Abstract
In this study we pursued a diagnostic target in Aspergillus fumigatus (AF) by using qualitative Realtime PCR combined with proprietary DNA primers and a hydrolysis probe specific for this fungal target. Qualitative Realtime PCR is a diagnostic tool that utilizes Realtime PCR technology and detects the presence or absence target specific DNA within a predetermined detection range. Respiratory tissue and fluids from experimentally infected guinea pigs were tested by extracting DNA from the samples which were amplified and detected using AF specific DNA primers and probe. This study included qualitative evaluations of all specimens for the presence of the DNA of AF. The findings in the tissues after AF infection were compared to the numbers of spores in aerosolized samples used to inoculate the animals. Results demonstrated that the specific probe and primer set could detect the presence or absence of AF DNA in the sample. The qualitative detection limit of the assay ranged from 6 × 104 copies to 6 copies. Since blood cultures are rarely positive for Aspergillosis, our data indicate that qualitative Realtime PCR, in combination with the appropriate DNA primers and probe can serve as an effective diagnostic tool in the early detection of fungal infections.
1. Introduction
Invasive fungal infections are the most common infections observed in immunocompromised patients []. The incidence of invasive aspergillosis (IA) and other fungal infections has increased significantly over recent decades because of the increasing numbers of surviving immunocompromised patients []. Fungal infections in adults and children have been extensively studied; however, the speed necessary to slow these infections and identify the causative organism has fallen short in the effort to reduce the mortality in these patients []. The incidence of infectious aspergillosis has increased dramatically over the past two decades in bone marrow transplant recipients [], yet the ability to diagnose these infections has not increased substantially in laboratory medicine. In the United States alone, greater than 27,000 solid organ transplant procedures were performed in 2008 []. Pappas et al., in 2010 [], reported a slight detectable increase in invasive fungal infections (IFIs) during a surveillance period of solid organ transplant patients in the U.S. during the period of 2002–2005. They further reported that a large number of IFIs occurred later than 1 year after transplantation, with as many as 25% of IFIs developing later than 3 years after transplantation. They suggested that these data indicated that prevention, diagnostic and treatment strategies need to be further developed to take these late developing infections into account.
At present, patients suspected of having a fungal infection are subjected to multiple blood, urine, tissue, and other body fluid cultures. Fluid specimens are placed in blood culture bottles and on various selective media and cultured at room temperature and 37 °C for various periods of time. The shortest period of time that various automated microbiological methods can detect fungal infections is 8–12 h, with the longest period of time in automated methods and/or culture media methods being as long as 2–4 weeks. Culture techniques such as these also lack sensitivity. Patterson [] provided a framework for evaluating genomic targets in animal models to improve the diagnosis and treatment of invasive aspergillosis. This framework evaluated an enzyme linked immunosorbent assay (ELISA) technique that allows for the detection of galactomannan from Aspergillus fumigatus (AF) [,] and incorporates other biomarkers, such as PCR-based diagnosis platforms. As Patterson pointed out, the galactomannan detection in serum and other body fluids, especially bronchoalveolar lavage (BAL) fluid, plays a major role at present in non-culture based diagnosis of IA []. However, the sensitivity and specificity of the galactomannan assay is low and fraught with many false negatives and false positives [,].
Methods utilizing biological molecular techniques, such as polymerase chain reaction (PCR) or various types of nucleic acid blots, which take time and have serious contamination problems, are currently available and used in many laboratories for this type of testing. However, Realtime Polymerase Chain Reaction (Realtime PCR) offers a clean, closed system which is not prone to contamination observed with other techniques. Realtime PCR instruments such as the Cepheid SmartCycler® and the Applied BiosystemsTM 7500 Fast were used for these studies and provided an instrument platform capable of determining the presence or absence of AF DNA and virtually eliminates contamination issues prevalent in open system instruments [–]. This paper describes work conducted using qualitative Realtime PCR which is a diagnostic tool that utilizes Realtime PCR technology and detects the presence or absence target specific DNA within a predetermined detection range. Qualitative detection assays have been previously described and are currently in use in clinical laboratories worldwide [–].
Realtime PCR based methods can be used to amplify, detect and identify specific fungal DNA by utilization of target specific primers and probes. Realtime PCR, using hydrolysis probes [–], combines amplification and simultaneous probe hybridization to achieve sensitive and specific detection of infectious fungi (e.g., molds) in Realtime, thereby providing rapid detection of opportunistic fungal pathogens such as AF, A. flavus, A. niger and A. terreus. Diagnostic tools available to a clinical lab in the mycology arena are extremely limited. Furthermore, successful treatment outcomes of fungal infections are predicated on effective, early, and sensitive/specific laboratory tests []. In the assay described here, primers and a probe were designed to amplify and detect a 136 base pair region on the 18S Ribosomal RNA gene of AF. The assay was optimized and validated as directed by the Clinical and Laboratory Institute (CLSI) [–]. Efficiency, accuracy, precision, analytical sensitivity (detection limit), and analytical specificity (interfering substances) were the performance characteristics determined in this test with the amplification efficiency of the assay being within the acceptable range of 90 to 105% and the linear standard curve (R2) greater than 0.980. Furthermore, the test was audited and approved for human diagnostic purposes by the Centers for Medicare and Medicaid Services (CMS) and the College of American Pathologists (CAP). After inspections of the laboratory, validations and procedures were found to be compliant with Clinical Laboratory Improvement Amendments (CLIA), the test was approved for human molecular diagnostic testing.
In this study, we examined one such promising and untapped diagnostic target in AF by using qualitative Realtime PCR combined with the proprietary DNA primers and a hydrolysis probe specific for this fungal target. Respiratory tissue and fluids from experimentally infected guinea pigs were tested by extracting DNA from the samples and amplified and detected using the AF specific DNA primers and probe described above.
This study provides data comparing the number of spores found in tissues after infection, with the numbers of conidia in aerosolized samples used to inoculate the animals by semi quantitative culture. In addition, this study includes qualitative evaluations of all specimens for the presence of the DNA of AF in the lung tissue and BALs of the tested animals.
2. Results and Discussion
Histopathology of the lungs of the guinea pigs demonstrated similar findings as reported by Vallor et al. [] in which conidia were found in the pulmonary alveolar spaces within 1 hour. post infection. As noted in that study, guinea pig lungs were visibly infected with Aspergillus lesions compared to lungs obtained from uninfected animals. At one hour post infection, the mean pulmonary fungal burden was assessed by semi-quantitative culture and revealed in this study to be a count of log10 4.23 +/− 0.7 CFU/g). There was a statistically significant decrease in the fungal burden demonstrated at day 3 through day 7, when compared to the 1 hour time point (Figure 1).
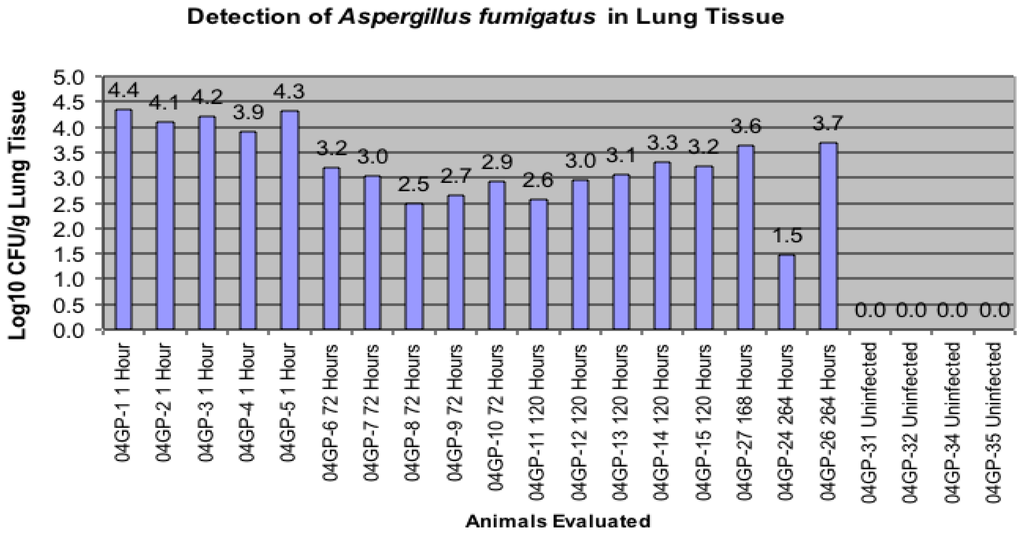
Figure 1.
Comparison of infected groups to numbers of Aspergillus fumigatus colony forming units (CFU) by culture present in the lung.
All data in Figure 1 are presented as the number of AF CFUs by culture and were detected (Log10/g Lung) upon sacrifice of the animal and removal of the lung tissue. This evaluation was conducted with each animal infected and sacrificed one hour later through animals being sacrificed 264 hours post-inoculation. Four uninfected animals were also used in this evaluation.
In addition, this study also demonstrated that qualitative Realtime PCR is capable of detecting very small amounts of target specific DNA (Figure 2 and Figure 3). The curves shown, demonstrate the amplification efficiency of the assay being within the acceptable range of 90 to 105% and the linear standard curve (R2) greater than 0.980. The AF assay limit of detection dilution series below shows the detection limit of the assay to be between approximately 6 × 104 copies to 6 copies of purchased AF genomic DNA (See explanation in Materials and Methods)
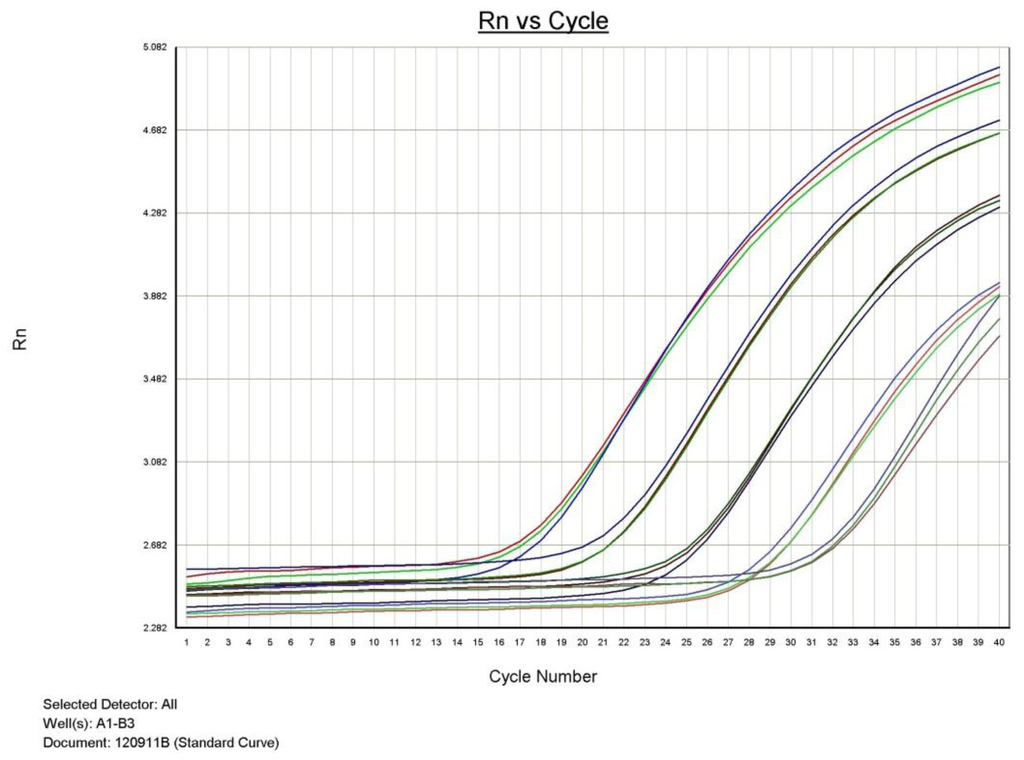
Figure 2.
Aspergillus fumigatus PCR dilution series demonstrating the qualitative detection limit.
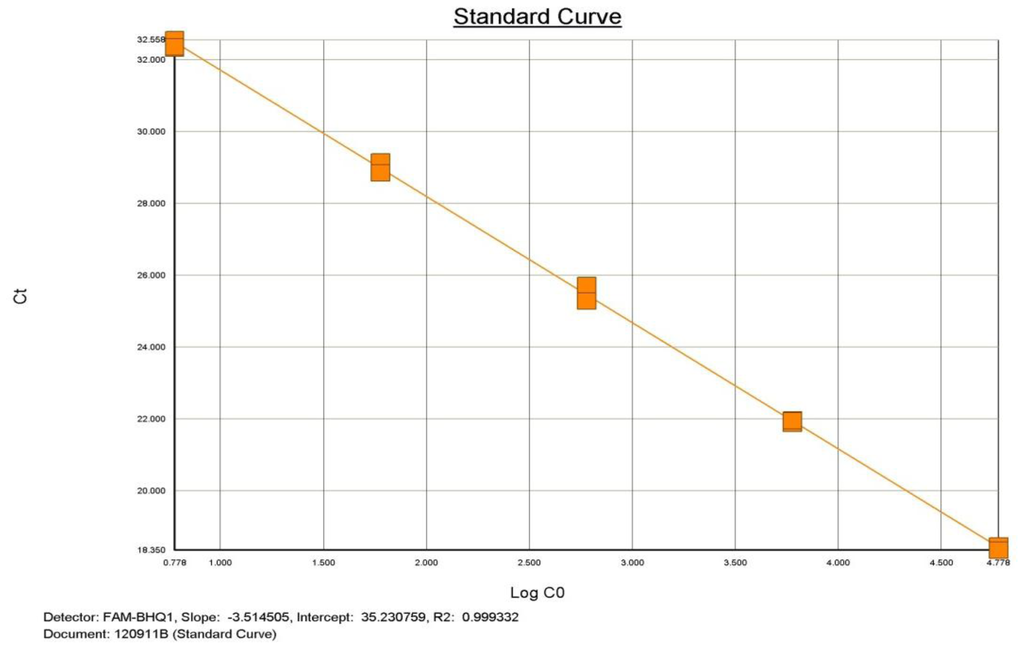
Figure 3.
Aspergillus fumigatus PCR dilution Standard Curve demonstrating efficiency and linearity.
Approximately 2.0 μg of AF DNA was acquired from the American Type Culture Collection (ATCC #1022 D-2). The approximate copy number was determined based on information provided by ATCC and the genome size of AF. The template was used in a five point dilution series ranging from 6 × 104 copies to 6 copies. All points of the serial dilution were detected by the ABI 7500 Fast with results demonstrated in Table 1.

Table 1.
Results by qualitative Realtime PCR assay of an Aspergillus fumigatus dilution series ranging from approximately 6 × 104 copies to 6 copies.
Utilizing the qualitative Realtime PCR assay above, the guinea pig bronchoalveolar lavages and lung tissue were evaluated for the presence of AF DNA. Results of these studies demonstrated that AF DNA could be detected in BALs and lung tissue within the detection limits described above (Table 2).
Table 2.
Detection of Aspergillus fumigatus DNA in lung homogenates and bronchoalveolar lavage (BAL).
Conidia were detected by histopathology in the pulmonary alveolar spaces of the guinea pigs within 1 hour post infection, confirming the results of Vallor et al. []. Also, as seen in the previous study [], by day 5 post infection, guinea pig lungs showed Aspergillus lesions when comparing them to lungs of non-infected animals. These same findings were noted in this study, thus demonstrating the reproducibility of the described animal model. Furthermore, this study determined that when an animal is infected with AF, the same organism can be detected by qualitative Realtime PCR in the BALs and lungs. This information could be extremely useful and important in determining the efficacy of many antifungals which may be used in patients who have fungal positive BALs. These studies may be used further to calculate the number of copies to determine prolonging survival when determining efficacy of drugs used in patients who have undergone transplantation or those immunocompromised patients who may succumb to infection by what was initially very few organisms. The results of this study may be expanded to observe the presence or absence of mycotoxins produced by Aspergillus sp. (not just AF), and to determine the number of copies of fungi needed to actually produce a significant, detectable amount of mycotoxin in a clinical specimen.
3. Experimental Section
3.1. Animals and Immunosuppression
The animal model assessment of AF burden in guinea pigs has been previously described []. Briefly, 8 male Harley guinea pigs (0.5 kg; Charles River Laboratories, Wilmington, MA) were made neutropenic with intraperitoneal cyclophosphamide (250 mg/kg; Cytoxan; Mead Johnson, Princeton, NJ, USA) and further immunosuppressed with cortisone acetate (250 mg/kg; Sigma, St Louis, MO, USA) given subcutaneously 2 days prior to inhalational challenge In addition, ceftazidime (100 mg/kg by subcutaneous injections; Glaxo SmithKline Beecham Pharmaceuticals, Philadelphia, PA, USA) was administered daily throughout the study for prevention of bacterial infections. In order to maintain immunosuppression throughout the study, additional doses of cyclophosphamide (200 mg/kg) and cortisone acetate (250 mg/kg) were administered on day 3 post inhalational challenge. All animal research procedures were approved by the Institutional Animal Care and Use Committee at the University of Texas Health Science Center at San Antonio, and animals were maintained in accordance with the American Association for Accreditation of Laboratory Animal Care.
3.2. Preparation of Inoculate and Inhalational Challenge
The preparation of AF (AF293) has been previously described [] as has the inhalational challenge [–]. Briefly, the organism was cultured; conidia were dislodged from potato dextrose plates, spun in Sorvall centrifuge and, filtered, by approved protocol []. A portable, acrylic chamber was used to establish the model of invasive pulmonary aspergillosis (IPA) [], and was used in this study. The acrylic chamber was used to simultaneously infect groups of guinea pigs with nebulized inocula of 108 CFU/mL of AF conidia (total volume was 12 mL). Eighteen animals were exposed to the aerosol mist for 1 hour. After one additional hour, five guinea pigs were sacrificed to confirm and assess the average conidial delivery of the infected run. Guinea pigs were sacrificed by terminal exsanguinations after being anesthetized with 44 mg/kg ketamine-HCl and 10 mg/kg xylazine. A cohort of four immunosuppressed guinea pigs was left uninfected but underwent the same immunosuppression and placebo therapeutic regimen as the infected cohort. After sacrifice, lungs, liver, kidney, brain, bladder, spleen, and serum were collected from each animal. Saline injections to obtain BALs were performed prior to exsanguinations from all animals. Only lung homogenates and BAL results were reported in this study.
3.3. Fungal Burden Assessment
Fungal burdens in the lungs of the 15 guinea pigs were determined by CFU counting using the same methods as previous reported []. Guinea pigs were infected with 2.3 × 107 CFU/mL of AF by aerosolization as previously described [].
3.4. AF Qualitative Realtime PCR Detection Limits
Lyophilized AF genomic DNA (strain NRRL 163) was acquired from ATCC to determine the detection limits of the AF Realtime PCR assay. The lyophilized genomic DNA (approximately 6.18 × 107 copies) was rehydrated in 100.0 μL of PCR grade water for a final concentration of approximately 6 × 105 copies/μL. This DNA solution was used to setup a dilution series from approximately 6 × 104 copies/μL to 6 copies/μL. Upon completion of the dilution series, 1.0 μL of each dilution series point was used in a Realtime PCR reaction.
3.5. Extraction and Qualitative Realtime PCR assessment of AF
To ensure maximum recovery of fungal DNA, lung homogenates and BALs were further homogenized using silica beads and a bead beating technique (Sigma, St. Louis, MO, USA) in order to ensure cellular release of genomic nucleic acid from all of the AF conidial and hyphal forms present. One milliliter (1.0 mL) aliquots of lung homogenates or BALs were subjected to sterile bead beating for 1.0 minute (Mini Bead Beater, BioSpec Products, Bartlesville, OK, USA) as previously described []. Homogenized samples were processed for DNA extraction using the QIAamp DNA Mini Kit (Qiagen, Valencia, CA, USA) according to the manufacturer’s directions. Purified DNA was obtained for all samples processed with the purified DNA used as templates for qualitative Realtime PCR analysis. Proprietary primers and a probe specific for AF plus an internal control assay were designed by RealTime Labs, Inc. and synthesized by Integrated DNA Technologies, Inc. (Coralville, IA, USA). Primers were synthesized at a scale of 25 nm and probes were synthesized at a scale of 100 nm.
3.6. Assay Controls and Reaction Details
The test was further controlled by using internal controls of Geotrichum sp. (GEO) and known positive controls of AF as well as known negative samples. The characteristics of the DNA probe and primers in this study were forward primer size: 20 base pairs (bp); reverse primer size 20 bp; and probe size 26 bp. All probes used for the assays were hydrolysis probes with the reporter fluorescent reporter dye, 6-carboxy-fluorescein (FAM) attached to the 5′ end and the quencher Black Hole Quencher (BHQ-1) attached to the 3′ end. Primers and probes were received lyophilized and re-suspended into 100 μM stocks. These stocks were then mixed into working stocks containing probe and primers at required concentrations. The enzyme and reaction buffers used in the reaction were purchased from Cepheid manufactured specifically for the SmartCycler® system and Applied Biosystems manufactured specifically for the 7500 FAST. The reactions were set up per manufacturer recommendations. An internal control assay was run for each extracted sample to insure the sample extracted generated purified DNA and was present in the reaction and contained no inhibitors. Additionally, a positive control and negative control was run for each assay to insure that each fungal Realtime PCR assay component was amplifying and detecting the target and that there were no problems with contamination. All assays and the internal control had a cycling profile containing an initial hot start followed by 45 denature and anneal cycles. All assays were designed and operated as qualitative detection assays and were able to determine the presence or absence of fungal DNA specific to the assay used in the reaction.
3.7. Histological Evaluation
Samples of guinea pig lungs were aseptically removed and fixed in 10% neutral buffered formalin. Paraffin-embedded tissue sections were then sectioned and stained with Grocott-Gomori methenamine-silver stain (GMS) and Hematoxylin-Eosin (H and E) stain (Tissue Techniques, Inc., Dallas, TX, USA) for evaluation of the inflammatory response in the immunosuppressed animals as well as for the presence of the fungal organisms.
3.8. BAL and Lung Homogenate Evaluation
Five hundred microliters of BALs and frozen tissue from lungs as well as 500 μL of homogenized lung tissue in saline from the guinea pigs were sent to RTL from the University of Texas Health Science Center at San Antonio on dry ice. Three hundred microliters of BAL and 300 μL of lung homogenate were centrifuged at 13,000 rpm for 10 minutes (AcuSpin, microcentrifuge, Fisher Scientific, Germany). The supernatant was removed and the pellet was re-suspended in 50 μL of normal saline and vortexed for 15 seconds. Fifty microliters of the sediment was placed on a glass microscopic slide and a drop of Lactophenol Cotton Blue was added to the sediment. A cover slip was placed on the sample and observed under low power. Spores were counted using a hemocytometer. Results of the spore count are seen in Figure 1. Hyphal elements were also noted.
4. Conclusions
This study shows that guinea pigs infected with AF by use of an aerosol mist had detectable fungal burden in their lungs up to 11 days following infection. It was also determined that AF DNA could be detected utilizing qualitative Realtime PCR in both bronchoalveolar lavage and lung tissue up to 11 days following infection. Lastly, the detection limit of the qualitative Realtime PCR assay was shown experimentally.
Realtime PCR is a powerful tool for determining the presence or absence of specific fungal targets in body fluids and tissue. Because blood cultures are rarely positive in Aspergillosis, the technology described here has the capability to detect down to one copy of DNA and has the practical ability to routinely detect down to 10 copies of DNA. This study has shown that specific Realtime PCR DNA probes and primers can be utilized in qualitative studies of body fluids and tissues, specifically BALs, to determine the presence or absence of AF DNA within the detection limits of the assay. Realtime PCR in combination with a well-designed target specific assay can and will be useful when evaluating transplant patients or immunocompromised patients for the presence of potentially lethal fungal organisms. This diagnostic ability will lead to more successful transplants accompanied by fewer patient complications and deaths.
References
- Patterson, T.F.; Kirkpatrick, W.R.; White, M.; Himenez, J.W.; Wingard, J.R.; Dupont, B.; Rinaldi, M.G.; Stevens, D.A.; Graybill, J.R. Invasive aspergillosis. Disease spectrum, treatment practices, and outcomes. Medicine 2000, 79, 250–260. [Google Scholar]
- Chandrasekar, P.H.; Alangaden, G.; Manavathu, E. Aspergillosis: An increasing problem in tertiary care hospitals? Clin. Infect. Dis 2000, 30, 984–985. [Google Scholar]
- Patterson, T.F. Advances and challenges in management of invasive mycoses. Lancet 2005, 366, 1013–1025. [Google Scholar]
- Singh, N.; Paterson, D.L. Aspergillus infections in transplant recipients. Clin. Microbiol. Rev 2005, 18, 44–69. [Google Scholar]
- Scientific Registry of Transplant Recipients. OPTN/SRTR Annual Report; US Department of Health & Human Services: Washington, DC, USA, 2009. Available online: http://www.ustransplant.org/annual_reports/current/chapter_I_AR_CD.htm#0 accessed on 22 February 2010.
- Pappas, P.G.; Alexander, B.D.; Andes, D.R.; Hadley, S.; Kauffman, C.A.; Freifeld, A.; Anaissie, E.J.; Brumble, L.M.; Herwaldt, L.; Ito, J.; et al. Invasive fungal infections among organ transplant recipients: Results of the transplant-associated infection surveillance network (TRANSNET). Clin. Infect. Dis 2010, 50, 1101–1111. [Google Scholar]
- Patterson, T.F. Clinical utility and development of biomarkers in invasive aspergillosis. Trans. Am. Clin. Climatol. Assoc 2010, 122, 174–183. [Google Scholar]
- Maertens, J.; Verhaegen, J.; Lagrou, K. Screening for circulating galactomannan as a noninvasive diagnostic tool for invasive aspergillosis in prolonged neutropenic patients and stem cell transplantation recipients: A prospective evaluation. Blood 2001, 97, 1604–1610. [Google Scholar]
- Boonsarngsuk, V.; Niyompattama, A.; Teosirimongkol, C.; Sriwanichrak, K. False-positive serum and bronchoalveolar lavage Aspergillus galactomannan assays caused by different antibiotics. Scand. J. Infect. Dis 2010, 42, 461–468. [Google Scholar]
- Pinel, C.; Fricker-Hidalgo, H.; Lebeau, B. Detection of circulating Aspergillus fumigatus galactomannan: Value and limits of the Platelia test for diagnosis invasive aspergillosis. J. Clin. Microbiol 2003, 41, 2184–2186. [Google Scholar]
- Wolk, D.; Mitchell, S.; Patel, R. Principles of molecular microbiology testing methods. Infect. Dis. Clin. N. Am 2001, 15, 1157–1204. [Google Scholar]
- Buchheidt, D. M.; Hummel, D.; Schleiermacher, B.; Spiess, H.; Skladny, R.; Schwerdtfeger, O. A.; Cornely, S.; Wilhelm, S.; Reuter, W. V.; Kern, T.; et al. Prospective multicenter clinical evaluation of a nested PCR assay, a lightcycler mediated PCR assay and a galactomannan ELISA for detection of invasive aspergillosis in neutropenic high risk patients. Abstr. M-2057. Proceedings of the 43rd Interscience Conference of Antimicrobial Agents and Chemotherapy, Chicago, IL, USA, 14–17 September 2003.
- Krajden, M.; Ziermann, R.; Khan, A.; Mak, A.; Leung, K.; Hendricks, D.; Comanor, L. Qualitative detection of hepatitis C virus RNA: Comparison of analytical sensitivity, clinical performance, and workflow of the Cobas Amplicor HCV test version 2.0 and the HCV RNA transcription-mediated amplification qualitative assay. J. Clin. Microbiol 2002, 40, 2903–2907. [Google Scholar]
- Caliendo, A.M.; Schuurman, R.; Yen-Lieberman, B.; Spector, S.A.; Andersen, J.; Manjiry, R.; Crumpacker, C.; Lurain, N.S.; Erice, A. Comparison of quantitative and qualitative PCR assays for Cytomegalovirus DNA in plasma. J. Clin Microbiol 2001, 39, 1334–1338. [Google Scholar]
- Qualitative assays. In Smart Cycler II, Operator Manual; Cepheid: Sunnyvale, CA, USA, 1999–2004; Volume Chapter 5, pp. 157–158.
- Arya, M.; Shergill, I.S.; Williamson, M.; Gommersall, L.; Arya, N.; Patel, H.R. Basic principles of Realtime PCR. Expert Rev. Mol. Diagn 2005, 5, 209–219. [Google Scholar]
- Watson, D.E.; Li, B. TaqMan applications in genetic and molecular toxicology. Int. J. Toxicol 2005, 24, 39–45. [Google Scholar]
- Protocols for Determination of Limits of Detection and Limits of Quantitation; Approved Guideline-Second Edition CLSI Document EP17; Clinical and Laboratory Standards Institute (CLSI): Wayne, PA, USA, 2004.
- Evaluation of Precision Performance of Quantitative Measurement Methods; Approved Guideline-Second Edition CLSI Document EP5-A2; Clinical and Laboratory Standards Institute (CLSI): Wayne, PA, USA, 2004.
- User Verification of Performance for Precision and Trueness; Approved Guideline-Second Edition CLSI Document EP15-A2; Clinical and laboratory standards institute (CLSI): Wayne, PA, USA, 2006.
- Vallor, A.E.; Kirkpatrick, W.R.; Najvar, L.K.; Bocanegra, R.; Kinney, M.C.; Fothergill, A.W.; Herrera, M.L.; Wickes, B.L.; Graybill, J.R.; Patterson, T.F. Assessment of Aspergillus fumigatus burden in pulmonary tissue of guinea pigs by quantitative PVR, galactomannan enzyme immunoassay, and quantitative culture. Antimicrobiol. Agents Chemother 2008, 52, 2593–2598. [Google Scholar]
- Sheppard, D.C.; Marr, K.A.; Fredricks, D.N.; Chiang, L.Y.; Doedt, T.; Filler, S.G. Comparison of three methodologies for the determination of pulmonary fungal burden in experimental murine aspergillosis. Clin. Microbiol. Infect 2006, 12, 376–380. [Google Scholar]
- Sheppard, D.C.; Rieg, G.; Chiang, L.Y.; Filler, S.G.; Edwards, J.E., Jr; Ibrahim, A.S. Novel inhalation murine model of invasive pulmonary aspergillosis. Antimicrob. Agents Chemother 2004, 48, 1908–1911. [Google Scholar]
- Steinbach, W.J.; Benjamin, D.K., Jr; Trasi, S.A.; Miller, J.L.; Schell, W.A.; Zaas, A.K.; Faster, W.M.; Perfect, J.R. Value of an inhalational l model of invasive aspergillosis. Med. Mycol 2004, 42, 417–425. [Google Scholar]
© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).