoulos, M.C.; Manley, G.T.; Krishna, S.; Verkman, A.S. Aquaporin-4 facilitates reabsorption of excess fluid in vasogenic brain edema. FASEB J 2004, 18, 1291–1293. [Google Scholar]
- Bloch, O.; Papadopoulos, M.C.; Manley, G.T.; Verkman, A.S. Aquaporin-4 gene deletion in mice increases focal edema associated with staphylococcal brain abscess. J. Neurochem 2005, 95, 254–262. [Google Scholar]
- Waters, P.; Leite, M.I.; Gray, B.; Vincent, A.; Jiang, Y.; Palace, J. Aquaporin-4 M23 isoform provides a more sensitive assay for aquaporin-4 antibodies. J. Neurol. Neurosurg. Psychiatry 2010, 81, E32. [Google Scholar]
- Ketelslegers, I.A.; Modderman, P.W.; Vennegoor, A.; Killestein, J.; Hamann, D.; Hintzen, R.Q. Antibodies against aquaporin-4 in neuromyelitis optica: Distinction between recurrent and monophasic patients. Mult. Scler 2011, 17, 1527–1530. [Google Scholar]
- Jarius, S.; Wildemann, B. AQP4 antibodies in neuromyelitis optica: Diagnostic and pathogenetic relevance. Nat. Rev. Neurol 2010, 6, 383–392. [Google Scholar]
- Kira, J. Autoimmunity in neuromyelitis optica and opticospinal multiple sclerosis: Astrocytopathy as a common denominator in demyelinating disorders. J. Neurol. Sci 2011, 311, 69–77. [Google Scholar]
- Hinson, S.R.; Pittock, S.J.; Lucchinetti, C.F.; Roemer, S.F.; Fryer, J.P.; Kryzer, T.J.; Lennon, V.A. Pathogenic potential of IgG binding to water channel extracellular domain in neuromyelitis optica. Neurology 2007, 69, 2221–2231. [Google Scholar]
- Jarius, S.; Franciotta, D.; Bergamaschi, R.; Wildemann, B.; Wandinger, K.P. Immunoglobulin M antibodies to aquaporin-4 in neuromyelitis optica and related disorders. Clin. Chem. Lab. Med 2010, 48, 659–663. [Google Scholar]
- Tani, T.; Sakimura, K.; Tsujita, M.; Nakada, T.; Tanaka, M.; Nishizawa, M.; Tanaka, K. Identification of binding sites for anti-aquaporin 4 antibodies in patients with neuromyelitis optica. J. Neuroimmunol 2009, 211, 110–113. [Google Scholar]
- Crane, J.M.; Lam, C.; Rossi, A.; Gupta, T.; Bennett, J.L.; Verkman, A.S. Binding affinity and specificity of neuromyelitis optica autoantibodies to aquaporin-4 M1/M23 isoforms and orthogonal arrays. J. Biol. Chem 2011, 286, 16516–16524. [Google Scholar]
- Nicchia, G.P.; Mastrototaro, M.; Rossi, A.; Pisani, F.; Tortorella, C.; Ruggieri, M.; Lia, A.; Trojano, M.; Frigeri, A.; Svelto, M. Aquaporin-4 orthogonal arrays of particles are the target for neuromyelitis optica autoantibodies. Glia 2009, 57, 1363–1373. [Google Scholar]
- Bradl, M.; Misu, T.; Takahashi, T.; Watanabe, M.; Mader, S.; Reindl, M.; Adzemovic, M.; Bauer, J.; Berger, T.; Fujihara, K.; et al. Neuromyelitis optica: Pathogenicity of patient immunoglobulin in vivo. Ann. Neurol 2009, 66, 630–643. [Google Scholar]
- Ratelade, J.; Bennett, J.L.; Verkman, A.S. Intravenous neuromyelitis optica autoantibody in mice targets aquaporin-4 in peripheral organs and area postrema. PLoS One 2011, 6, e27412. [Google Scholar]
- Takahashi, T.; Fujihara, K.; Nakashima, I.; Misu, T.; Miyazawa, I.; Nakamura, M.; Watanabe, S.; Shiga, Y.; Kanaoka, C.; Fujimori, J.; et al. Anti-aquaporin-4 antibody is involved in the pathogenesis of NMO: A study on antibody titre. Brain 2007, 130, 1235–1243. [Google Scholar]
- Kalluri, S.R.; Illes, Z.; Srivastava, R.; Cree, B.; Menge, T.; Bennett, J.L.; Berthele, A.; Hemmer, B. Quantification and functional characterization of antibodies to native aquaporin 4 in neuromyelitis optica. Arch. Neurol 2010, 67, 1201–1208. [Google Scholar]
- Klawiter, E.C.; Alvarez, E., 3rd; Xu, J.; Paciorkowski, A.R.; Zhu, L.; Parks, B.J.; Cross, A.H.; Naismith, R.T. NMO-IgG detected in CSF in seronegative neuromyelitis optica. Neurology 2009, 72, 1101–1103. [Google Scholar]
- Bennett, J.L.; Lam, C.; Kalluri, S.R.; Saikali, P.; Bautista, K.; Dupree, C.; Glogowska, M.; Case, D.; Antel, J.P.; Owens, G.P.; et al. Intrathecal pathogenic anti-aquaporin-4 antibodies in early neuromyelitis optica. Ann. Neurol 2009, 66, 617–629. [Google Scholar]
- Chihara, N.; Aranami, T.; Sato, W.; Miyazaki, Y.; Miyake, S.; Okamoto, T.; Ogawa, M.; Toda, T.; Yamamura, T. Interleukin 6 signaling promotes anti-aquaporin 4 autoantibody production from plasmablasts in neuromyelitis optica. Proc. Natl. Acad. Sci. USA 2011, 108, 3701–3706. [Google Scholar]
- Kira, J. Multiple sclerosis in the japanese population. Lancet Neurol 2003, 2, 117–127. [Google Scholar]
- Osuntokun, B.O. The pattern of neurological illness in tropical Africa. Experience at Ibadan, Nigeria. J. Neurol. Sci 1971, 12, 417–442. [Google Scholar]
- Chopra, J.S.; Radhakrishnan, K.; Sawhney, B.B.; Pal, S.R.; Banerjee, A.K. Multiple sclerosis in north-west India. Acta Neurol. Scand 1980, 62, 312–321. [Google Scholar]
- Papais-Alvarenga, R.M.; Miranda-Santos, C.M.; Puccioni-Sohler, M.; de Almeida, A.M.; Oliveira, S.; Basilio De Oliveira, C.A.; Alvarenga, H.; Poser, C.M. Optic neuromyelitis syndrome in Brazilian patients. J. Neurol. Neurosurg. Psychiatry 2002, 73, 429–435. [Google Scholar]
- McAlpine, D. Familial neuromyelitis optica: Its occurrence in identical twins. Brain 1938, 61, 430–448. [Google Scholar]
- Matiello, M.; Kim, H.J.; Kim, W.; Brum, D.G.; Barreira, A.A.; Kingsbury, D.J.; Plant, G.T.; Adoni, T.; Weinshenker, B.G. Familial neuromyelitis optica. Neurology 2010, 75, 310–315. [Google Scholar]
- Zephir, H.; Fajardy, I.; Outteryck, O.; Blanc, F.; Roger, N.; Fleury, M.; Rudolf, G.; Marignier, R.; Vukusic, S.; Confavreux, C.; et al. Is neuromyelitis optica associated with human leukocyte antigen? Mult. Scler 2009, 15, 571–579. [Google Scholar]
- Brum, D.G.; Barreira, A.A.; dos Santos, A.C.; Kaimen-Maciel, D.R.; Matiello, M.; Costa, R.M.; Deghaide, N.H.; Costa, L.S.; Louzada-Junior, P.; Diniz, P.R.; et al. HLA-DRB association in neuromyelitis optica is different from that observed in multiple sclerosis. Mult. Scler 2010, 16, 21–29. [Google Scholar]
- Matsushita, T.; Matsuoka, T.; Isobe, N.; Kawano, Y.; Minohara, M.; Shi, N.; Nishimura, Y.; Ochi, H.; Kira, J. Association of the HLA-DPB1*0501 allele with anti-aquaporin-4 antibody positivity in japanese patients with idiopathic central nervous system demyelinating disorders. Tissue Antigens 2009, 73, 171–176. [Google Scholar]
- Wang, H.; Dai, Y.; Qiu, W.; Zhong, X.; Wu, A.; Wang, Y.; Lu, Z.; Bao, J.; Hu, X. HLA-DPB1 0501 is associated with susceptibility to anti-aquaporin-4 antibodies positive neuromyelitis optica in southern han chinese. J. Neuroimmunol 2011, 233, 181–184. [Google Scholar]
- Tsao, B.P. The genetics of human systemic lupus erythematosus. Trends Immunol 2003, 24, 595–602. [Google Scholar]
- Onuma, H.; Ota, M.; Sugenoya, A.; Inoko, H. Association of HLA-DPB1*0501 with early-onset Graves’ disease in Japanese. Hum. Immunol 1994, 39, 195–201. [Google Scholar]
- Matiello, M.; Kantarci, O.; Brum, D.; Schaefer-Klein, J.; Weinshenker, B.; Consortium, N.G. HLA DRB1*1501 tagging rs3135388 polymorphism associated with multiple sclerosis is inversely associated with NMO. Mult. Scler 2009, 15, S69. [Google Scholar]
- Deschamps, R.; Paturel, L.; Jeannin, S.; Chausson, N.; Olindo, S.; Bera, O.; Bellance, R.; Smadja, D.; Cesaire, D.; Cabre, P. Different HLA class II (DRB1 and DQB1) alleles determine either susceptibility or resistance to nmo and multiple sclerosis among the French Afro-Caribbean population. Mult. Scler 2011, 17, 24–31. [Google Scholar]
- Stewart, G.J.; Teutsch, S.M.; Castle, M.; Heard, R.N.; Bennetts, B.H. HLA-DR, -DQA1 and -DQB1 associations in Australian multiple sclerosis patients. Eur. J. Immunogenet 1997, 24, 81–92. [Google Scholar]
- Mealy, M.A.; Wingerchuk, D.M.; Greenberg, B.M.; Levy, M. Epidemiology of neuromyelitis optica in the United States: A multicenter analysis epidemiology of NMO. Arch. Neurol 2012, 69, 1176–1180. [Google Scholar]
- Matiello, M.; Schaefer-Klein, J.L.; Hebrink, D.D.; Kingsbury, D.J.; Atkinson, E.J.; Weinshenker, B.G. NMO genetics collaborators, genetic analysis of aquaporin-4 in neuromyelitis optica. Neurology 2011, 77, 1149–1155. [Google Scholar]
- Rossi, A.; Pisani, F.; Nicchia, G.P.; Svelto, M.; Frigeri, A. Evidences for a leaky scanning mechanism for the synthesis of the shorter M23 protein isoform of aquaporin-4 implication in orthogonal array formation and neuromyelitis optica antibody interaction. J. Biol. Chem 2010, 285, 4562–4569. [Google Scholar]
- Sellner, J.; Hemmer, B.; Muhlau, M. The clinical spectrum and immunobiology of parainfectious neuromyelitis optica (devic) syndromes. J. Autoimmunity 2010, 34, 371–379. [Google Scholar]
- Menge, T.; Cree, B.; Saleh, A.; Waterboer, T.; Berthele, A.; Kalluri, S.R.; Hemmer, B.; Aktas, O.; Hartung, H.P.; Methner, A.; et al. Neuromyelitis optica following human papillomavirus vaccination. Neurology 2012, 79, 285–287. [Google Scholar]
- Pittock, S.J.; Lennon, V.A. Aquaporin-4 autoantibodies in a paraneoplastic context. Arch. Neurol 2008, 65, 629–632. [Google Scholar]
- Varrin-Doyer, M.; Spencer, C.M.; Schulze-Topphoff, U.; Nelson, P.A.; Stroud, R.M.; BA, C.C.; Zamvil, S.S. Aquaporin 4-specific T cells in neuromyelitis optica exhibit a Th17 bias and recognize Clostridium ABC transporter. Ann. Neurol 2012, 72, 53–64. [Google Scholar]
- Hatheway, C.L. Toxigenic clostridia. Clin. Microbiol. Rev 1990, 3, 66–98. [Google Scholar]
- Egg, R.; Reindl, M.; Deisenhammer, F.; Linington, C.; Berger, T. Anti-MOG and anti-MBP antibody subclasses in multiple sclerosis. Mult. Scler 2001, 7, 285–289. [Google Scholar]
- Angelucci, F.; Mirabella, M.; Frisullo, G.; Caggiula, M.; Tonali, P.A.; Batocchi, A.P. Serum levels of anti-myelin antibodies in relapsing-remitting multiple sclerosis patients during different phases of disease activity and immunomodulatory therapy. Dis. Markers 2005, 21, 49–55. [Google Scholar]
- Li, L.; Zhang, H.; Varrin-Doyer, M.; Zamvil, S.S.; Verkman, A.S. Proinflammatory role of aquaporin-4 in autoimmune neuroinflammation. FASEB J 2011, 25, 1556–1566. [Google Scholar]
- Kinoshita, M.; Nakatsuji, Y.; Kimura, T.; Moriya, M.; Takata, K.; Okuno, T.; Kumanogoh, A.; Kajiyama, K.; Yoshikawa, H.; Sakoda, S. Neuromyelitis optica: Passive transfer to rats by human immunoglobulin. Biochem. Biophys. Res. Commun 2009, 386, 623–627. [Google Scholar]
- Zhang, H.; Bennett, J.L.; Verkman, A.S. Ex vivo spinal cord slice model of neuromyelitis optica reveals novel immunopathogenic mechanisms. Ann. Neurol 2011, 70, 943–954. [Google Scholar]
- Kinoshita, M.; Nakatsuji, Y.; Kimura, T.; Moriya, M.; Takata, K.; Okuno, T.; Kumanogoh, A.; Kajiyama, K.; Yoshikawa, H.; Sakoda, S. Anti-aquaporin-4 antibody induces astrocytic cytotoxicity in the absence of CNS antigen-specific T cells. Biochem. Biophys. Res. Commun 2010, 394, 205–210. [Google Scholar]
- Lennon, V.A.; Kryzer, T.J.; Pittock, S.J.; Verkman, A.S.; Hinson, S.R. IgG marker of optic-spinal multiple sclerosis binds to the aquaporin-4 water channel. J. Exp. Med 2005, 202, 473–477. [Google Scholar]
- Jarius, S.; Aboul-Enein, F.; Waters, P.; Kuenz, B.; Hauser, A.; Berger, T.; Lang, W.; Reindl, M.; Vincent, A.; Kristoferitsch, W. Antibody to aquaporin-4 in the long-term course of neuromyelitis optica. Brain 2008, 131, 3072–3080. [Google Scholar]
- Hinson, S.R.; McKeon, A.; Fryer, J.P.; Apiwattanakul, M.; Lennon, V.A.; Pittock, S.J. Prediction of neuromyelitis optica attack severity by quantitation of complement-mediated injury to aquaporin-4-expressing cells. Arch. Neurol 2009, 66, 1164–1167. [Google Scholar]
- Matsushita, T.; Isobe, N.; Matsuoka, T.; Shi, N.; Kawano, Y.; Wu, X.M.; Yoshiura, T.; Nakao, Y.; Ishizu, T.; Kira, J.I. Aquaporin-4 autoimmune syndrome and anti-aquaporin-4 antibody-negative opticospinal multiple sclerosis in Japanese. Mult. Scler 2009, 15, 834–847. [Google Scholar]
- Nishiyama, S.; Ito, T.; Misu, T.; Takahashi, T.; Kikuchi, A.; Suzuki, N.; Jin, K.; Aoki, M.; Fujihara, K.; Itoyama, Y. A case of NMO seropositive for aquaporin-4 antibody more than 10 years before onset. Neurology 2009, 72, 1960–1961. [Google Scholar]
- Kinoshita, M.; Nakatsuji, Y.; Moriya, M.; Okuno, T.; Kumanogoh, A.; Nakano, M.; Takahashi, T.; Fujihara, K.; Tanaka, K.; Sakoda, S. Astrocytic necrosis is induced by anti-aquaporin-4 antibody-positive serum. Neuroreport 2009, 20, 508–512. [Google Scholar]
- Kim, Y.U.; Kinoshita, T.; Molina, H.; Hourcade, D.; Seya, T.; Wagner, L.M.; Holers, V.M. Mouse complement regulatory protein Crry/p65 uses the specific mechanisms of both human decay-accelerating factor and membrane cofactor protein. J. Exp. Med 1995, 181, 151–159. [Google Scholar]
- Lener, M.; Vinci, G.; Duponchel, C.; Meo, T.; Tosi, M. Molecular cloning, gene structure and expression profile of mouse C1 inhibitor. Eur. J. Biochem 1998, 254, 117–122. [Google Scholar]
- Pinter, C.; Beltrami, S.; Caputo, D.; Ferrante, P.; Clivio, A. Presence of autoantibodies against complement regulatory proteins in relapsing-remitting multiple sclerosis. J. Neurovirol 2000, 6, S42–S46. [Google Scholar]
- Vincent, T.; Saikali, P.; Cayrol, R.; Roth, A.D.; Bar-Or, A.; Prat, A.; Antel, J.P. Functional consequences of neuromyelitis optica-IgG astrocyte interactions on blood-brain barrier permeability and granulocyte recruitment. J. Immunol 2008, 181, 5730–5737. [Google Scholar]
- Hinson, S.R.; Romero, M.F.; Popescu, B.F.; Lucchinetti, C.F.; Fryer, J.P.; Wolburg, H.; Fallier-Becker, P.; Noell, S.; Lennon, V.A. Molecular outcomes of neuromyelitis optica (NMO)-IgG binding to aquaporin-4 in astrocytes. Proc. Natl. Acad. Sci. USA 2012, 109, 1245–1250. [Google Scholar]
- Melamud, L.; Fernandez, J.M.; Rivarola, V.; Di Giusto, G.; Ford, P.; Villa, A.; Capurro, C. Neuromyelitis optica immunoglobulin G present in sera from neuromyelitis optica patients affects aquaporin-4 expression and water permeability of the astrocyte plasma membrane. J. Neurosci. Res 2012, 90, 1240–1248. [Google Scholar]
- Rossi, A.; Ratelade, J.; Papadopoulos, M.C.; Bennett, J.L.; Verkman, A.S. Consequences of NMO-IgG binding to aquaporin-4 in neuromyelitis optica. Proc. Natl. Acad. Sci. USA 2012, 109. [Google Scholar] [CrossRef]
- Eng, L.F.; Ghirnikar, R.S.; Lee, Y.L. Glial fibrillary acidic protein: GFAP-thirty-one years (1969–2000). Neurochem. Res 2000, 25, 1439–1451. [Google Scholar]
- Takano, R.; Misu, T.; Takahashi, T.; Sato, S.; Fujihara, K.; Itoyama, Y. Astrocytic damage is far more severe than demyelination in NMO a clinical CSF biomarker study. Neurology 2010, 75, 208–216. [Google Scholar]
- Sofroniew, M.V.; Vinters, H.V. Astrocytes: Biology and pathology. Acta Neuropathol 2010, 119, 7–35. [Google Scholar]
- Uzawa, A.; Mori, M.; Arai, K.; Sato, Y.; Hayakawa, S.; Masuda, S.; Taniguchi, J.; Kuwabara, S. Cytokine and chemokine profiles in neuromyelitis optica: Significance of interleukin-6. Mult. Scler 2010, 16, 1443–1452. [Google Scholar]
- Tanaka, M.; Matsushita, T.; Tateishi, T.; Ochi, H.; Kawano, Y.; Mei, F.J.; Minohara, M.; Murai, H.; Kira, J.I. Distinct CSF cytokine/chemokine profiles in atopic myelitis and other causes of myelitis. Neurology 2008, 71, 974–981. [Google Scholar]
- Ishizu, T.; Osoegawa, M.; Mei, F.J.; Kikuchi, H.; Tanaka, M.; Takakura, Y.; Minohara, M.; Murai, H.; Mihara, F.; Taniwaki, T.; et al. Intrathecal activation of the IL-17/IL-8 axis in opticospinal multiple sclerosis. Brain 2005, 128, 988–1002. [Google Scholar]
- Correale, J.; Fiol, M. Chitinase effects on immune cell response in neuromyelitis optica and multiple sclerosis. Mult. Scler 2011, 17, 521–531. [Google Scholar]
- Mandler, R.N.; Davis, L.E.; Jeffery, D.R.; Kornfeld, M. Devic’s neuromyelitis optica: A clinicopathological study of 8 patients. Ann. Neurol 1993, 34, 162–168. [Google Scholar]
- Zhong, X.; Wang, H.; Dai, Y.; Wu, A.; Bao, J.; Xu, W.; Cheng, C.; Lu, Z.; Qiu, W.; Hu, X. Cerebrospinal fluid levels of CXCL13 are elevated in neuromyelitis optica. J. Neuroimmunol 2011, 240–241, 104–108. [Google Scholar]
- Manzo, A.; Vitolo, B.; Humby, F.; Caporali, R.; Jarrossay, D.; Dell’accio, F.; Ciardelli, L.; Uguccioni, M.; Montecucco, C.; Pitzalis, C. Mature antigen-experienced T helper cells synthesize and secrete the B cell chemoattractant CXCL13 in the inflammatory environment of the rheumatoid joint. Arthritis Rheum 2008, 58, 3377–3387. [Google Scholar]
- Carlsen, H.S.; Baekkevold, E.S.; Morton, H.C.; Haraldsen, G.; Brandtzaeg, P. Monocyte-like and mature macrophages produce CXCL13 (B cell-attracting chemokine 1) in inflammatory lesions with lymphoid neogenesis. Blood 2004, 104, 3021–3027. [Google Scholar]
- Vaknin-Dembinsky, A.; Brill, L.; Orpaz, N.; Abramsky, O.; Karussis, D. Preferential increase of B-cell activating factor in the cerebrospinal fluid of neuromyelitis optica in a white population. Mult. Scler 2010, 16, 1453–1457. [Google Scholar]
- Mackay, F.; Schneider, P. Cracking the BAFF code. Nat. Rev. Immunol 2009, 9, 491–502. [Google Scholar]
- Krumbholz, M.; Theil, D.; Derfuss, T.; Rosenwald, A.; Schrader, F.; Monoranu, C.M.; Kalled, S.L.; Hess, D.M.; Serafini, B.; Aloisi, F.; et al. BAFF is produced by astrocytes and up-regulated in multiple sclerosis lesions and primary central nervous system lymphoma. J. Exp. Med 2005, 201, 195–200. [Google Scholar]
- Kayagaki, N.; Yan, M.; Seshasayee, D.; Wang, H.; Lee, W.; French, D.M.; Grewal, I.S.; Cochran, A.G.; Gordon, N.C.; Yin, J.; et al. BAFF/BLyS receptor 3 binds the B cell survival factor BAFF ligand through a discrete surface loop and promotes processing of NF-kappaB2. Immunity 2002, 17, 515–524. [Google Scholar]
- Bergamaschi, R. Glatiramer acetate treatment in Devic’s neuromyelitis optica. Brain 2003, 126. [Google Scholar] [CrossRef]
- Gartzen, K.; Limmroth, V.; Putzki, N. Relapsing neuromyelitis optica responsive to glatiramer acetate treatment. Eur. J. Neurol 2007, 14, e12–e13. [Google Scholar]
- Icoz, S.; Tuzun, E.; Kurtuncu, M.; Durmus, H.; Mutlu, M.; Eraksoy, M.; Akman-Demir, G. Enhanced IL-6 production in aquaporin-4 antibody positive neuromyelitis optica patients. Int. J. Neurosci 2010, 120, 71–75. [Google Scholar]
- Farina, C.; Aloisi, F.; Meinl, E. Astrocytes are active players in cerebral innate immunity. Trends Immunol 2007, 28, 138–145. [Google Scholar]
- Ulusoy, C.; Tuzun, E.; Kurtuncu, M.; Turkoglu, R.; Akman-Demir, G.; Eraksoy, M. Comparison of the cytokine profiles of patients with neuronal-antibody-associated central nervous system disorders. Int. J. Neurosci 2012, 122, 284–289. [Google Scholar]
- Bettelli, E.; Carrier, Y.; Gao, W.; Korn, T.; Strom, T.B.; Oukka, M.; Weiner, H.L.; Kuchroo, V.K. Reciprocal developmental pathways for the generation of pathogenic effector Th17 and regulatory T cells. Nature 2006, 441, 235–238. [Google Scholar]
- Wilke, C.M.; Bishop, K.; Fox, D.; Zou, W. Deciphering the role of Th17 cells in human disease. Trends Immunol 2011, 32, 603–611. [Google Scholar]
- Wang, H.H.; Dai, Y.Q.; Qiu, W.; Lu, Z.Q.; Peng, F.H.; Wang, Y.G.; Bao, J.; Li, Y.; Hu, X.Q. Interleukin-17-secreting T cells in neuromyelitis optica and multiple sclerosis during relapse. J. Clin. Neurosci 2011, 18, 1313–1317. [Google Scholar]
- Li, Y.; Wang, H.; Long, Y.; Lu, Z.; Hu, X. Increased memory Th17 cells in patients with neuromyelitis optica and multiple sclerosis. J. Neuroimmunol 2011, 234, 155–160. [Google Scholar]
- Murphy, A.C.; Lalor, S.J.; Lynch, M.A.; Mills, K.H. Infiltration of Th1 and Th17 cells and activation of microglia in the CNS during the course of experimental autoimmune encephalomyelitis. Brain Behav. Immun 2010, 24, 641–651. [Google Scholar]
- Matsuya, N.; Komori, M.; Nomura, K.; Nakane, S.; Fukudome, T.; Goto, H.; Shiraishi, H.; Wandinger, K.P.; Matsuo, H.; Kondo, T. Increased T-cell immunity against aquaporin-4 and proteolipid protein in neuromyelitis optica. Int. Immunol 2011, 23, 565–573. [Google Scholar]
- Vaknin-Dembinsky, A.; Brill, L.; Kassis, I.; Petrou, P.; Ovadia, H.; Ben-Hur, T.; Abramsky, O.; Karussis, D. T-cell reactivity against AQP4 in neuromyelitis optica. Neurology 2012, 79, 945–946. [Google Scholar]
- Nakashima, I.; Takahashi, T.; Cree, B.A.; Kim, H.J.; Suzuki, C.; Genain, C.P.; Vincent, T.; Fujihara, K.; Itoyama, Y.; Bar-Or, A. Transient increases in anti-aquaporin-4 antibody titers following rituximab treatment in neuromyelitis optica, in association with elevated serum BAFF levels. J. Clin. Neurosci 2011, 18, 997–998. [Google Scholar]
- Tedder, T.F.; Engel, P. Cd20: A regulator of cell-cycle progression of B lymphocytes. Immunol. Today 1994, 15, 450–454. [Google Scholar]
- Manz, R.A.; Arce, S.; Cassese, G.; Hauser, A.E.; Hiepe, F.; Radbruch, A. Humoral immunity and long-lived plasma cells. Curr. Opin. Immunol 2002, 14, 517–521. [Google Scholar]
- Uzawa, A.; Mori, M.; Masuda, S.; Kuwabara, S. Markedly elevated soluble intercellular adhesion molecule 1, soluble vascular cell adhesion molecule 1 levels, and blood-brain barrier breakdown in neuromyelitis optica. Arch. Neurol 2011, 68, 913–917. [Google Scholar]
- Shimizu, F.; Sano, Y.; Takahashi, T.; Haruki, H.; Saito, K.; Koga, M.; Kanda, T. Sera from neuromyelitis optica patients disrupt the blood-brain barrier. J. Neurol. Neurosurg. Psychiatry 2012, 83, 288–297. [Google Scholar]
- Roemer, S.F.; Parisi, J.E.; Lennon, V.A.; Benarroch, E.E.; Lassmann, H.; Bruck, W.; Mandler, R.N.; Weinshenker, B.G.; Pittock, S.J.; Wingerchuk, D.M.; et al. Pattern-specific loss of aquaporin-4 immunoreactivity distinguishes neuromyelitis optica from multiple sclerosis. Brain 2007, 130, 1194–1205. [Google Scholar]
- Lucchinetti, C.F.; Mandler, R.N.; McGavern, D.; Bruck, W.; Gleich, G.; Ransohoff, R.M.; Trebst, C.; Weinshenker, B.; Wingerchuk, D.; Parisi, J.E.; et al. A role for humoral mechanisms in the pathogenesis of Devic’s neuromyelitis optica. Brain 2002, 125, 1450–1461. [Google Scholar]
- Barnett, M.H.; Parratt, J.D.; Cho, E.S.; Prineas, J.W. Immunoglobulins and complement in postmortem multiple sclerosis tissue. Ann. Neurol 2009, 65, 32–46. [Google Scholar]
- Barnett, M.H.; Prineas, J.W.; Buckland, M.E.; Parratt, J.D.; Pollard, J.D. Massive astrocyte destruction in neuromyelitis optica despite natalizumab therapy. Mult. Scler 2012, 18, 108–112. [Google Scholar]
- Kobayashi, Z.; Tsuchiya, K.; Uchihara, T.; Nakamura, A.; Haga, C.; Yokota, O.; Ishizu, H.; Taki, K.; Arai, T.; Akiyama, H.; et al. Intractable hiccup caused by medulla oblongata lesions: A study of an autopsy patient with possible neuromyelitis optica. J. Neurol. Sci 2009, 285, 241–245. [Google Scholar]
- Matsuoka, T.; Suzuki, S.O.; Suenaga, T.; Iwaki, T.; Kira, J. Reappraisal of aquaporin-4 astrocytopathy in asian neuromyelitis optica and multiple sclerosis patients. Brain Pathol 2011, 21, 516–532. [Google Scholar]
- Lee, D.H.; Metz, I.; Berthele, A.; Stadelmann, C.; Bruck, W.; Linker, R.A.; Gold, R.; Schroeder, A. Supraspinal demyelinating lesions in neuromyelitis optica display a typical astrocyte pathology. Neuropathol. Appl. Neurobiol 2010, 36, 685–687. [Google Scholar]
- Tradtrantip, L.; Zhang, H.; Anderson, M.O.; Saadoun, S.; Phuan, P.W.; Papadopoulos, M.C.; Bennett, J.L.; Verkman, A.S. Small-molecule inhibitors of NMO-IgG binding to aquaporin-4 reduce astrocyte cytotoxicity in neuromyelitis optica. FASEB J 2012, 26, 2197–2208. [Google Scholar]
- Hutas, G. Ocrelizumab, a humanized monoclonal antibody against CD20 for inflammatory disorders and B-cell malignancies. Curr. Opin. Investig. Drugs 2008, 9, 1206–1215. [Google Scholar]
- Herbst, R.; Wang, Y.; Gallagher, S.; Mittereder, N.; Kuta, E.; Damschroder, M.; Woods, R.; Rowe, D.C.; Cheng, L.; Cook, K.; et al. B-cell depletion in vitro and in vivo with an afucosylated anti-CD19 antibody. J. Pharmacol. Exp. Ther 2010, 335, 213–222. [Google Scholar]
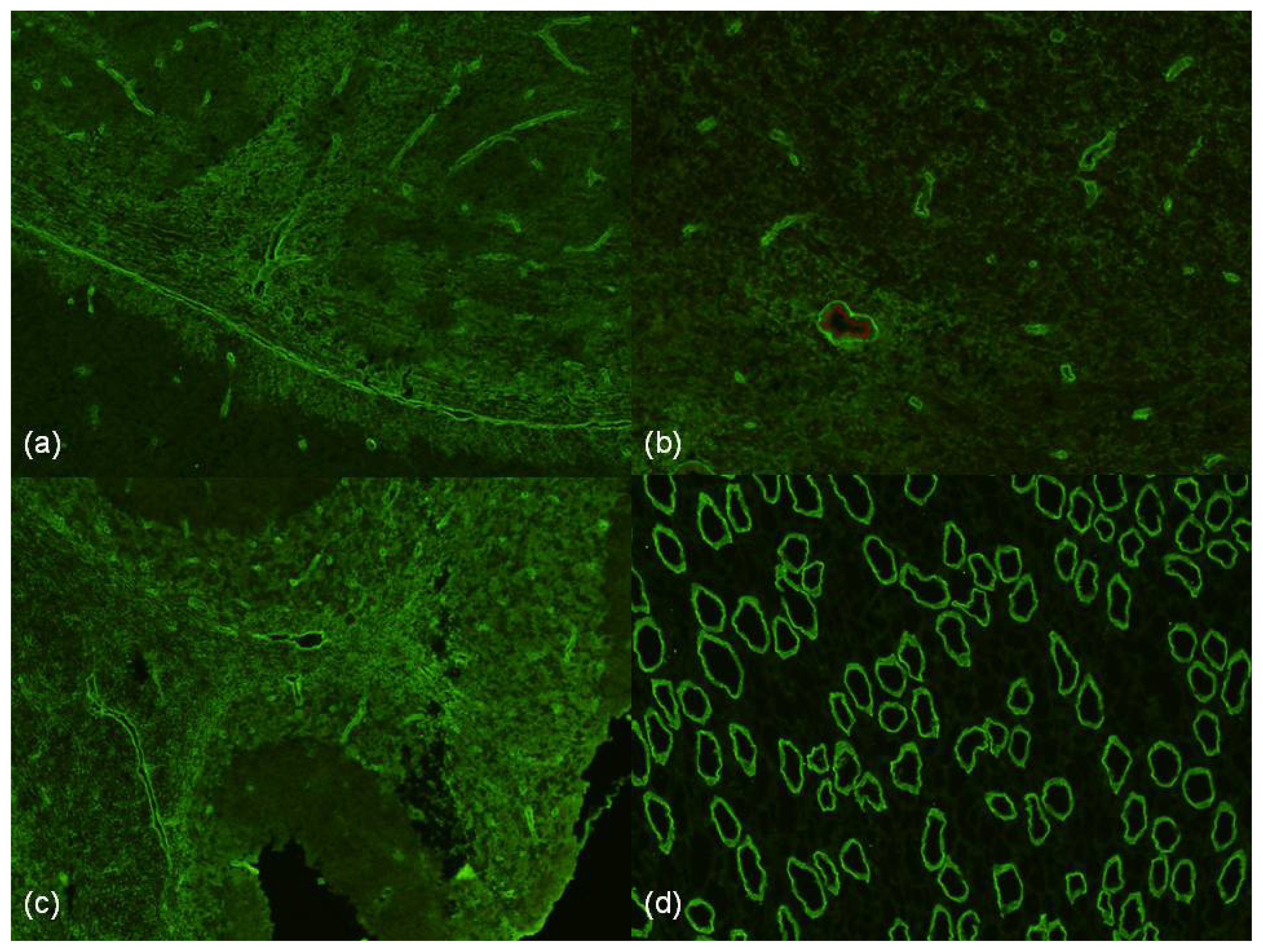

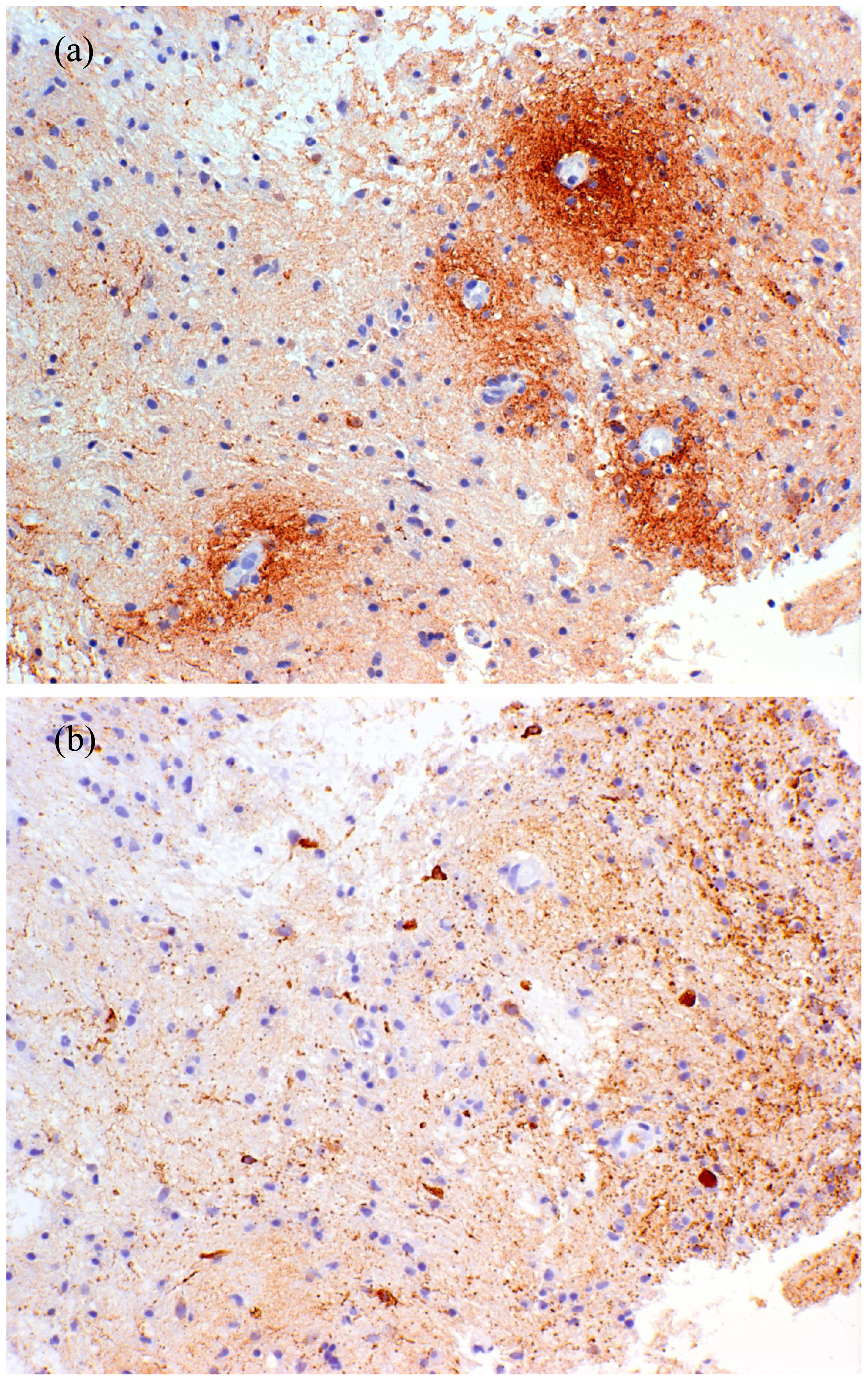

{kind=link}
{kind=link}
{kind=link}
| Clinical feature | NMO | MS |
|---|---|---|
| Optic neuritis | +++ | ++ |
| Severe, bilateral | +++ | (+) |
| Myelitis | +++ | ++ |
| Partial | (+) | +++ |
| Extensive (>3 segments) | +++ | − |
| CSF analysis | ||
| Oligoclonal bands | (+) | ++ |
| Elevated protein | + | (+) |
| Pleiocytosis | ++ | + |
| Lymphocytes | ++ | − |
| MRI brain | ||
| Normal at onset | ++ | + |
| Hypothalamic/thalamic | + | − |
| Large hemispheric | + | + |
| Brainstem | + | + |
| “Barkhof” abnormal MRI | + | +++ |
| NMO IgG | ++ | (−) |
| Gender ratio (F:M) | 9:1 | 3:1 |
© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Bukhari, W.; Barnett, M.H.; Prain, K.; Broadley, S.A. Molecular Pathogenesis of Neuromyelitis Optica. Int. J. Mol. Sci. 2012, 13, 12970-12993. https://doi.org/10.3390/ijms131012970
Bukhari W, Barnett MH, Prain K, Broadley SA. Molecular Pathogenesis of Neuromyelitis Optica. International Journal of Molecular Sciences. 2012; 13(10):12970-12993. https://doi.org/10.3390/ijms131012970
Chicago/Turabian StyleBukhari, Wajih, Michael H Barnett, Kerri Prain, and Simon A Broadley. 2012. "Molecular Pathogenesis of Neuromyelitis Optica" International Journal of Molecular Sciences 13, no. 10: 12970-12993. https://doi.org/10.3390/ijms131012970
APA StyleBukhari, W., Barnett, M. H., Prain, K., & Broadley, S. A. (2012). Molecular Pathogenesis of Neuromyelitis Optica. International Journal of Molecular Sciences, 13(10), 12970-12993. https://doi.org/10.3390/ijms131012970