E2F1 and p53 Transcription Factors as Accessory Factors for Nucleotide Excision Repair

{kind=link}
{kind=link}
Abstract
:1. Introduction
2. The Nucleotide Excision Repair Pathway
2.1. The Nucleotide Excision Repair Model
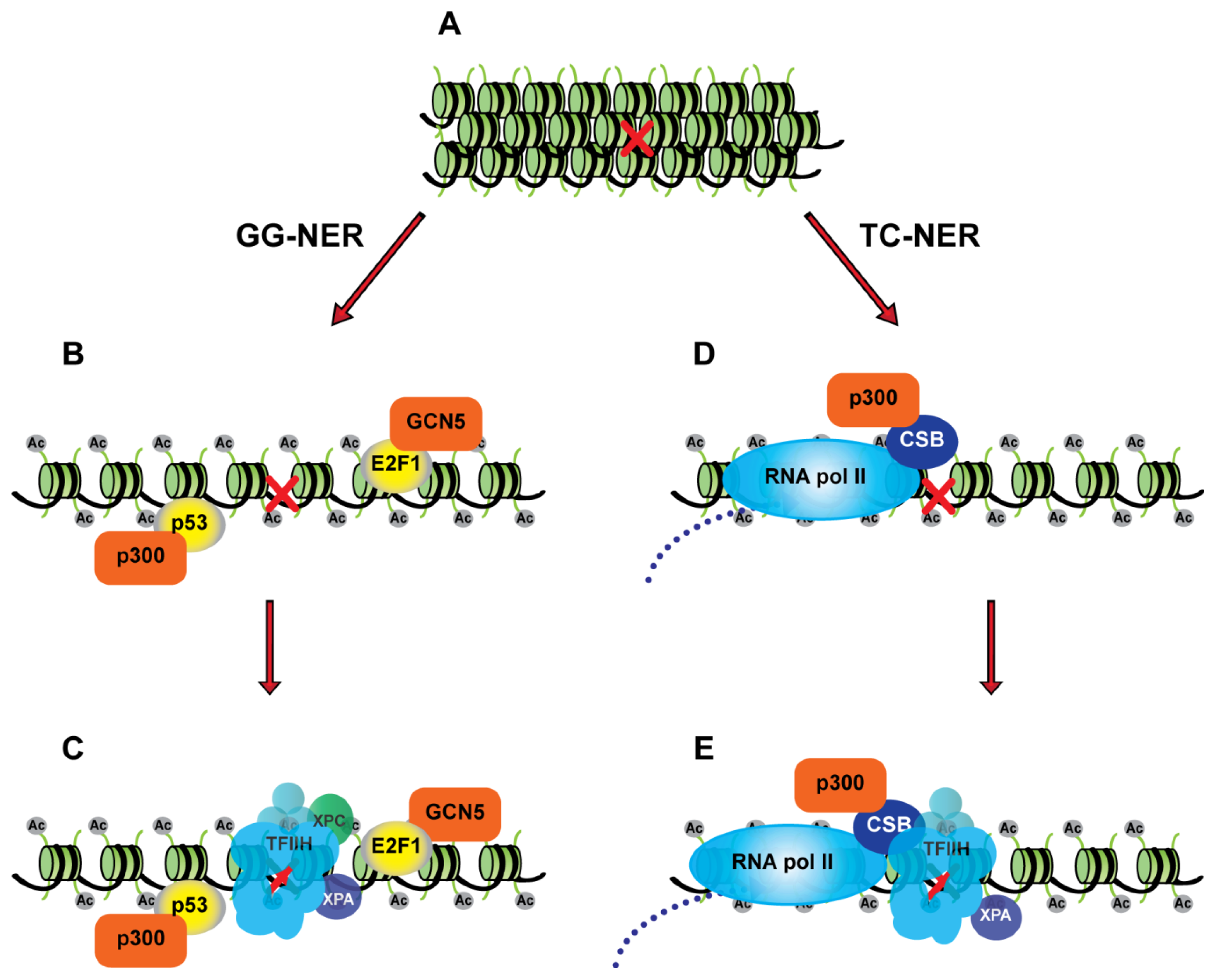
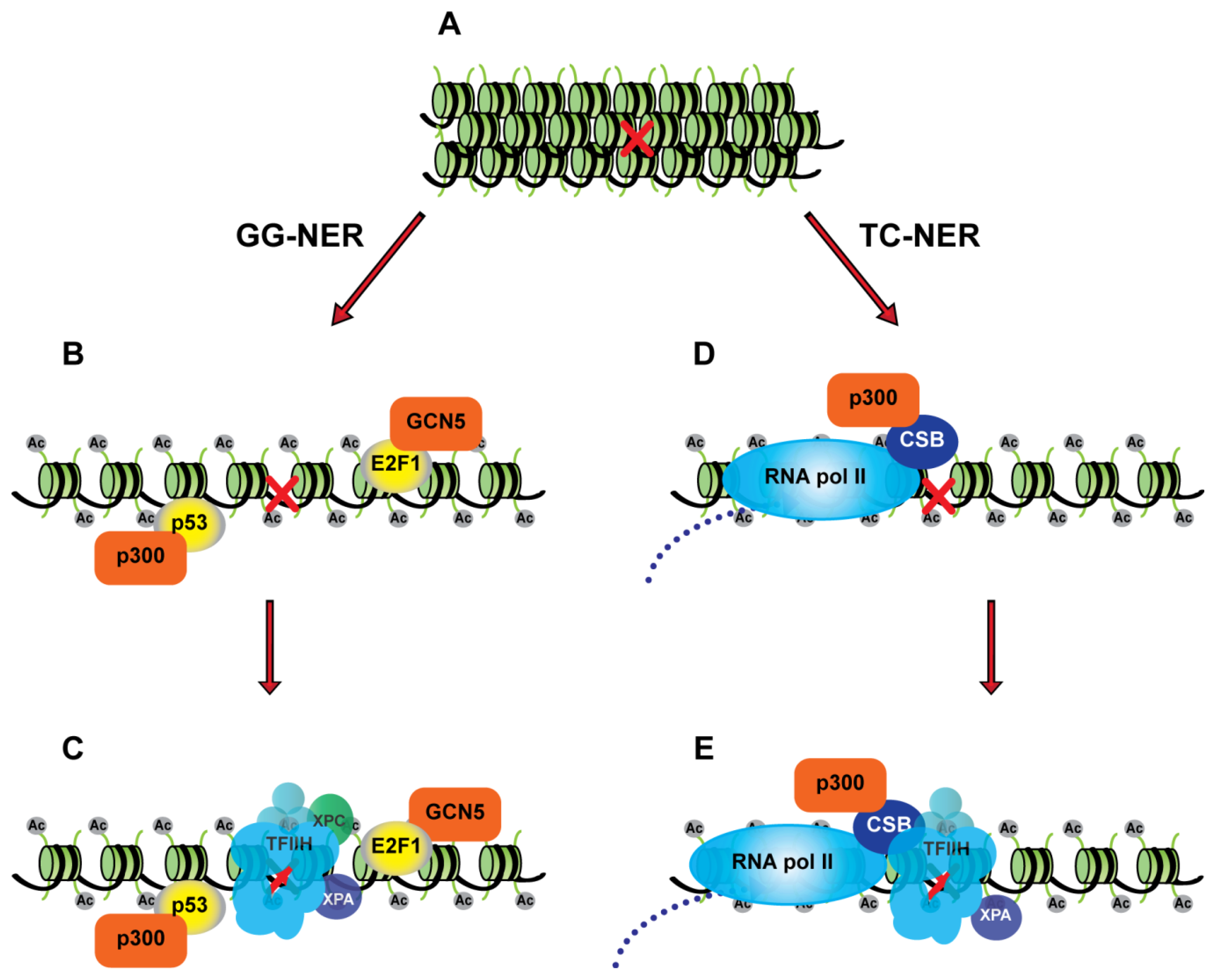
2.2. Nucleotide Excision Repair in Chromatin
3. Transcription Factors and the Nucleotide Excision Repair Pathway
3.1. p53 as a Chromatin Accessibility Factor for NER
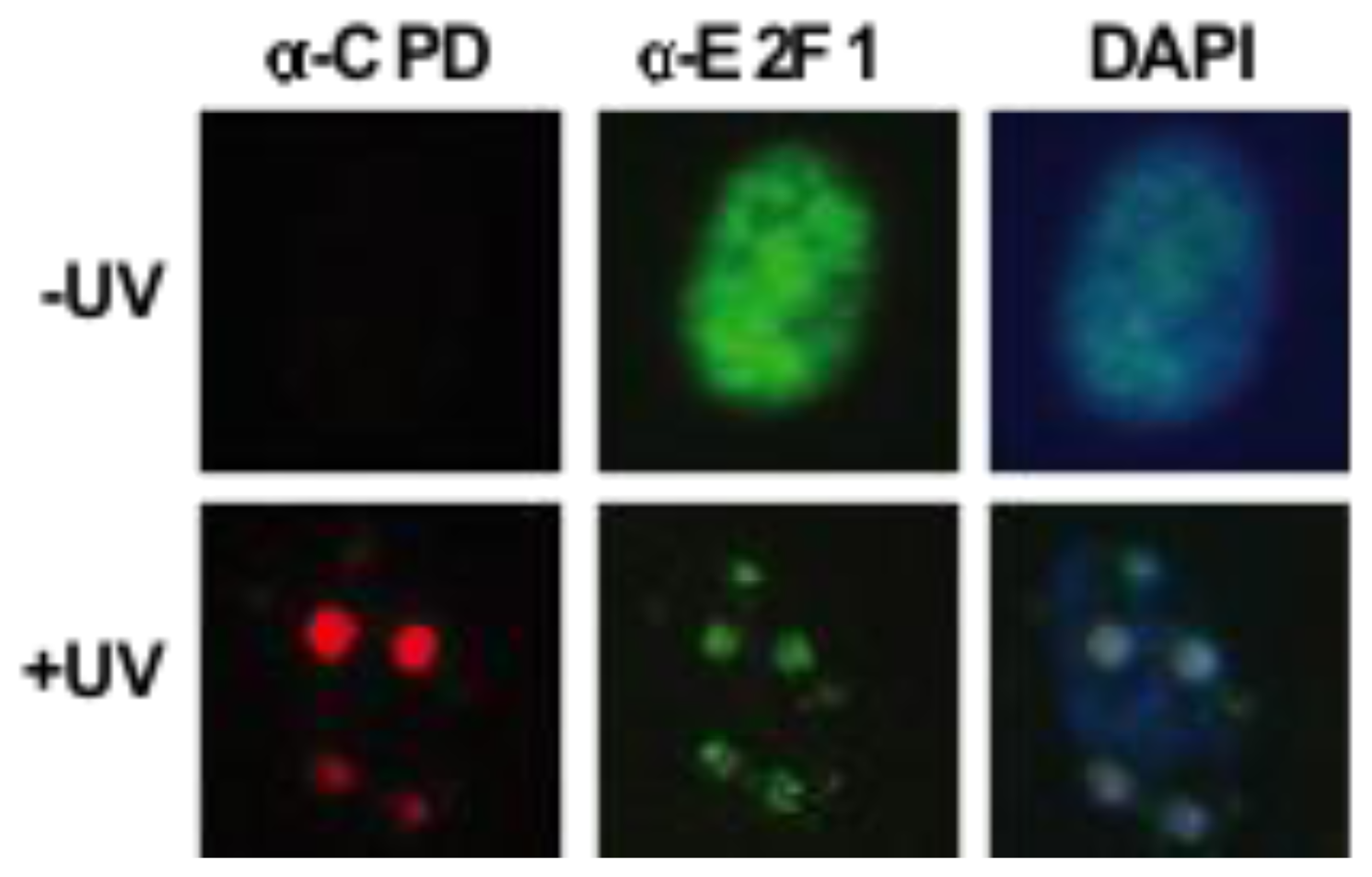
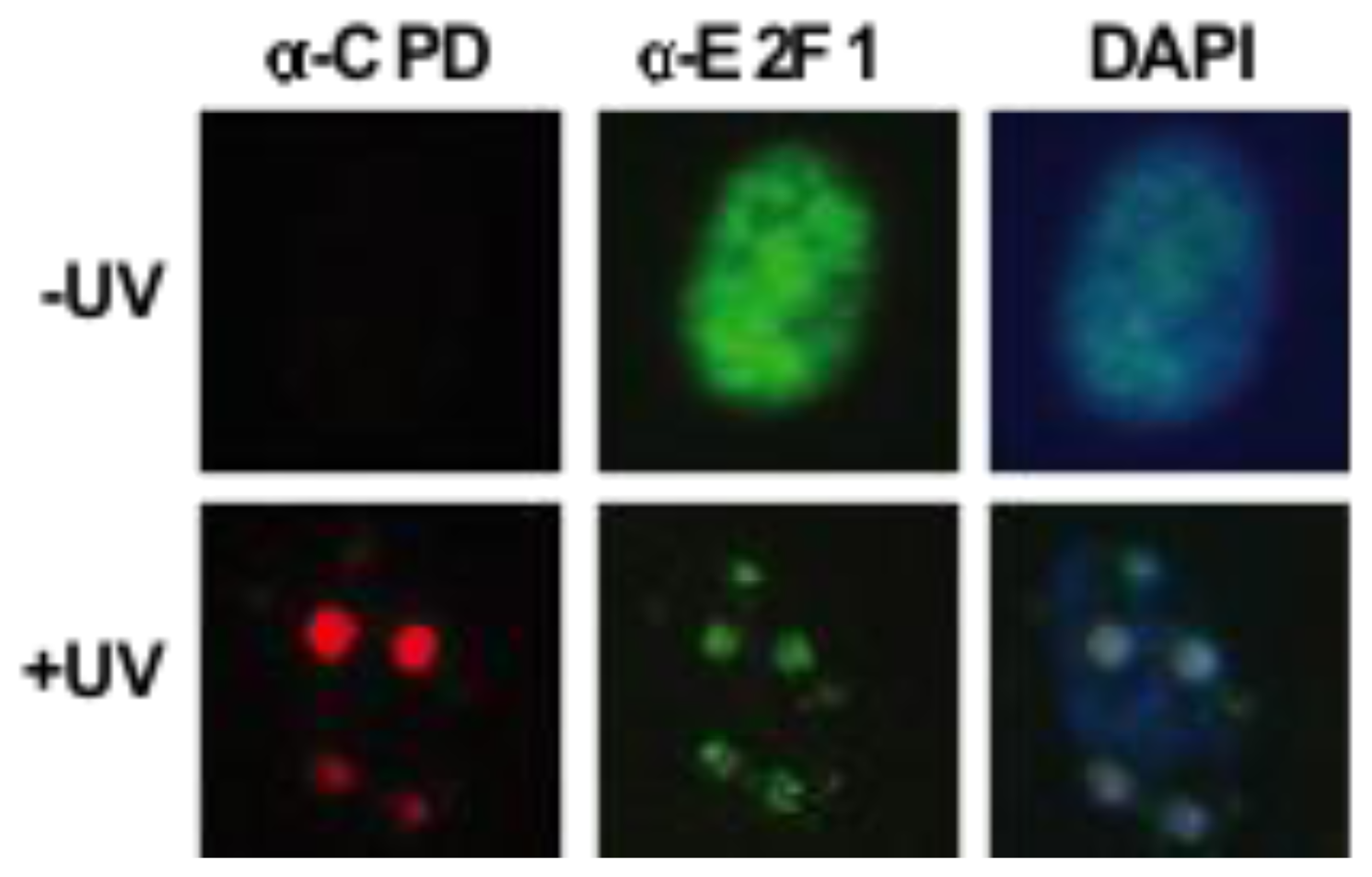
3.2. Transcription-Independent Functions of E2F1 in DNA Repair
4. Conclusion
4.1. Unanswered Questions and Future Directions
Acknowledgments
References
- Kornberg, R.D. Structure of chromatin. Annu. Rev. Biochem 1977, 46, 931–954. [Google Scholar]
- Kornberg, R.D.; Lorch, Y. Chromatin structure and transcription. Annu. Rev. Cell Biol 1992, 8, 563–587. [Google Scholar]
- Luger, K.; Dechassa, M.L.; Tremethick, D.J. New insights into nucleosome and chromatin structure: An ordered state or a disordered affair? Nat. Rev. Mol. Cell Biol 2012, 13, 436–447. [Google Scholar]
- Chi, P.; Allis, C.D.; Wang, G.G. Covalent histone modifications-miswritten, misinterpreted and mis-erased in human cancers. Nat. Rev. Cancer 2010, 10, 457–469. [Google Scholar]
- Baylin, S.B.; Jones, P.A. A decade of exploring the cancer epigenome—Biological and translational implications. Nat. Rev. Cancer 2011, 11, 726–734. [Google Scholar]
- Füllgrabe, J.; Kavanagh, E.; Joseph, B. Histone onco-modifications. Oncogene 2011, 30, 3391–3403. [Google Scholar]
- Varier, R.A.; Timmers, H.T.M. Histone lysine methylation and demethylation pathways in cancer. Biochim. Biophys. Acta 2011, 1815, 75–89. [Google Scholar]
- Bycroft, M. Recognition of non-methyl histone marks. Curr. Opin. Struct. Biol 2011, 21, 761–766. [Google Scholar]
- Yun, M.; Wu, J.; Workman, J.L.; Li, B. Readers of histone modifications. Cell Res 2011, 21, 564–578. [Google Scholar]
- Cleaver, J.E. Cancer in xeroderma pigmentosum and related disorders of DNA repair. Nat. Rev. Cancer 2005, 5, 564–573. [Google Scholar]
- Serrano, M.; Blasco, M.A. Cancer and ageing: Convergent and divergent mechanisms. Nat. Rev. Mol. Cell Biol 2007, 8, 715–722. [Google Scholar]
- Garinis, G.A.; van der Horst, G.T.J.; Vijg, J.; Hoeijmakers, J.H.J. DNA damage and ageing: New-age ideas for an age-old problem. Nat. Cell Biol 2008, 10, 1241–1247. [Google Scholar]
- Feser, J.; Tyler, J. Chromatin structure as a mediator of aging. FEBS Lett 2011, 585, 2041–2048. [Google Scholar]
- Cruickshanks, H.A.; Adams, P.D. Chromatin: A molecular interface between cancer and aging. Curr. Opin. Genet. Dev 2011, 21, 100–106. [Google Scholar]
- Le May, N.; Mota-Fernandes, D.; Vélez-Cruz, R.; Iltis, I.; Biard, D.; Egly, J.M. NER factors are recruited to active promoters and facilitate chromatin modification for transcription in the absence of exogenous genotoxic attack. Mol. Cell 2010, 38, 54–66. [Google Scholar]
- Fong, Y.W.; Inouye, C.; Yamaguchi, T.; Cattoglio, C.; Grubisic, I.; Tjian, R. A DNA Repair Complex Functions as an Oct4/Sox2 Coactivator in Embryonic Stem Cells. Cell 2011, 147, 120–131. [Google Scholar]
- Le May, N.; Fradin, D.; Iltis, I.; Bougnères, P.; Egly, J.-M. XPG and XPF Endonucleases Trigger Chromatin Looping and DNA Demethylation for Accurate Expression of Activated Genes. Mol. Cell 2012, 47, 622–632. [Google Scholar]
- Friedberg, E.C.; Walker, G.C.; Siede, W. DNA Repair and Mutagenesis; ASM Press: Washington DC, USA, 1995. [Google Scholar]
- Hoeijmakers, J.H.J. DNA damage, aging, and cancer. N. Engl. J. Med 2009, 361, 1475–1485. [Google Scholar]
- Aboussekhra, A.; Biggerstaff, M.; Shivji, M.K.; Vilpo, J.A.; Moncollin, V.; Podust, V.N.; Protić, M.; Hübscher, U.; Egly, J.M.; Wood, R.D. Mammalian DNA nucleotide excision repair reconstituted with purified protein components. Cell 1995, 80, 859–868. [Google Scholar]
- Araújo, S.J.; Tirode, F.; Coin, F.; Pospiech, H.; Syväoja, J.E.; Stucki, M.; Hübscher, U.; Egly, J.M.; Wood, R.D. Nucleotide excision repair of DNA with recombinant human proteins: Definition of the minimal set of factors, active forms of TFIIH, and modulation by CAK. Genes Dev 2000, 14, 349–359. [Google Scholar]
- Kuper, J.; Kisker, C. Damage recognition in nucleotide excision DNA repair. Curr. Opin. Struct. Biol 2012, 22, 88–93. [Google Scholar]
- Evans, E.; Moggs, J.G.; Hwang, J.R.; Egly, J.M.; Wood, R.D. Mechanism of open complex and dual incision formation by human nucleotide excision repair factors. EMBO J 1997, 16, 6559–6573. [Google Scholar]
- Riedl, T.; Hanaoka, F.; Egly, J.-M. The comings and goings of nucleotide excision repair factors on damaged DNA. EMBO J 2003, 22, 5293–5303. [Google Scholar]
- Shivji, M.K.; Podust, V.N.; Hübscher, U.; Wood, R.D. Nucleotide excision repair DNA synthesis by DNA polymerase epsilon in the presence of PCNA, RFC, and RPA. Biochemistry 1995, 34, 5011–5017. [Google Scholar]
- Ogi, T.; Limsirichaikul, S.; Overmeer, R.M.; Volker, M.; Takenaka, K.; Cloney, R.; Nakazawa, Y.; Niimi, A.; Miki, Y.; Jaspers, N.G.; et al. Three DNA polymerases, recruited by different mechanisms, carry out NER repair synthesis in human cells. Mol. Cell 2010, 37, 714–727. [Google Scholar]
- Zotter, A.; Luijsterburg, M.S.; Warmerdam, D.O.; Ibrahim, S.; Nigg, A.; van Cappellen, W.A.; Hoeijmakers, J.H.J.; van Driel, R.; Vermeulen, W.; Houtsmuller, A.B. Recruitment of the nucleotide excision repair endonuclease XPG to sites of UV-induced dna damage depends on functional TFIIH. Mol. Cell Biol 2006, 26, 8868–8879. [Google Scholar]
- Mocquet, V.; Lainé, J.P.; Riedl, T.; Yajin, Z.; Lee, M.Y.; Egly, J.M. Sequential recruitment of the repair factors during NER: The role of XPG in initiating the resynthesis step. EMBO J 2008, 27, 155–167. [Google Scholar]
- Staresincic, L.; Fagbemi, A.F.; Enzlin, J.H.; Gourdin, A.M.; Wijgers, N.; Dunand-Sauthier, I.; Giglia-Mari, G.; Clarkson, S.G.; Vermeulen, W.; Schärer, O.D. Coordination of dual incision and repair synthesis in human nucleotide excision repair. EMBO J 2009, 28, 1111–1120. [Google Scholar]
- Vermeulen, W. Dynamics of mammalian NER proteins. DNA Repair 2011, 10, 760–771. [Google Scholar]
- Mellon, I.; Bohr, V.A.; Smith, C.A.; Hanawalt, P.C. Preferential DNA repair of an active gene in human cells. Proc. Natl. Acad. Sci. USA 1986, 83, 8878–8882. [Google Scholar]
- Hanawalt, P.C.; Spivak, G. Transcription-coupled DNA repair: Two decades of progress and surprises. Nat. Rev. Mol. Cell Biol 2008, 9, 958–970. [Google Scholar]
- Fousteri, M.; Vermeulen, W.; van Zeeland, A.A.; Mullenders, L.H.F. Cockayne syndrome A and B proteins differentially regulate recruitment of chromatin remodeling and repair factors to stalled RNA polymerase II in vivo. Mol. Cell 2006, 23, 471–482. [Google Scholar]
- Lainé, J.-P.; Egly, J.-M. Initiation of DNA repair mediated by a stalled RNA polymerase IIO. EMBO J 2006, 25, 387–397. [Google Scholar]
- Tornaletti, S. DNA repair in mammalian cells: Transcription-coupled DNA repair: Directing your effort where it’s most needed. Cell Mol. Life Sci 2009, 66, 1010–1020. [Google Scholar]
- Lehmann, A.R. The xeroderma pigmentosum group D (XPD) gene: One gene, two functions, three diseases. Genes Dev 2001, 15, 15–23. [Google Scholar]
- Cleaver, J.E.; Lam, E.T.; Revet, I. Disorders of nucleotide excision repair: The genetic and molecular basis of heterogeneity. Nat. Rev. Genet 2009, 10, 756–768. [Google Scholar]
- Kraemer, K.H.; Patronas, N.J.; Schiffmann, R.; Brooks, B.P.; Tamura, D.; DiGiovanna, J.J. Xeroderma pigmentosum, trichothiodystrophy and Cockayne syndrome: A complex genotype-phenotype relationship. Neuroscience 2007, 145, 1388–1396. [Google Scholar]
- Cleaver, J.E.; Revet, I. Clinical implications of the basic defects in Cockayne syndrome and xeroderma pigmentosum and the DNA lesions responsible for cancer, neurodegeneration and aging. Mech. Ageing Dev 2008, 129, 492–497. [Google Scholar]
- Van Den Boom, V.; Jaspers, N.G.J.; Vermeulen, W. When machines get stuck-obstructed RNA polymerase II: Displacement, degradation or suicide. Bioessays 2002, 24, 780–784. [Google Scholar]
- Svejstrup, J.Q. Rescue of arrested RNA polymerase II complexes. J. Cell Sci 2003, 116, 447–451. [Google Scholar]
- Bradsher, J.; Auriol, J.; Proietti De Santis, L.; Iben, S.; Vonesch, J.L.; Grummt, I.; Egly, J.M. CSB is a component of RNA pol I transcription. Mol. Cell 2002, 10, 819–829. [Google Scholar]
- Drané, P.; Compe, E.; Catez, P.; Chymkowitch, P.; Egly, J.-M. Selective regulation of vitamin D receptor-responsive genes by TFIIH. Mol. Cell 2004, 16, 187–197. [Google Scholar]
- Compe, E.; Drané, P.; Laurent, C.; Diderich, K.; Braun, C.; Hoeijmakers, J.H.J.; Egly, J.-M. Dysregulation of the peroxisome proliferator-activated receptor target genes by XPD mutations. Mol. Cell Biol 2005, 25, 6065–6076. [Google Scholar]
- Proietti-De-Santis, L.; Drané, P.; Egly, J.-M. Cockayne syndrome B protein regulates the transcriptional program after UV irradiation. EMBO J 2006, 25, 1915–1923. [Google Scholar]
- Compe, E.; Malerba, M.; Soler, L.; Marescaux, J.; Borrelli, E.; Egly, J.-M. Neurological defects in trichothiodystrophy reveal a coactivator function of TFIIH. Nat. Neurosci 2007, 10, 1414–1422. [Google Scholar]
- Etchegaray, J.-P.; Mostoslavsky, R. eNERgizing Pluripotent Gene Transcription. Cell Stem Cell 2011, 9, 285–286. [Google Scholar]
- Chymkowitch, P.; Le May, N.; Charneau, P.; Compe, E.; Egly, J.-M. The phosphorylation of the androgen receptor by TFIIH directs the ubiquitin/proteasome process. EMBO J 2011, 30, 468–479. [Google Scholar]
- Ramanathan, B.; Smerdon, M.J. Changes in nuclear protein acetylation in u.v.-damaged human cells. Carcinogenesis 1986, 7, 1087–1094. [Google Scholar]
- Smerdon, M.J.; Lieberman, M.W. Nucleosome rearrangement in human chromatin during UV-induced DNA-repair synthesis. Proc. Natl. Acad. Sci. USA 1978, 75, 4238–4241. [Google Scholar]
- Smerdon, M.J.; Lan, S.Y.; Calza, R.E.; Reeves, R. Sodium butyrate stimulates DNA repair in UV-irradiated normal and xeroderma pigmentosum human fibroblasts. J. Biol. Chem 1982, 257, 13441–13447. [Google Scholar]
- Beckerman, R.; Prives, C. Transcriptional Regulation by P53. CSH Perspect. Biol 2010, 2, a000935. [Google Scholar]
- Ford, J.M.; Hanawalt, P.C. Li-Fraumeni syndrome fibroblasts homozygous for p53 mutations are deficient in global DNA repair but exhibit normal transcription-coupled repair and enhanced UV resistance. Proc. Natl. Acad. Sci. USA 1995, 92, 8876–8880. [Google Scholar]
- Wang, Q.-E.; Zhu, Q.; Wani, M.A.; Wani, G.; Chen, J.; Wani, A.A. Tumor suppressor p53 dependent recruitment of nucleotide excision repair factors XPC and TFIIH to DNA damage. DNA Repair 2003, 2, 483–499. [Google Scholar]
- Smith, M.L.; Chen, I.T.; Zhan, Q.; O’Connor, P.M.; Fornace, A.J. Involvement of the p53 tumor suppressor in repair of u.v.-type DNA damage. Oncogene 1995, 10, 1053–1059. [Google Scholar]
- Wani, M.A.; Zhu, Q.Z.; El-Mahdy, M.; Wani, A.A. Influence of p53 tumor suppressor protein on bias of DNA repair and apoptotic response in human cells. Carcinogenesis 1999, 20, 765–772. [Google Scholar]
- Rubbi, C.P.; Milner, J. p53 is a chromatin accessibility factor for nucleotide excision repair of DNA damage. EMBO J 2003, 22, 975–986. [Google Scholar]
- Fitch, M.E.; Cross, I.V.; Ford, J.M. p53 responsive nucleotide excision repair gene products p48 and XPC, but not p53, localize to sites of UV-irradiation-induced DNA damage, in vivo. Carcinogenesis 2003, 24, 843–850. [Google Scholar]
- Cheung, K.J.; Mitchell, D.; Lin, P.; Li, G. The tumor suppressor candidate p33(ING1) mediates repair of UV-damaged DNA. Cancer Res 2001, 61, 4974–4977. [Google Scholar]
- Wang, J.; Chin, M.Y.; Li, G. The novel tumor suppressor p33ING2 enhances nucleotide excision repair via inducement of histone H4 acetylation and chromatin relaxation. Cancer Res 2006, 66, 1906–1911. [Google Scholar]
- Kuo, W.-H.W.; Wang, Y.; Wong, R.P.C.; Campos, E.I.; Li, G. The ING1b tumor suppressor facilitates nucleotide excision repair by promoting chromatin accessibility to XPA. Exp. Cell Res 2007, 313, 1628–1638. [Google Scholar]
- Smith, M.L.; Ford, J.M.; Hollander, M.C.; Bortnick, R.A.; Amundson, S.A.; Seo, Y.R.; Deng, C.X.; Hanawalt, P.C.; Fornace, A.J. p53-Mediated DNA Repair Responses to UV Radiation: Studies of Mouse Cells Lacking p53, p21, and/or gadd45 Genes. Mol. Cell Biol 2000, 20, 3705–3714. [Google Scholar]
- Carrier, F.; Georgel, P.T.; Pourquier, P.; Blake, M.; Kontny, H.U.; Antinore, M.J.; Gariboldi, M.; Myers, T.G.; Weinstein, J.N.; Pommier, Y.; Fornace, A.J. Gadd45, a p53-responsive stress protein, modifies DNA accessibility on damaged chromatin. Mol. Cell Biol 1999, 19, 1673–1685. [Google Scholar]
- Schmitz, K.-M.; Schmitt, N.; Hoffmann-Rohrer, U.; Schäfer, A.; Grummt, I.; Mayer, C. TAF12 recruits Gadd45a and the nucleotide excision repair complex to the promoter of rRNA genes leading to active DNA demethylation. Mol. Cell 2009, 33, 344–353. [Google Scholar]
- Jin, S.-G.; Guo, C.; Pfeifer, G.P.; Reik, W. GADD45A does not promote DNA demethylation. PLoS Genet 2008, 4, e1000013. [Google Scholar]
- Degregori, J.; Johnson, D.G. Distinct and Overlapping Roles for E2F Family Members in Transcription, Proliferation and Apoptosis. Curr. Mol. Med 2006, 6, 739–748. [Google Scholar]
- Biswas, A.K.; Johnson, D.G. Transcriptional and Nontranscriptional Functions of E2F1 in Response to DNA Damage. Cancer Res 2012, 72, 13–17. [Google Scholar]
- Lin, W.C.; Lin, F.T.; Nevins, J.R. Selective induction of E2F1 in response to DNA damage, mediated by ATM-dependent phosphorylation. Genes Dev 2001, 15, 1833–1844. [Google Scholar]
- Liu, K.; Lin, F.-T.; Ruppert, J.M.; Lin, W.-C. Regulation of E2F1 by BRCT domain-containing protein TopBP1. Mol. Cell Biol 2003, 23, 3287–3304. [Google Scholar]
- Liu, K.; Luo, Y.; Lin, F.-T.; Lin, W.-C. TopBP1 recruits Brg1/Brm to repress E2F1-induced apoptosis, a novel pRb-independent and E2F1-specific control for cell survival. Genes Dev 2004, 18, 673–686. [Google Scholar]
- Chen, J.; Zhu, F.; Weaks, R.L.; Biswas, A.K.; Guo, R.; Li, Y.; Johnson, D.G. E2F1 promotes the recruitment of DNA repair factors to sites of DNA double-strand breaks. Cell Cycle 2011, 10, 1287–1294. [Google Scholar]
- Berton, T.R.; Mitchell, D.L.; Guo, R.; Johnson, D.G. Regulation of epidermal apoptosis and DNA repair by E2F1 in response to ultraviolet B radiation. Oncogene 2005, 24, 2449–2460. [Google Scholar]
- Guo, R.; Chen, J.; Zhu, F.; Biswas, A.K.; Berton, T.R.; Mitchell, D.L.; Johnson, D.G. E2F1 localizes to sites of UV-induced DNA damage to enhance nucleotide excision repair. J. Biol. Chem 2010, 285, 19308–19315. [Google Scholar]
- Guo, R.; Chen, J.; Mitchell, D.L.; Johnson, D.G. GCN5 and E2F1 stimulate nucleotide excision repair by promoting H3K9 acetylation at sites of damage. Nucleic Acids Res 2011, 39, 1390–1397. [Google Scholar]
- Costelloe, T.; Fitzgerald, J.; Murphy, N.J.; Flaus, A.; Lowndes, N.F. Chromatin modulation and the DNA damage response. Exp. Cell Res 2006, 312, 2677–2686. [Google Scholar]
- Yu, Y.; Teng, Y.; Liu, H.; Reed, S.H.; Waters, R. UV irradiation stimulates histone acetylation and chromatin remodeling at a repressed yeast locus. Proc. Natl. Acad. Sci. USA 2005, 102, 8650–8655. [Google Scholar]
- Teng, Y.; Yu, Y.; Waters, R. The Saccharomyces cerevisiae histone acetyltransferase Gcn5 has a role in the photoreactivation and nucleotide excision repair of UV-induced cyclobutane pyrimidine dimers in the MFA2 gene. J. Mol. Biol 2002, 316, 489–499. [Google Scholar]
- Bhoumik, A.; Takahashi, S.; Breitweiser, W.; Shiloh, Y.; Jones, N.; Ronai, Z. ATM-dependent phosphorylation of ATF2 is required for the DNA damage response. Mol. Cell 2005, 18, 577–587. [Google Scholar]
- Bhoumik, A.; Singha, N.; O’Connell, M.J.; Ronai, Z.A. Regulation of TIP60 by ATF2 modulates ATM activation. J. Biol. Chem 2008, 283, 17605–17614. [Google Scholar]
- Tsai, W.-B.; Chung, Y.M.; Takahashi, Y.; Xu, Z.; Hu, M.C.T. Functional interaction between FOXO3a and ATM regulates DNA damage response. Nature 2008, 10, 460–467. [Google Scholar]
- Bowen, C.; Gelmann, E.P. NKX3.1 activates cellular response to DNA damage. Cancer Res 2010, 70, 3089–3097. [Google Scholar]
- Malewicz, M.; Kadkhodaei, B.; Kee, N.; Volakakis, N.; Hellman, U.; Viktorsson, K.; Leung, C.Y.; Chen, B.; Lewensohn, R.; van Gent, D.C.; et al. Essential role for DNA-PKmediated phosphorylation of NR4A nuclear orphan receptors in DNA double-strand break repair. Genes Dev 2011, 25, 2031–2040. [Google Scholar]
- Volcic, M.; Karl, S.; Baumann, B.; Salles, D.; Daniel, P.; Fulda, S.; Wiesmüller, L. NF-κB regulates DNA double-strand break repair in conjunction with BRCA1-CtIP complexes. Nucleic Acids Res 2012, 40, 181–195. [Google Scholar]
- Al Rashid, S.T.; Dellaire, G.; Cuddihy, A.; Jalali, F.; Vaid, M.; Coackley, C.; Folkard, M.; Xu, Y.; Chen, B.P.C.; Chen, D.J.; et al. Evidence for the direct binding of phosphorylated p53 to sites of DNA breaks in vivo. Cancer Res 2005, 65, 10810–10821. [Google Scholar]
- Dinant, C.; Luijsterburg, M.S. The emerging role of HP1 in the DNA damage response. Mol. Cell Biol 2009, 29, 6335–6340. [Google Scholar]
- Chou, D.M.; Adamson, B.; Dephoure, N.E.; Tan, X.; Nottke, A.C.; Hurov, K.E.; Gygi, S.P.; Colaiácovo, M.P.; Elledge, S.J. A chromatin localization screen reveals poly (ADP ribose)-regulated recruitment of the repressive polycomb and NuRD complexes to sites of DNA damage. Proc. Natl. Acad. Sci. USA 2010, 107, 18475–18480. [Google Scholar]
- Smerdon, M.J. DNA repair and the role of chromatin structure. Curr. Opin. Cell Biol 1991, 3, 422–428. [Google Scholar]
- Green, C.M.; Almouzni, G. When repair meets chromatin. First in series on chromatin dynamics. EMBO Rep 2002, 3, 28–33. [Google Scholar]
- Soria, G.; Polo, S.E.; Almouzni, G. Prime, repair, restore: The active role of chromatin in the DNA damage response. Mol. Cell 2012, 46, 722–734. [Google Scholar]
- Hayes, S.; Shiyanov, P.; Chen, X.; Raychaudhuri, P. DDB, a putative DNA repair protein, can function as a transcriptional partner of E2F1. Mol. Cell Biol 1998, 18, 240–249. [Google Scholar]
- Maser, R.S.; Mirzoeva, O.K.; Wells, J.; Olivares, H.; Williams, B.R.; Zinkel, R.A.; Farnham, P.J.; Petrini, J.H. Mre11 complex and DNA replication: Linkage to E2F and sites of DNA synthesis. Mol. Cell Biol 2001, 21, 6006–6016. [Google Scholar]
- De Siervi, A.; de Luca, P.; Byun, J.S.; Di, L.J.; Fufa, T.; Haggerty, C.M.; Vazquez, E.; Moiola, C.; Longo, D.L.; Gardner, K. Transcriptional autoregulation by BRCA1. Cancer Res 2010, 70, 532–542. [Google Scholar]
- Xu, X.; Stern, D.F. NFBD1/MDC1 regulates ionizing radiation-induced focus formation by DNA checkpoint signaling and repair factors. FASEB J 2003, 17, 1842–1848. [Google Scholar]
- Luijsterburg, M.S.; Lindh, M.; Acs, K.; Vrouwe, M.G.; Pines, A.; van Attikum, H.; Mullenders, L.H.; Dantuma, N.P. DDB2 promotes chromatin decondensation at UV-induced DNA damage. J. Cell Biol 2012, 197, 267–281. [Google Scholar]
- Datta, A.; Bagchi, S.; Nag, A.; Shiyanov, P.; Adami, G.R.; Yoon, T.; Raychaudhuri, P. The p48 subunit of the damaged-DNA binding protein DDB associates with the CBP/p300 family of histone acetyltransferase. Mutat. Res 2001, 486, 89–97. [Google Scholar]
- Gu, B.; Zhu, W.-G. Surf the post-translational modification network of p53 regulation. Int. J. Biol. Sci 2012, 8, 672–684. [Google Scholar]
- Ozaki, T.; Okoshi, R.; Sang, M.; Kubo, N.; Nakagawara, A. Acetylation status of E2F-1 has an important role in the regulation of E2F-1-mediated transactivation of tumor suppressor p73. Biochem. Biophys. Res. Commun 2009, 386, 207–211. [Google Scholar]
- Pediconi, N.; Guerrieri, F.; Vossio, S.; Bruno, T.; Belloni, L.; Schinzari, V.; Scisciani, C.; Fanciulli, M.; Levrero, M. hSirT1-dependent regulation of the PCAF-E2F1-p73 apoptotic pathway in response to DNA damage. Mol. Cell Biol 2009, 29, 1989–1998. [Google Scholar]
- Pediconi, N.; Ianari, A.; Costanzo, A.; Belloni, L.; Gallo, R.; Cimino, L.; Porcellini, A.; Screpanti, I.; Balsano, C.; Alesse, E.; et al. Differential regulation of E2F1 apoptotic target genes in response to DNA damage. Nat. Cell Biol 2003, 5, 552–558. [Google Scholar]
© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Vélez-Cruz, R.; Johnson, D.G. E2F1 and p53 Transcription Factors as Accessory Factors for Nucleotide Excision Repair. Int. J. Mol. Sci. 2012, 13, 13554-13568. https://doi.org/10.3390/ijms131013554
Vélez-Cruz R, Johnson DG. E2F1 and p53 Transcription Factors as Accessory Factors for Nucleotide Excision Repair. International Journal of Molecular Sciences. 2012; 13(10):13554-13568. https://doi.org/10.3390/ijms131013554
Chicago/Turabian StyleVélez-Cruz, Renier, and David G. Johnson. 2012. "E2F1 and p53 Transcription Factors as Accessory Factors for Nucleotide Excision Repair" International Journal of Molecular Sciences 13, no. 10: 13554-13568. https://doi.org/10.3390/ijms131013554