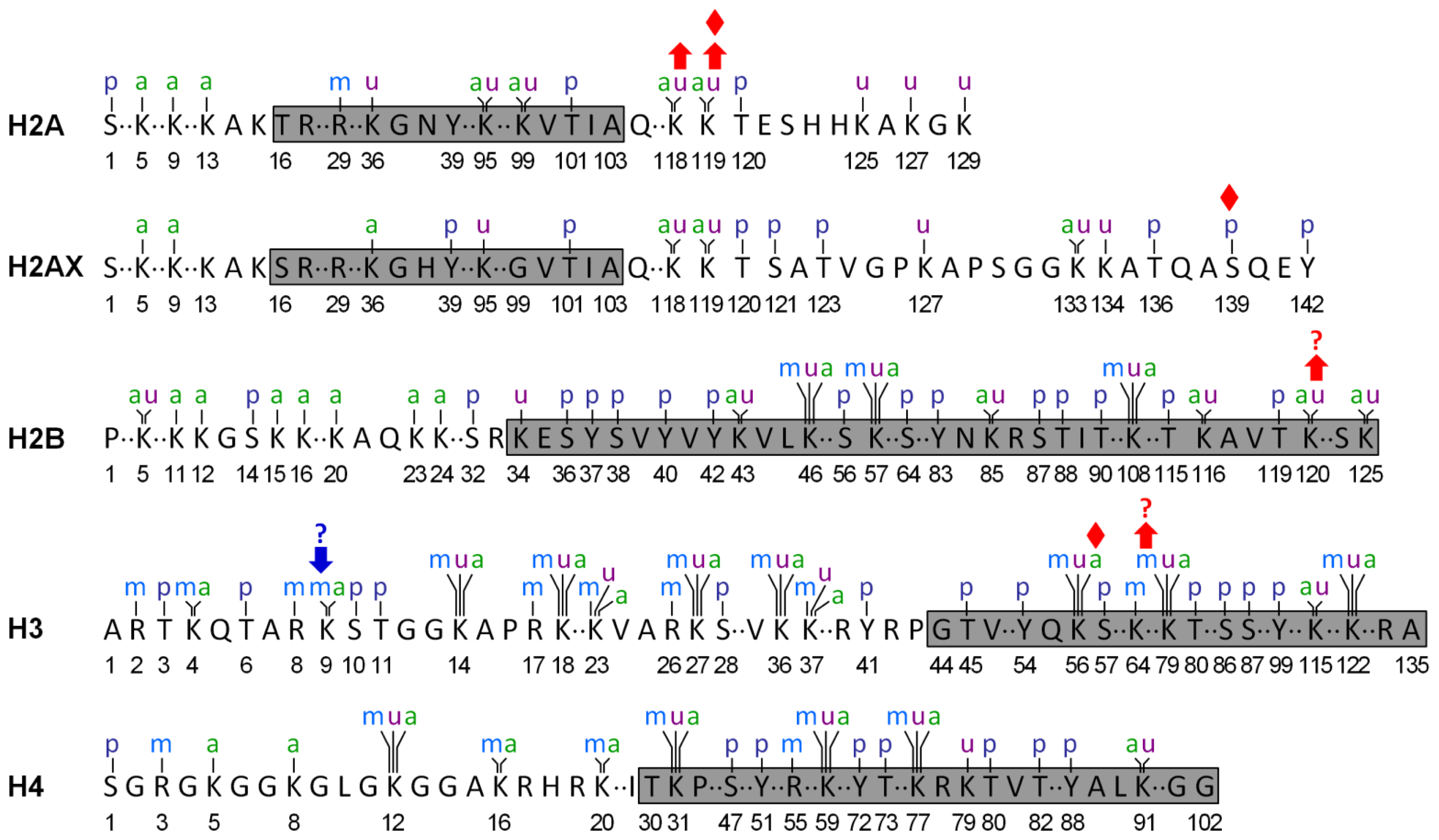
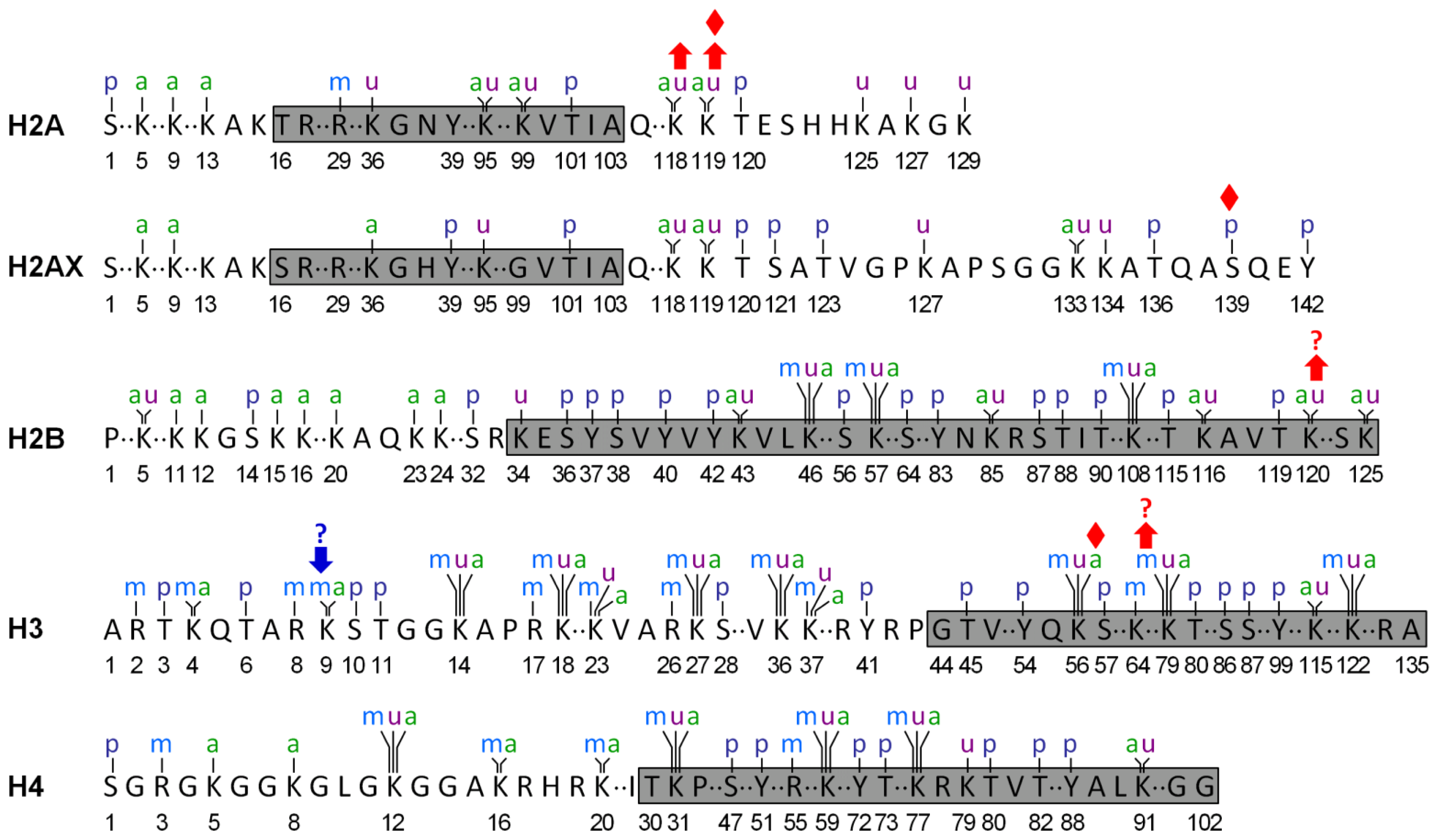
Implication of Posttranslational Histone Modifications in Nucleotide Excision Repair

Abstract
:1. Introduction
2. Histone Acetylation and NER
2.1. Gcn5 and Related Complexes in NER
2.2. p300/CBP and Related HATs in NER
2.3. TIP60 and Related HATs in NER
3. Histone Methylation and NER
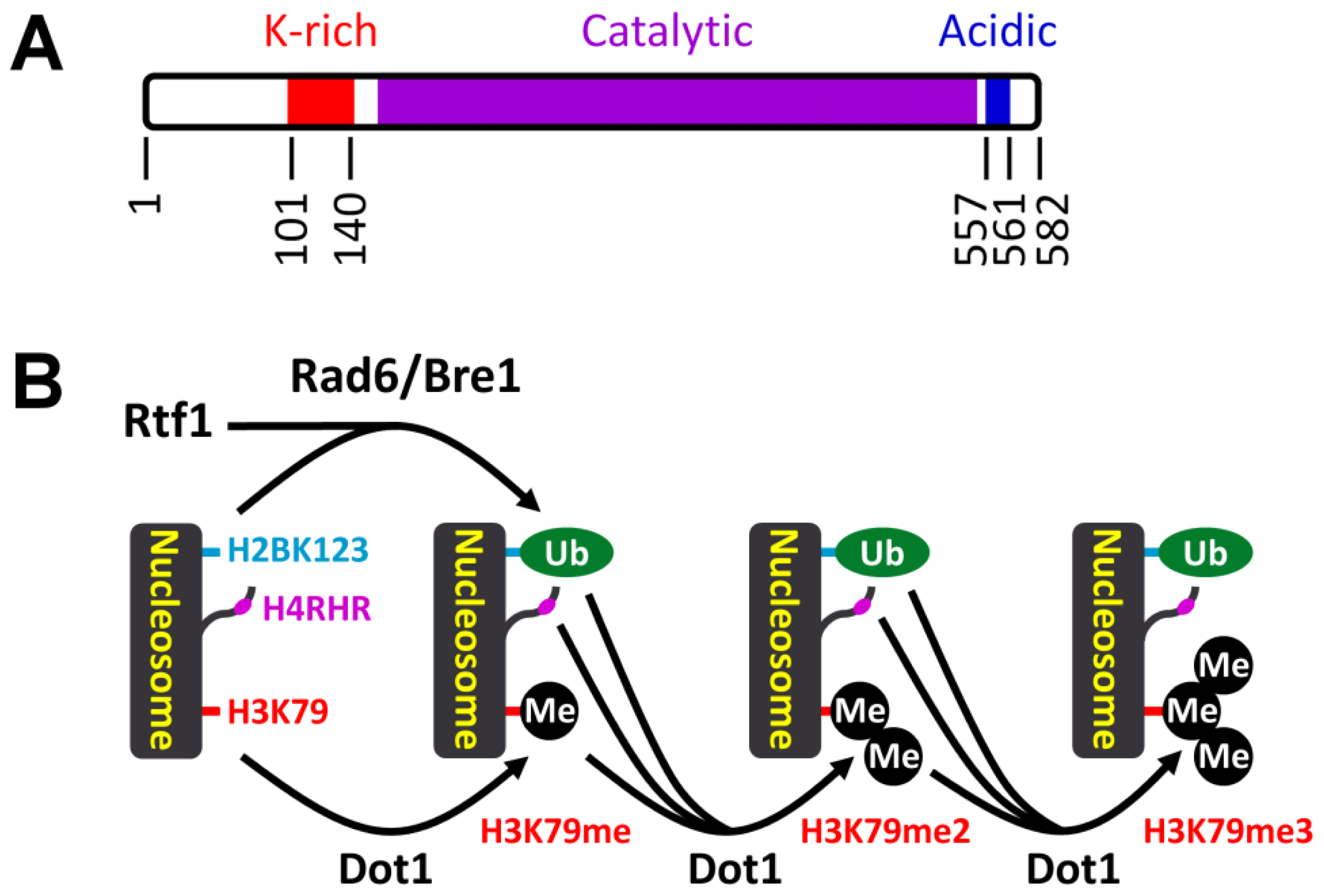
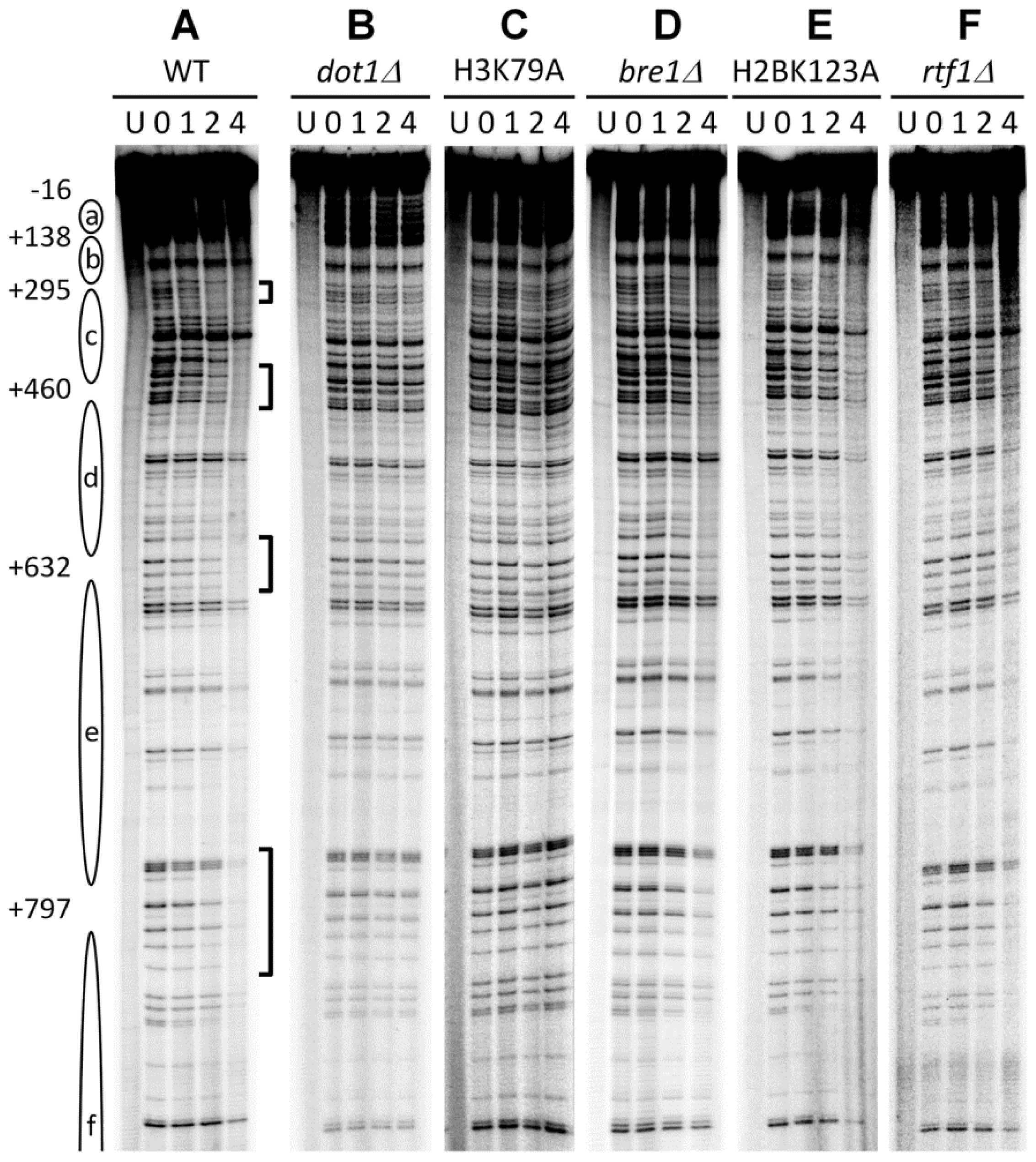
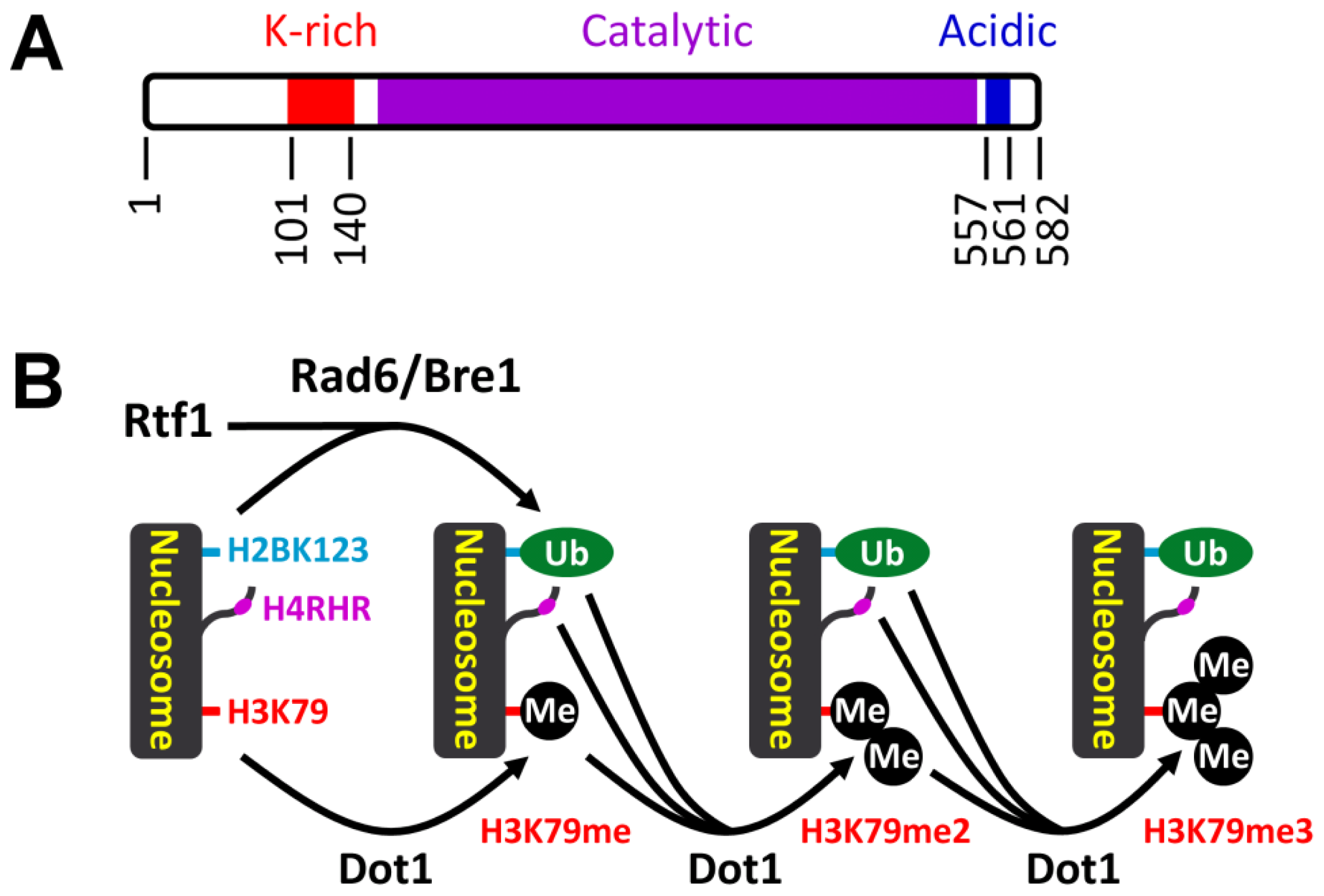
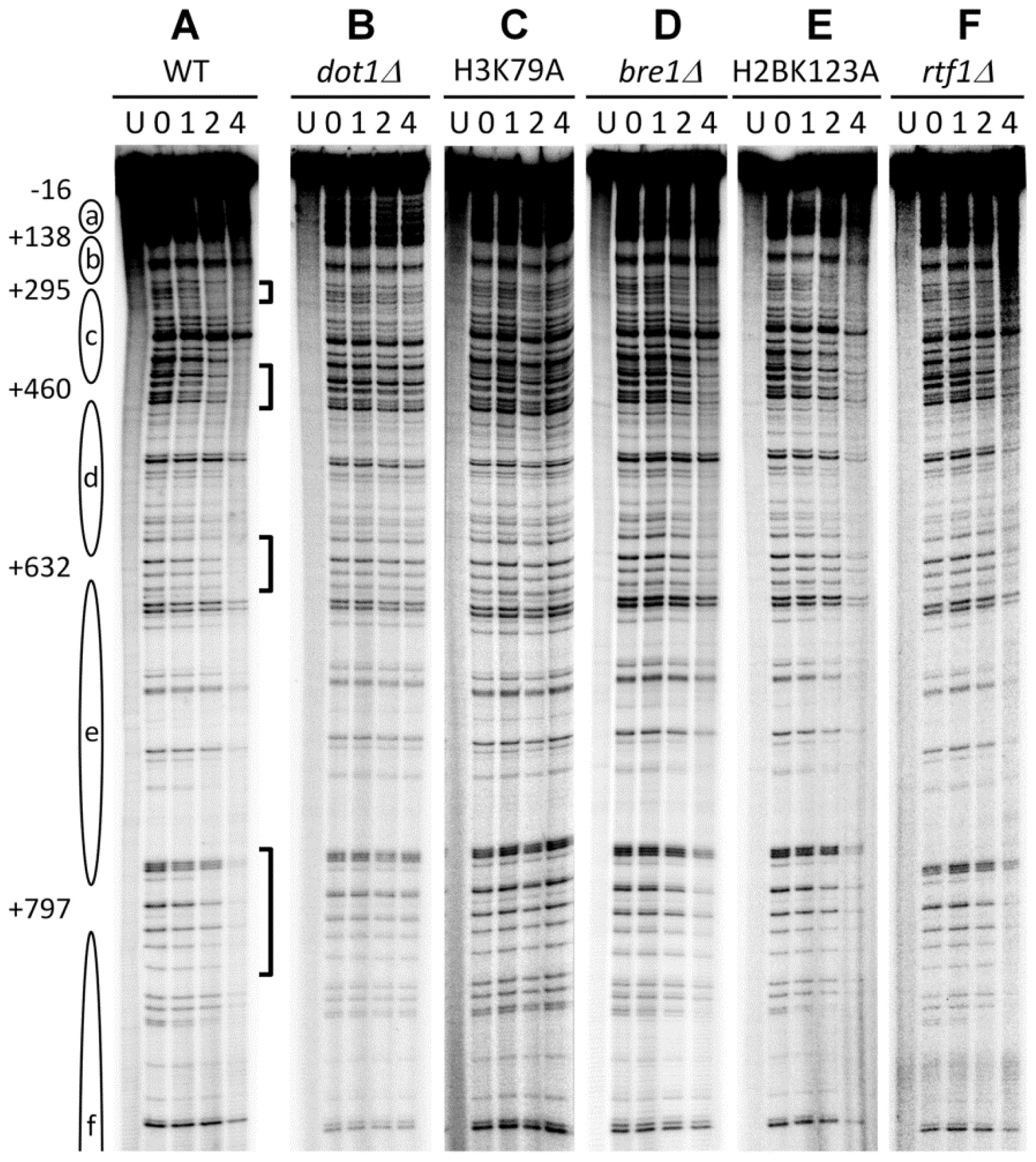
3.1. Histone H3 K79 Methylation in NER
3.2. Implication of Other Histone Methylations in NER
4. Histone Phosphorylation and NER
5. Histone Ubiquitination and NER
6. Concluding Remarks
Acknowledgments
References
- Luger, K.; Mader, A.W.; Richmond, R.K.; Sargent, D.F.; Richmond, T.J. Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature 1997, 389, 251–260. [Google Scholar]
- Kouzarides, T. Chromatin modifications and their function. Cell 2007, 128, 693–705. [Google Scholar]
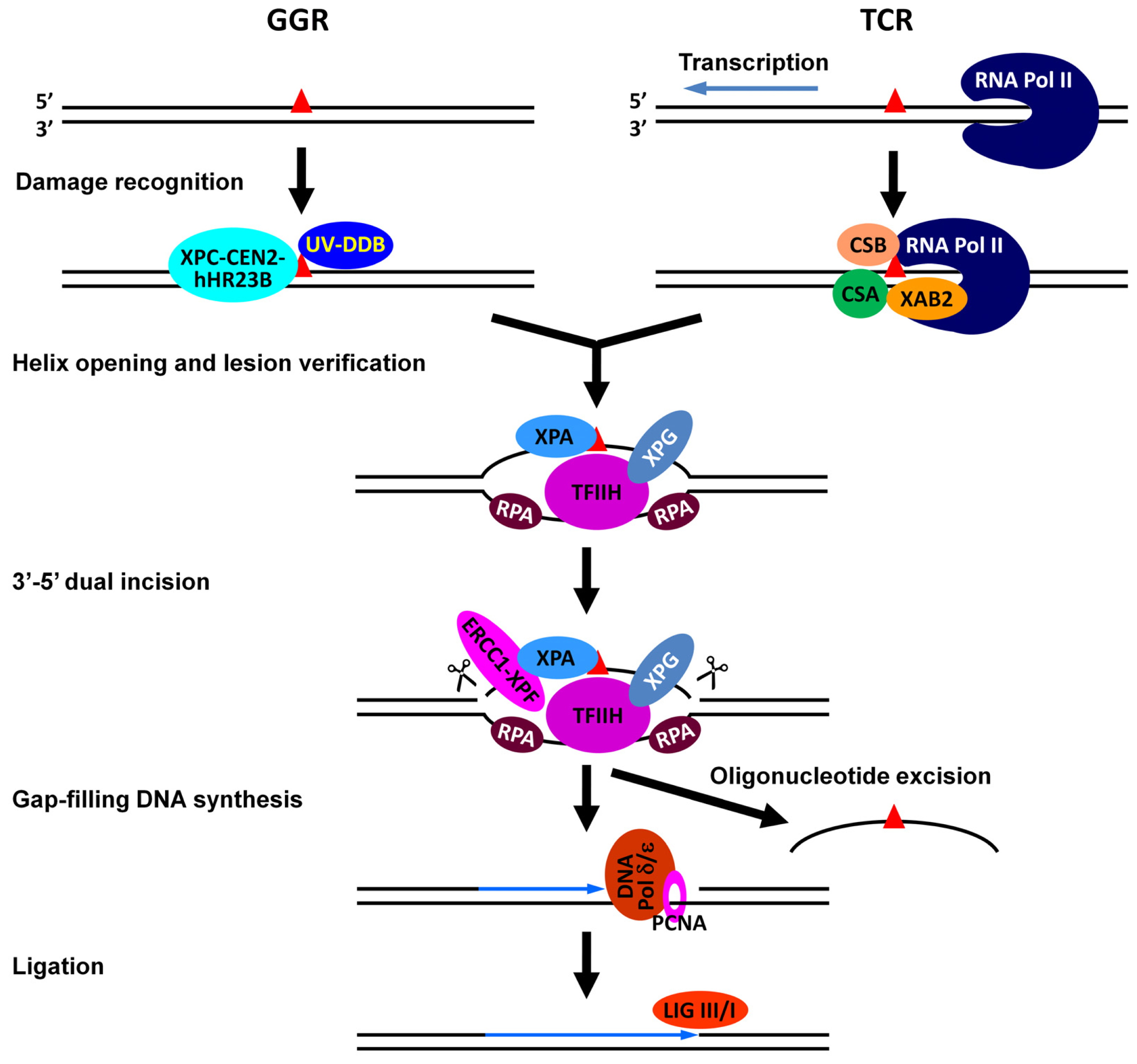
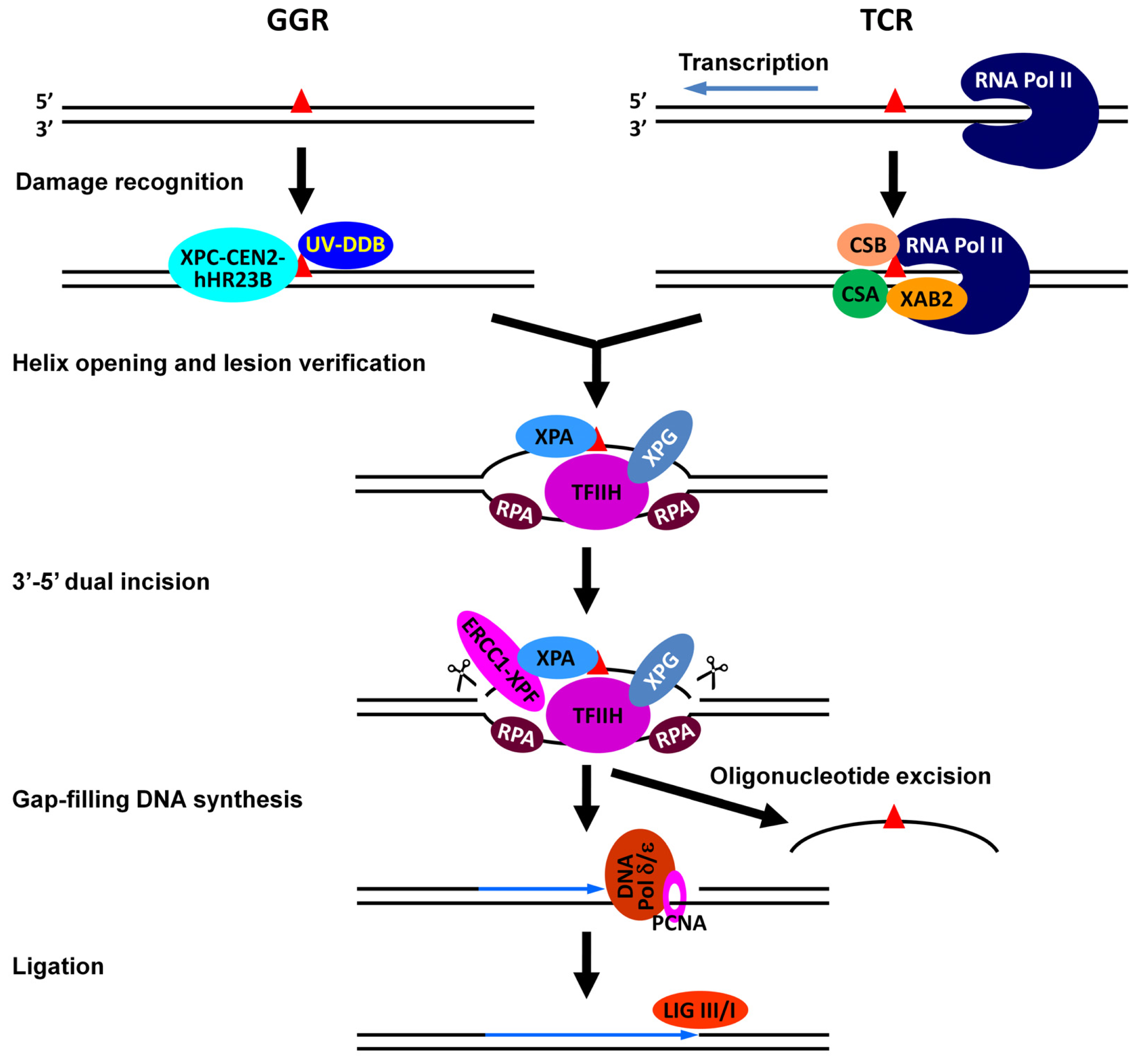
- Reardon, J.T.; Sancar, A. Nucleotide excision repair. Prog. Nucleic Acid Res. Mol. Biol 2005, 79, 183–235. [Google Scholar]
- Gillet, L.C.; Scharer, O.D. Molecular mechanisms of mammalian global genome nucleotide excision repair. Chem. Rev 2006, 106, 253–276. [Google Scholar]
- Nouspikel, T. DNA repair in mammalian cells: Nucleotide excision repair: Variations on versatility. Cell. Mol. Life Sci 2009, 66, 994–1009. [Google Scholar]
- Tatum, D.; Li, S. Nucleotide Excision Repair in S. cerevisiae. DNA Repair/Book 1; Storici, F., Ed.; InTech Open Access Publisher, 2011. available online: http://www.intechopen.com/books/dna-repair-on-the-pathways-to-fixing-dna-damage-and-errors/nucleotide-excision-repair-in-s-cerevisiae accessed on 28 September 2012.
- Waters, R.; Teng, Y.; Yu, Y.; Yu, S.; Reed, S.H. Tilting at windmills? The nucleotide excision repair of chromosomal DNA. DNA Repair (Amst.) 2009, 8, 146–152. [Google Scholar]
- Fousteri, M.; Mullenders, L.H. Transcription-coupled nucleotide excision repair in mammalian cells: Molecular mechanisms and biological effects. Cell Res 2008, 18, 73–84. [Google Scholar]
- Hanawalt, P.C.; Spivak, G. Transcription-coupled DNA repair: Two decades of progress and surprises. Nat. Rev. Mol. Cell Biol 2008, 9, 958–970. [Google Scholar]
- Nag, R.; Smerdon, M.J. Altering the chromatin landscape for nucleotide excision repair. Mutat. Res 2009, 682, 13–20. [Google Scholar]
- Reed, S.H. Nucleotide excision repair in chromatin: Damage removal at the drop of a HAT. DNA Repair (Amst.) 2011, 10, 734–742. [Google Scholar]
- Zhang, L.; Jones, K.; Gong, F. The molecular basis of chromatin dynamics during nucleotide excision repair. Biochem. Cell Biol 2009, 87, 265–272. [Google Scholar]
- Cao, J.; Yan, Q. Histone ubiquitination and deubiquitination in transcription, DNA damage response, and cancer. Front. Oncol 2012, 2, 26. [Google Scholar]
- Chatterjee, S.; Senapati, P.; Kundu, T.K. Post-translational modifications of lysine in DNA-damage repair. Essays Biochem 2012, 52, 93–111. [Google Scholar]
- Dinant, C.; Houtsmuller, A.B.; Vermeulen, W. Chromatin structure and DNA damage repair. Epigenet. Chromatin 2008, 1, 9. [Google Scholar]
- Escargueil, A.E.; Soares, D.G.; Salvador, M.; Larsen, A.K.; Henriques, J.A. What histone code for DNA repair? Mutat. Res 2008, 658, 259–270. [Google Scholar]
- Greenberg, R.A. Histone tails: Directing the chromatin response to DNA damage. FEBS Lett 2011, 585, 2883–2890. [Google Scholar]
- Mendez-Acuna, L.; Di Tomaso, M.V.; Palitti, F.; Martinez-Lopez, W. Histone post-translational modifications in DNA damage response. Cytogenet. Genome Res 2010, 128, 28–36. [Google Scholar]
- Thomson, T.M.; Guerra-Rebollo, M. Ubiquitin and SUMO signalling in DNA repair. Biochem. Soc. Trans 2010, 38, 116–131. [Google Scholar]
- Zhu, Q.; Wani, A.A. Histone modifications: Crucial elements for damage response and chromatin restoration. J. Cell. Physiol 2010, 223, 283–288. [Google Scholar]
- Smerdon, M.J.; Lan, S.Y.; Calza, R.E.; Reeves, R. Sodium butyrate stimulates DNA repair in UV-irradiated normal and xeroderma pigmentosum human fibroblasts. J. Biol. Chem 1982, 257, 13441–13447. [Google Scholar]
- Ramanathan, B.; Smerdon, M.J. Changes in nuclear protein acetylation in u.v.-damaged human cells. Carcinogenesis 1986, 7, 1087–1094. [Google Scholar]
- Ramanathan, B.; Smerdon, M.J. Enhanced DNA repair synthesis in hyperacetylated nucleosomes. J. Biol. Chem 1989, 264, 11026–11034. [Google Scholar]
- Lee, K.K.; Workman, J.L. Histone acetyltransferase complexes: One size doesn’t fit all. Nat. Rev. Mol. Cell Biol 2007, 8, 284–295. [Google Scholar]
- Hodawadekar, S.C.; Marmorstein, R. Chemistry of acetyl transfer by histone modifying enzymes: Structure, mechanism and implications for effector design. Oncogene 2007, 26, 5528–5540. [Google Scholar]
- Berton, T.R.; Mitchell, D.L.; Guo, R.; Johnson, D.G. Regulation of epidermal apoptosis and DNA repair by E2F1 in response to ultraviolet B radiation. Oncogene 2005, 24, 2449–2460. [Google Scholar]
- Guo, R.; Chen, J.; Mitchell, D.L.; Johnson, D.G. GCN5 and E2F1 stimulate nucleotide excision repair by promoting H3K9 acetylation at sites of damage. Nucleic Acids Res 2011, 39, 1390–1397. [Google Scholar]
- Guo, R.; Chen, J.; Zhu, F.; Biswas, A.K.; Berton, T.R.; Mitchell, D.L.; Johnson, D.G. E2F1 localizes to sites of UV-induced DNA damage to enhance nucleotide excision repair. J. Biol. Chem 2010, 285, 19308–19315. [Google Scholar]
- Brand, M.; Moggs, J.G.; Oulad-Abdelghani, M.; Lejeune, F.; Dilworth, F.J.; Stevenin, J.; Almouzni, G.; Tora, L. UV-damaged DNA-binding protein in the TFTC complex links DNA damage recognition to nucleosome acetylation. EMBO J 2001, 20, 3187–3196. [Google Scholar]
- Martinez, E.; Palhan, V.B.; Tjernberg, A.; Lymar, E.S.; Gamper, A.M.; Kundu, T.K.; Chait, B.T.; Roeder, R.G. Human STAGA complex is a chromatin-acetylating transcription coactivator that interacts with pre-mRNA splicing and DNA damage-binding factors in vivo. Mol. Cell. Biol 2001, 21, 6782–6795. [Google Scholar]
- Teng, Y.; Liu, H.; Gill, H.W.; Yu, Y.; Waters, R.; Reed, S.H. Saccharomyces cerevisiae Rad16 mediates ultraviolet-dependent histone H3 acetylation required for efficient global genome nucleotide-excision repair. EMBO Rep 2008, 9, 97–102. [Google Scholar]
- Yu, S.; Teng, Y.; Waters, R.; Reed, S.H. How chromatin is remodelled during DNA repair of UV-induced DNA damage in Saccharomyces cerevisiae. PLoS Genet 2011, 7, e1002124. [Google Scholar]
- Yu, Y.; Teng, Y.; Liu, H.; Reed, S.H.; Waters, R. UV irradiation stimulates histone acetylation and chromatin remodeling at a repressed yeast locus. Proc. Natl. Acad. Sci. USA 2005, 102, 8650–8655. [Google Scholar]
- Battu, A.; Ray, A.; Wani, A.A. ASF1A and ATM regulate H3K56-mediated cell-cycle checkpoint recovery in response to UV irradiation. Nucleic Acids Res 2011, 39, 7931–7945. [Google Scholar]
- Cazzalini, O.; Perucca, P.; Savio, M.; Necchi, D.; Bianchi, L.; Stivala, L.A.; Ducommun, B.; Scovassi, A.I.; Prosperi, E. Interaction of p21(CDKN1A) with PCNA regulates the histone acetyltransferase activity of p300 in nucleotide excision repair. Nucleic Acids Res 2008, 36, 1713–1722. [Google Scholar]
- Hasan, S.; Hassa, P.O.; Imhof, R.; Hottiger, M.O. Transcription coactivator p300 binds PCNA and may have a role in DNA repair synthesis. Nature 2001, 410, 387–391. [Google Scholar]
- Kuo, W.H.; Wang, Y.; Wong, R.P.; Campos, E.I.; Li, G. The ING1b tumor suppressor facilitates nucleotide excision repair by promoting chromatin accessibility to XPA. Exp. Cell Res 2007, 313, 1628–1638. [Google Scholar]
- Pedeux, R.; Sengupta, S.; Shen, J.C.; Demidov, O.N.; Saito, S.; Onogi, H.; Kumamoto, K.; Wincovitch, S.; Garfield, S.H.; McMenamin, M.; et al. ING2 regulates the onset of replicative senescence by induction of p300-dependent p53 acetylation. Mol. Cell. Biol 2005, 25, 6639–6648. [Google Scholar]
- Rubbi, C.P.; Milner, J. p53 is a chromatin accessibility factor for nucleotide excision repair of DNA damage. EMBO J 2003, 22, 975–986. [Google Scholar]
- Scott, M.; Bonnefin, P.; Vieyra, D.; Boisvert, F.M.; Young, D.; Bazett-Jones, D.P.; Riabowol, K. UV-induced binding of ING1 to PCNA regulates the induction of apoptosis. J. Cell Sci 2001, 114, 3455–3462. [Google Scholar]
- Vieyra, D.; Loewith, R.; Scott, M.; Bonnefin, P.; Boisvert, F.M.; Cheema, P.; Pastyryeva, S.; Meijer, M.; Johnston, R.N.; Bazett-Jones, D.P.; et al. Human ING1 proteins differentially regulate histone acetylation. J. Biol. Chem 2002, 277, 29832–29839. [Google Scholar]
- Wang, J.; Chin, M.Y.; Li, G. The novel tumor suppressor p33ING2 enhances nucleotide excision repair via inducement of histone H4 acetylation and chromatin relaxation. Cancer Res 2006, 66, 1906–1911. [Google Scholar]
- Driscoll, R.; Hudson, A.; Jackson, S.P. Yeast Rtt109 promotes genome stability by acetylating histone H3 on lysine 56. Science 2007, 315, 649–652. [Google Scholar]
- Han, J.; Zhou, H.; Horazdovsky, B.; Zhang, K.; Xu, R.M.; Zhang, Z. Rtt109 acetylates histone H3 lysine 56 and functions in DNA replication. Science 2007, 315, 653–655. [Google Scholar]
- Schneider, J.; Bajwa, P.; Johnson, F.C.; Bhaumik, S.R.; Shilatifard, A. Rtt109 is required for proper H3K56 acetylation: A chromatin mark associated with the elongating RNA polymerase II. J. Biol. Chem 2006, 281, 37270–37274. [Google Scholar]
- Tsubota, T.; Berndsen, C.E.; Erkmann, J.A.; Smith, C.L.; Yang, L.; Freitas, M.A.; Denu, J.M.; Kaufman, P.D. Histone H3-K56 acetylation is catalyzed by histone chaperone-dependent complexes. Mol. Cell 2007, 25, 703–712. [Google Scholar]
- Cheng, Z.; Ke, Y.; Ding, X.; Wang, F.; Wang, H.; Wang, W.; Ahmed, K.; Liu, Z.; Xu, Y.; Aikhionbare, F.; et al. Functional characterization of TIP60 sumoylation in UV-irradiated DNA damage response. Oncogene 2008, 27, 931–941. [Google Scholar]
- Decker, P.V.; Yu, D.Y.; Iizuka, M.; Qiu, Q.; Smith, M.M. Catalytic-site mutations in the MYST family histone Acetyltransferase Esa1. Genetics 2008, 178, 1209–1220. [Google Scholar]
- Campi, M.; D’Andrea, L.; Emiliani, J.; Casati, P. Participation of chromatin-remodeling proteins in the repair of ultraviolet-B-damaged DNA. Plant Physiol 2012, 158, 981–995. [Google Scholar]
- Bostelman, L.J.; Keller, A.M.; Albrecht, A.M.; Arat, A.; Thompson, J.S. Methylation of histone H3 lysine-79 by Dot1p plays multiple roles in the response to UV damage in Saccharomyces cerevisiae. DNA Repair (Amst.) 2007, 6, 383–395. [Google Scholar]
- Chaudhuri, S.; Wyrick, J.J.; Smerdon, M.J. Histone H3 Lys79 methylation is required for efficient nucleotide excision repair in a silenced locus of Saccharomyces cerevisiae. Nucleic Acids Res 2009, 37, 1690–1700. [Google Scholar]
- Tatum, D.; Li, S. Evidence that the histone methyltransferase Dot1 mediates global genomic repair by methylating histone H3 on lysine 79. J. Biol. Chem 2011, 286, 17530–17535. [Google Scholar]
- Palomera-Sanchez, Z.; Bucio-Mendez, A.; Valadez-Graham, V.; Reynaud, E.; Zurita, M. Drosophila p53 is required to increase the levels of the dKDM4B demethylase after UV-induced DNA damage to demethylate histone H3 lysine 9. J. Biol. Chem 2010, 285, 31370–31379. [Google Scholar]
- Sanders, S.L.; Portoso, M.; Mata, J.; Bahler, J.; Allshire, R.C.; Kouzarides, T. Methylation of histone H4 lysine 20 controls recruitment of Crb2 to sites of DNA damage. Cell 2004, 119, 603–614. [Google Scholar]
- Schotta, G.; Sengupta, R.; Kubicek, S.; Malin, S.; Kauer, M.; Callen, E.; Celeste, A.; Pagani, M.; Opravil, S.; De La Rosa-Velazquez, I.A.; et al. A chromatin-wide transition to H4K20 monomethylation impairs genome integrity and programmed DNA rearrangements in the mouse. Genes Dev 2008, 22, 2048–2061. [Google Scholar]
- Hanasoge, S.; Ljungman, M. H2AX phosphorylation after UV irradiation is triggered by DNA repair intermediates and is mediated by the ATR kinase. Carcinogenesis 2007, 28, 2298–2304. [Google Scholar]
- Marti, T.M.; Hefner, E.; Feeney, L.; Natale, V.; Cleaver, J.E. H2AX phosphorylation within the G1 phase after UV irradiation depends on nucleotide excision repair and not DNA double-strand breaks. Proc. Natl. Acad. Sci. USA 2006, 103, 9891–9896. [Google Scholar]
- Matsumoto, M.; Yaginuma, K.; Igarashi, A.; Imura, M.; Hasegawa, M.; Iwabuchi, K.; Date, T.; Mori, T.; Ishizaki, K.; Yamashita, K.; et al. Perturbed gap-filling synthesis in nucleotide excision repair causes histone H2AX phosphorylation in human quiescent cells. J. Cell Sci 2007, 120, 1104–1112. [Google Scholar]
- Stiff, T.; Walker, S.A.; Cerosaletti, K.; Goodarzi, A.A.; Petermann, E.; Concannon, P.; O’Driscoll, M.; Jeggo, P.A. ATR-dependent phosphorylation and activation of ATM in response to UV treatment or replication fork stalling. EMBO J 2006, 25, 5775–5782. [Google Scholar]
- Sen, S.P.; De Benedetti, A. TLK1B promotes repair of UV-damaged DNA through chromatin remodeling by Asf1. BMC Mol. Biol 2006, 7, 37. [Google Scholar]
- Shimada, M.; Niida, H.; Zineldeen, D.H.; Tagami, H.; Tanaka, M.; Saito, H.; Nakanishi, M. Chk1 is a histone H3 threonine 11 kinase that regulates DNA damage-induced transcriptional repression. Cell 2008, 132, 221–232. [Google Scholar]
- Moore, J.D.; Yazgan, O.; Ataian, Y.; Krebs, J.E. Diverse roles for histone H2A modifications in DNA damage response pathways in yeast. Genetics 2007, 176, 15–25. [Google Scholar]
- Tatum, D.; Li, W.; Placer, M.; Li, S. Diverse roles of RNA polymerase II-associated factor 1 complex in different subpathways of nucleotide excision repair. J. Biol. Chem 2011, 286, 30304–30313. [Google Scholar]
- Guerrero-Santoro, J.; Kapetanaki, M.G.; Hsieh, C.L.; Gorbachinsky, I.; Levine, A.S.; Rapic-Otrin, V. The cullin 4B-based UV-damaged DNA-binding protein ligase binds to UV-damaged chromatin and ubiquitinates histone H2A. Cancer Res 2008, 68, 5014–5022. [Google Scholar]
- Kapetanaki, M.G.; Guerrero-Santoro, J.; Bisi, D.C.; Hsieh, C.L.; Rapic-Otrin, V.; Levine, A.S. The DDB1-CUL4ADDB2 ubiquitin ligase is deficient in xeroderma pigmentosum group E and targets histone H2A at UV-damaged DNA sites. Proc. Natl. Acad. Sci. USA 2006, 103, 2588–2593. [Google Scholar]
- Takedachi, A.; Saijo, M.; Tanaka, K. DDB2 complex-mediated ubiquitylation around DNA damage is oppositely regulated by XPC and Ku and contributes to the recruitment of XPA. Mol. Cell. Biol 2010, 30, 2708–2723. [Google Scholar]
- Wang, H.; Zhai, L.; Xu, J.; Joo, H.Y.; Jackson, S.; Erdjument-Bromage, H.; Tempst, P.; Xiong, Y.; Zhang, Y. Histone H3 and H4 ubiquitylation by the CUL4-DDB-ROC1 ubiquitin ligase facilitates cellular response to DNA damage. Mol. Cell 2006, 22, 383–394. [Google Scholar]
- Sugasawa, K.; Okuda, Y.; Saijo, M.; Nishi, R.; Matsuda, N.; Chu, G.; Mori, T.; Iwai, S.; Tanaka, K.; Tanaka, K.; et al. UV-induced ubiquitylation of XPC protein mediated by UV-DDB-ubiquitin ligase complex. Cell 2005, 121, 387–400. [Google Scholar]
- Groisman, R.; Polanowska, J.; Kuraoka, I.; Sawada, J.; Saijo, M.; Drapkin, R.; Kisselev, A.F.; Tanaka, K.; Nakatani, Y. The ubiquitin ligase activity in the DDB2 and CSA complexes is differentially regulated by the COP9 signalosome in response to DNA damage. Cell 2003, 113, 357–367. [Google Scholar]
- Bergink, S.; Salomons, F.A.; Hoogstraten, D.; Groothuis, T.A.; de Waard, H.; Wu, J.; Yuan, L.; Citterio, E.; Houtsmuller, A.B.; Neefjes, J.; et al. DNA damage triggers nucleotide excision repair-dependent monoubiquitylation of histone H2A. Genes Dev 2006, 20, 1343–1352. [Google Scholar]
- Marteijn, J.A.; Bekker-Jensen, S.; Mailand, N.; Lans, H.; Schwertman, P.; Gourdin, A.M.; Dantuma, N.P.; Lukas, J.; Vermeulen, W. Nucleotide excision repair-induced H2A ubiquitination is dependent on MDC1 and RNF8 and reveals a universal DNA damage response. J. Cell Biol 2009, 186, 835–847. [Google Scholar]
- Zhu, Q.; Wani, G.; Arab, H.H.; El-Mahdy, M.A.; Ray, A.; Wani, A.A. Chromatin restoration following nucleotide excision repair involves the incorporation of ubiquitinated H2A at damaged genomic sites. DNA Repair (Amst.) 2009, 8, 262–273. [Google Scholar]
- Grant, P.A.; Duggan, L.; Cote, J.; Roberts, S.M.; Brownell, J.E.; Candau, R.; Ohba, R.; Owen-Hughes, T.; Allis, C.D.; Winston, F.; et al. Yeast Gcn5 functions in two multisubunit complexes to acetylate nucleosomal histones: Characterization of an Ada complex and the SAGA (Spt/Ada) complex. Genes Dev 1997, 11, 1640–1650. [Google Scholar]
- Ruiz-Garcia, A.B.; Sendra, R.; Pamblanco, M.; Tordera, V. Gcn5p is involved in the acetylation of histone H3 in nucleosomes. FEBS Lett 1997, 403, 186–190. [Google Scholar]
- Suka, N.; Suka, Y.; Carmen, A.A.; Wu, J.; Grunstein, M. Highly specific antibodies determine histone acetylation site usage in yeast heterochromatin and euchromatin. Mol. Cell 2001, 8, 473–479. [Google Scholar]
- Zhang, W.; Bone, J.R.; Edmondson, D.G.; Turner, B.M.; Roth, S.Y. Essential and redundant functions of histone acetylation revealed by mutation of target lysines and loss of the Gcn5p acetyltransferase. EMBO J 1998, 17, 3155–3167. [Google Scholar]
- Ferreiro, J.A.; Powell, N.G.; Karabetsou, N.; Mellor, J.; Waters, R. Roles for Gcn5p and Ada2p in transcription and nucleotide excision repair at the Saccharomyces cerevisiae MET16 gene. Nucleic Acids Res 2006, 34, 976–985. [Google Scholar]
- Teng, Y.; Yu, Y.; Waters, R. The Saccharomyces cerevisiae histone acetyltransferase Gcn5 has a role in the photoreactivation and nucleotide excision repair of UV-induced cyclobutane pyrimidine dimers in the MFA2 gene. J. Mol. Biol 2002, 316, 489–499. [Google Scholar]
- Waters, R.; Evans, K.; Bennett, M.; Yu, S.; Reed, S.H. Nucleotide excision repair in cellular chromatin: Studies with yeast from nucleotide to gene to genome. Int. J. Mol. Sci 2012, 13, 11141–11164. [Google Scholar]
- Sterner, D.E.; Berger, S.L. Acetylation of histones and transcription-related factors. Microbiol. Mol. Biol. Rev 2000, 64, 435–459. [Google Scholar]
- Ogryzko, V.V.; Schiltz, R.L.; Russanova, V.; Howard, B.H.; Nakatani, Y. The transcriptional coactivators p300 and CBP are histone acetyltransferases. Cell 1996, 87, 953–959. [Google Scholar]
- Zhao, Q.; Barakat, B.M.; Qin, S.; Ray, A.; El-Mahdy, M.A.; Wani, G.; Arafa el, S.; Mir, S.N.; Wang, Q.E.; Wani, A.A. The p38 mitogen-activated protein kinase augments nucleotide excision repair by mediating DDB2 degradation and chromatin relaxation. J. Biol. Chem 2008, 283, 32553–32561. [Google Scholar]
- Yin, D.T.; Wang, Q.; Chen, L.; Liu, M.Y.; Han, C.; Yan, Q.; Shen, R.; He, G.; Duan, W.; Li, J.J.; et al. Germline stem cell gene PIWIL2 mediates DNA repair through relaxation of chromatin. PLoS One 2011, 6, e27154. [Google Scholar]
- Das, C.; Lucia, M.S.; Hansen, K.C.; Tyler, J.K. CBP/p300-mediated acetylation of histone H3 on lysine 56. Nature 2009, 459, 113–117. [Google Scholar]
- Tang, Y.; Holbert, M.A.; Wurtele, H.; Meeth, K.; Rocha, W.; Gharib, M.; Jiang, E.; Thibault, P.; Verreault, A.; Cole, P.A.; et al. Fungal Rtt109 histone acetyltransferase is an unexpected structural homolog of metazoan p300/CBP. Nat. Struct. Mol. Biol 2008, 15, 738–745. [Google Scholar]
- Tillhon, M.; Cazzalini, O.; Nardo, T.; Necchi, D.; Sommatis, S.; Stivala, L.A.; Scovassi, A.I.; Prosperi, E. p300/CBP acetyl transferases interact with and acetylate the nucleotide excision repair factor XPG. DNA Repair (Amst.) 2012. [Google Scholar] [CrossRef]
- Marmorstein, R. Structure and function of histone acetyltransferases. Cell. Mol. Life Sci 2001, 58, 693–703. [Google Scholar]
- Ikura, T.; Ogryzko, V.V.; Grigoriev, M.; Groisman, R.; Wang, J.; Horikoshi, M.; Scully, R.; Qin, J.; Nakatani, Y. Involvement of the TIP60 histone acetylase complex in DNA repair and apoptosis. Cell 2000, 102, 463–473. [Google Scholar]
- Bird, A.W.; Yu, D.Y.; Pray-Grant, M.G.; Qiu, Q.; Harmon, K.E.; Megee, P.C.; Grant, P.A.; Smith, M.M.; Christman, M.F. Acetylation of histone H4 by Esa1 is required for DNA double-strand break repair. Nature 2002, 419, 411–415. [Google Scholar]
- Casati, P.; Campi, M.; Chu, F.; Suzuki, N.; Maltby, D.; Guan, S.; Burlingame, A.L.; Walbot, V. Histone acetylation and chromatin remodeling are required for UV-B-dependent transcriptional activation of regulated genes in maize. Plant Cell 2008, 20, 827–842. [Google Scholar]
- Greer, E.L.; Shi, Y. Histone methylation: A dynamic mark in health, disease and inheritance. Nat. Rev. Genet 2012, 13, 343–357. [Google Scholar]
- Nguyen, A.T.; Zhang, Y. The diverse functions of Dot1 and H3K79 methylation. Genes Dev 2011, 25, 1345–1358. [Google Scholar]
- van Leeuwen, F.; Gafken, P.R.; Gottschling, D.E. Dot1p modulates silencing in yeast by methylation of the nucleosome core. Cell 2002, 109, 745–756. [Google Scholar]
- Shanower, G.A.; Muller, M.; Blanton, J.L.; Honti, V.; Gyurkovics, H.; Schedl, P. Characterization of the grappa gene, the Drosophila histone H3 lysine 79 methyltransferase. Genetics 2005, 169, 173–184. [Google Scholar]
- Jones, B.; Su, H.; Bhat, A.; Lei, H.; Bajko, J.; Hevi, S.; Baltus, G.A.; Kadam, S.; Zhai, H.; Valdez, R.; et al. The histone H3K79 methyltransferase Dot1L is essential for mammalian development and heterochromatin structure. PLoS Genet 2008, 4, e1000190. [Google Scholar]
- Janzen, C.J.; Hake, S.B.; Lowell, J.E.; Cross, G.A. Selective di- or trimethylation of histone H3 lysine 76 by two DOT1 homologs is important for cell cycle regulation in Trypanosoma brucei. Mol. Cell 2006, 23, 497–507. [Google Scholar]
- Feng, Q.; Wang, H.; Ng, H.H.; Erdjument-Bromage, H.; Tempst, P.; Struhl, K.; Zhang, Y. Methylation of H3-lysine 79 is mediated by a new family of HMTases without a SET domain. Curr. Biol 2002, 12, 1052–1058. [Google Scholar]
- Lacoste, N.; Utley, R.T.; Hunter, J.M.; Poirier, G.G.; Cote, J. Disruptor of telomeric silencing-1 is a chromatin-specific histone H3 methyltransferase. J. Biol. Chem 2002, 277, 30421–30424. [Google Scholar]
- Shahbazian, M.D.; Zhang, K.; Grunstein, M. Histone H2B ubiquitylation controls processive methylation but not monomethylation by Dot1 and Set1. Mol. Cell 2005, 19, 271–277. [Google Scholar]
- Altaf, M.; Utley, R.T.; Lacoste, N.; Tan, S.; Briggs, S.D.; Cote, J. Interplay of chromatin modifiers on a short basic patch of histone H4 tail defines the boundary of telomeric heterochromatin. Mol. Cell 2007, 28, 1002–1014. [Google Scholar]
- Fingerman, I.M.; Li, H.C.; Briggs, S.D. A charge-based interaction between histone H4 and Dot1 is required for H3K79 methylation and telomere silencing: Identification of a new trans-histone pathway. Genes Dev 2007, 21, 2018–2029. [Google Scholar]
- Oh, S.; Jeong, K.; Kim, H.; Kwon, C.S.; Lee, D. A lysine-rich region in Dot1p is crucial for direct interaction with H2B ubiquitylation and high level methylation of H3K79. Biochem. Biophys. Res. Commun 2010, 399, 512–517. [Google Scholar]
- Robzyk, K.; Recht, J.; Osley, M.A. Rad6-dependent ubiquitination of histone H2B in yeast. Science 2000, 287, 501–504. [Google Scholar]
- Wood, A.; Krogan, N.J.; Dover, J.; Schneider, J.; Heidt, J.; Boateng, M.A.; Dean, K.; Golshani, A.; Zhang, Y.; Greenblatt, J.F.; et al. Bre1, an E3 ubiquitin ligase required for recruitment and substrate selection of Rad6 at a promoter. Mol. Cell 2003, 11, 267–274. [Google Scholar]
- Krogan, N.J.; Dover, J.; Wood, A.; Schneider, J.; Heidt, J.; Boateng, M.A.; Dean, K.; Ryan, O.W.; Golshani, A.; Johnston, M.; et al. The Paf1 complex is required for histone H3 methylation by COMPASS and Dot1p: Linking transcriptional elongation to histone methylation. Mol. Cell 2003, 11, 721–729. [Google Scholar]
- Ng, H.H.; Dole, S.; Struhl, K. The Rtf1 component of the Paf1 transcriptional elongation complex is required for ubiquitination of histone H2B. J. Biol. Chem 2003, 278, 33625–33628. [Google Scholar]
- Wood, A.; Schneider, J.; Dover, J.; Johnston, M.; Shilatifard, A. The Paf1 complex is essential for histone monoubiquitination by the Rad6-Bre1 complex, which signals for histone methylation by COMPASS and Dot1p. J. Biol. Chem 2003, 278, 34739–34742. [Google Scholar]
- Piro, A.S.; Mayekar, M.K.; Warner, M.H.; Davis, C.P.; Arndt, K.M. Small region of Rtf1 protein can substitute for complete Paf1 complex in facilitating global histone H2B ubiquitylation in yeast. Proc. Natl. Acad. Sci. USA 2012, 109, 10837–10842. [Google Scholar]
- Sawada, K.; Yang, Z.; Horton, J.R.; Collins, R.E.; Zhang, X.; Cheng, X. Structure of the conserved core of the yeast Dot1p, a nucleosomal histone H3 lysine 79 methyltransferase. J. Biol. Chem 2004, 279, 43296–43306. [Google Scholar]
- Friedberg, E.C.; Walker, G.C.; Siede, W.; Wood, R.D.; Schultz, R.A.; Ellenberger, T. DNA Repair and Mutagenesis, 2nd ed; ASM Press: Washington, DC, USA, 2006. [Google Scholar]
- Struhl, K. Transcriptional noise and the fidelity of initiation by RNA polymerase II. Nat. Struct. Mol. Biol 2007, 14, 103–105. [Google Scholar]
- Li, S.; Ding, B.; Lejeune, D.; Ruggiero, C.; Chen, X.; Smerdon, M.J. The roles of Rad16 and Rad26 in repairing repressed and actively transcribed genes in yeast. DNA Repair (Amst.) 2007, 6, 1596–1606. [Google Scholar]
- Ng, H.H.; Ciccone, D.N.; Morshead, K.B.; Oettinger, M.A.; Struhl, K. Lysine-79 of histone H3 is hypomethylated at silenced loci in yeast and mammalian cells: A potential mechanism for position-effect variegation. Proc. Natl. Acad. Sci. USA 2003, 100, 1820–1825. [Google Scholar]
- Hughes, T.R.; Marton, M.J.; Jones, A.R.; Roberts, C.J.; Stoughton, R.; Armour, C.D.; Bennett, H.A.; Coffey, E.; Dai, H.; He, Y.D.; et al. Functional discovery via a compendium of expression profiles. Cell 2000, 102, 109–126. [Google Scholar]
- Giannattasio, M.; Lazzaro, F.; Plevani, P.; Muzi-Falconi, M. The DNA damage checkpoint response requires histone H2B ubiquitination by Rad6-Bre1 and H3 methylation by Dot1. J. Biol. Chem 2005, 280, 9879–9886. [Google Scholar]
- Siede, W.; Allen, J.B.; Elledge, S.J.; Friedberg, E.C. The Saccharomyces cerevisiae MEC1 gene, which encodes a homolog of the human ATM gene product, is required for G1 arrest following radiation treatment. J. Bacteriol 1996, 178, 5841–5843. [Google Scholar]
- Lu, X.; Simon, M.D.; Chodaparambil, J.V.; Hansen, J.C.; Shokat, K.M.; Luger, K. The effect of H3K79 dimethylation and H4K20 trimethylation on nucleosome and chromatin structure. Nat. Struct. Mol. Biol 2008, 15, 1122–1124. [Google Scholar]
- Fink, M.; Thompson, J.S.; Thoma, F. Contributions of histone H3 nucleosome core surface mutations to chromatin structures, silencing and DNA repair. PLoS One 2011, 6, e26210. [Google Scholar]
- Fernandez-Capetillo, O.; Lee, A.; Nussenzweig, M.; Nussenzweig, A. H2AX: The histone guardian of the genome. DNA Repair (Amst.) 2004, 3, 959–967. [Google Scholar]
- Zou, L.; Elledge, S.J. Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science 2003, 300, 1542–1548. [Google Scholar]
- Revet, I.; Feeney, L.; Bruguera, S.; Wilson, W.; Dong, T.K.; Oh, D.H.; Dankort, D.; Cleaver, J.E. Functional relevance of the histone gammaH2Ax in the response to DNA damaging agents. Proc. Natl. Acad. Sci. USA 2011, 108, 8663–8667. [Google Scholar]
- Fink, M.; Imholz, D.; Thoma, F. Contribution of the serine 129 of histone H2A to chromatin structure. Mol. Cell. Biol 2007, 27, 3589–3600. [Google Scholar]
- Oh, K.S.; Bustin, M.; Mazur, S.J.; Appella, E.; Kraemer, K.H. UV-induced histone H2AX phosphorylation and DNA damage related proteins accumulate and persist in nucleotide excision repair-deficient XP-B cells. DNA Repair (Amst.) 2011, 10, 5–15. [Google Scholar]
- Vrouwe, M.G.; Pines, A.; Overmeer, R.M.; Hanada, K.; Mullenders, L.H. UV-induced photolesions elicit ATR-kinase-dependent signaling in non-cycling cells through nucleotide excision repair-dependent and -independent pathways. J. Cell Sci 2011, 124, 435–446. [Google Scholar]
- Fernandez-Capetillo, O.; Chen, H.T.; Celeste, A.; Ward, I.; Romanienko, P.J.; Morales, J.C.; Naka, K.; Xia, Z.; Camerini-Otero, R.D.; Motoyama, N.; et al. DNA damage-induced G2-M checkpoint activation by histone H2AX and 53BP1. Nat. Cell Biol 2002, 4, 993–997. [Google Scholar]
- Clements, A.; Poux, A.N.; Lo, W.S.; Pillus, L.; Berger, S.L.; Marmorstein, R. Structural basis for histone and phosphohistone binding by the GCN5 histone acetyltransferase. Mol. Cell 2003, 12, 461–473. [Google Scholar]
- Ivaldi, M.S.; Karam, C.S.; Corces, V.G. Phosphorylation of histone H3 at Ser10 facilitates RNA polymerase II release from promoter-proximal pausing in Drosophila. Genes Dev 2007, 21, 2818–2831. [Google Scholar]
- Welchman, R.L.; Gordon, C.; Mayer, R.J. Ubiquitin and ubiquitin-like proteins as multifunctional signals. Nat. Rev. Mol. Cell Biol 2005, 6, 599–609. [Google Scholar]
- Sun, Z.W.; Allis, C.D. Ubiquitination of histone H2B regulates H3 methylation and gene silencing in yeast. Nature 2002, 418, 104–108. [Google Scholar]
- Chandrasekharan, M.B.; Huang, F.; Sun, Z.W. Ubiquitination of histone H2B regulates chromatin dynamics by enhancing nucleosome stability. Proc. Natl. Acad. Sci. USA 2009, 106, 16686–16691. [Google Scholar]
- Chandrasekharan, M.B.; Huang, F.; Sun, Z.W. Histone H2B ubiquitination and beyond: Regulation of nucleosome stability, chromatin dynamics and the trans-histone H3 methylation. Epigenetics 2010, 5, 460–468. [Google Scholar]
- Hannah, J.; Zhou, P. Regulation of DNA damage response pathways by the cullin-RING ubiquitin ligases. DNA Repair (Amst.) 2009, 8, 536–543. [Google Scholar]
- Sugasawa, K. UV-DDB: A molecular machine linking DNA repair with ubiquitination. DNA Repair (Amst.) 2009, 8, 969–972. [Google Scholar]
- Lan, L.; Nakajima, S.; Kapetanaki, M.G.; Hsieh, C.L.; Fagerburg, M.; Thickman, K.; Rodriguez-Collazo, P.; Leuba, S.H.; Levine, A.S.; Rapic-Otrin, V. Monoubiquitinated histone H2A destabilizes photolesion-containing nucleosomes with concomitant release of UV-damaged DNA-binding protein E3 ligase. J. Biol. Chem 2012, 287, 12036–12049. [Google Scholar]
- Bertolaet, B.L.; Clarke, D.J.; Wolff, M.; Watson, M.H.; Henze, M.; Divita, G.; Reed, S.I. UBA domains of DNA damage-inducible proteins interact with ubiquitin. Nat. Struct. Boil 2001, 8, 417–422. [Google Scholar]
- Ryu, K.S.; Lee, K.J.; Bae, S.H.; Kim, B.K.; Kim, K.A.; Choi, B.S. Binding surface mapping of intra- and interdomain interactions among hHR23B, ubiquitin, and polyubiquitin binding site 2 of S5a. J. Biol. Chem 2003, 278, 36621–36627. [Google Scholar]
- Parada, L.A.; McQueen, P.G.; Misteli, T. Tissue-specific spatial organization of genomes. Genome Biol 2004, 5, R44. [Google Scholar]
- Ernst, J.; Kheradpour, P.; Mikkelsen, T.S.; Shoresh, N.; Ward, L.D.; Epstein, C.B.; Zhang, X.; Wang, L.; Issner, R.; Coyne, M.; et al. Mapping and analysis of chromatin state dynamics in nine human cell types. Nature 2011, 473, 43–49. [Google Scholar]
- Teng, Y.; Bennett, M.; Evans, K.E.; Zhuang-Jackson, H.; Higgs, A.; Reed, S.H.; Waters, R. A novel method for the genome-wide high resolution analysis of DNA damage. Nucleic Acids Res 2011, 39, e10. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Modification enzymes | Modulators | Histone modifications | Organisms | Stage(s) of NER implicated | Ref. |
|---|---|---|---|---|---|
| Histone acetylation | |||||
| GCN5 | UV-DDB, E2F1 | H3, H4 | H. sapiens, M. musculus | After damage recognition by UVDDB | [26–30] |
| Gcn5 | Rad7, Rad16 | H2B, H3, H4 | S. cerevisiae | After damage recognition by Rad7/Rad16 | [31–33] |
| p300/CBP | p53, p21, ING1b, ING2, ASF1 | H2A, H2B, H3, H4 | H. sapiens, M. musculus | Chromatin relaxation prior to and after lesion detection; repair synthesis; chromatin restoration after NER | [34–42] |
| Rtt109 | Asf1, Vps75 | H3 (K9, K56) | S. cerevisiae | ? | [43–46] |
| TIP60 | H2A, H4 | H. sapiens | Chromatin relaxation prior to and after lesion detection? | [47] | |
| Esa1 | H2A, H4 | S. cerevisiae | ? | [48] | |
| HAM1 | A. thaliana | A stage before incision | [49] | ||
| HAM2 | A. thaliana | A stage before incision | [49] | ||
| Histone methylation | |||||
| Dot1 | Bre1, Rad6, Rtf1 | H3 (K79) | S. cerevisiae | A stage before incision | [50–52] |
| dKDM4B | Dmp53 | H3 (K9) demethylation | D. melanogaster | A stage before incision | [53] |
| Set9 | H4 (K20) | S. pombe | ? | [54] | |
| Suv4-20h | H4 (K20) | M. musculus | ? | [55] | |
| Histone phosphorylation | |||||
| ATR | H2AX (S139) | H. sapiens, M. musculus | After incision; chromatin restoration after NER | [56–59] | |
| Multiple kinases (e.g., TPL-2 and DAP kinase 3) | H3 (S10, T11) | H. sapiens, M. musculus | ? | [60,61] | |
| ? | H2A (S122, T126) | S. cerevisiae | ? | [62] | |
| Histone ubiquitination | |||||
| Bre1 | Rad6, Rtf1 | H3 (K123) | S. cerevisiae | A stage before incision | [52,63] |
| UV-DDBcullin- RING ubiquitin ligase | H2A (K118, 119), H2B, H3, H4 | H. sapiens, M. musculus | Chromatin destabilization and possible recruitment of lesion recognition factors after initial lesion detection by UV-DDB | [64–69] | |
| RNF8 | UBC13, CAF-1 | H2A (K119) | H. sapiens | During or after repair synthesis; chromatin restoration after NER | [70–72] |
© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Li, S. Implication of Posttranslational Histone Modifications in Nucleotide Excision Repair. Int. J. Mol. Sci. 2012, 13, 12461-12486. https://doi.org/10.3390/ijms131012461
Li S. Implication of Posttranslational Histone Modifications in Nucleotide Excision Repair. International Journal of Molecular Sciences. 2012; 13(10):12461-12486. https://doi.org/10.3390/ijms131012461
Chicago/Turabian StyleLi, Shisheng. 2012. "Implication of Posttranslational Histone Modifications in Nucleotide Excision Repair" International Journal of Molecular Sciences 13, no. 10: 12461-12486. https://doi.org/10.3390/ijms131012461
APA StyleLi, S. (2012). Implication of Posttranslational Histone Modifications in Nucleotide Excision Repair. International Journal of Molecular Sciences, 13(10), 12461-12486. https://doi.org/10.3390/ijms131012461