Biodegradable Nanoparticles of mPEG-PLGA-PLL Triblock Copolymers as Novel Non-Viral Vectors for Improving siRNA Delivery and Gene Silencing

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
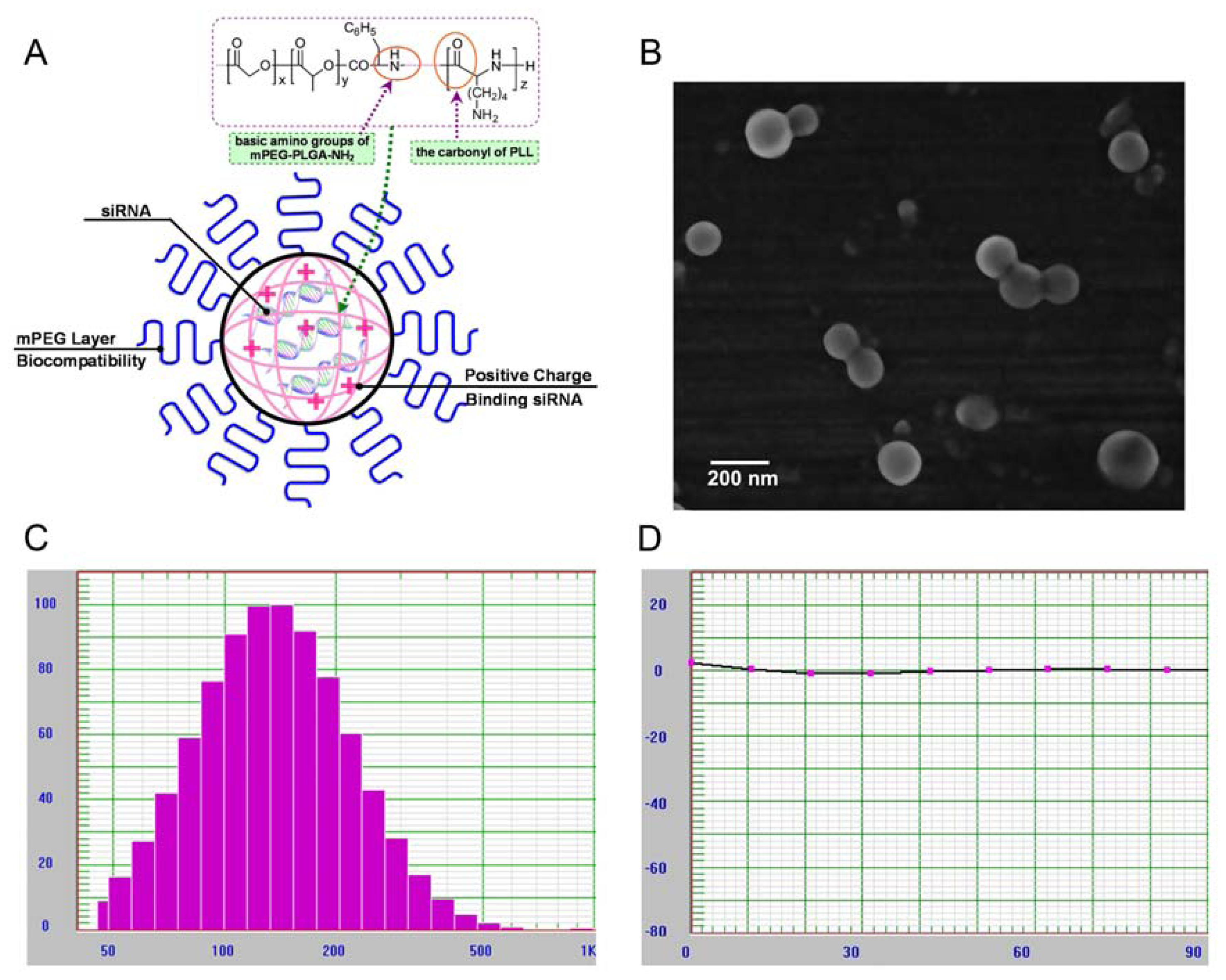
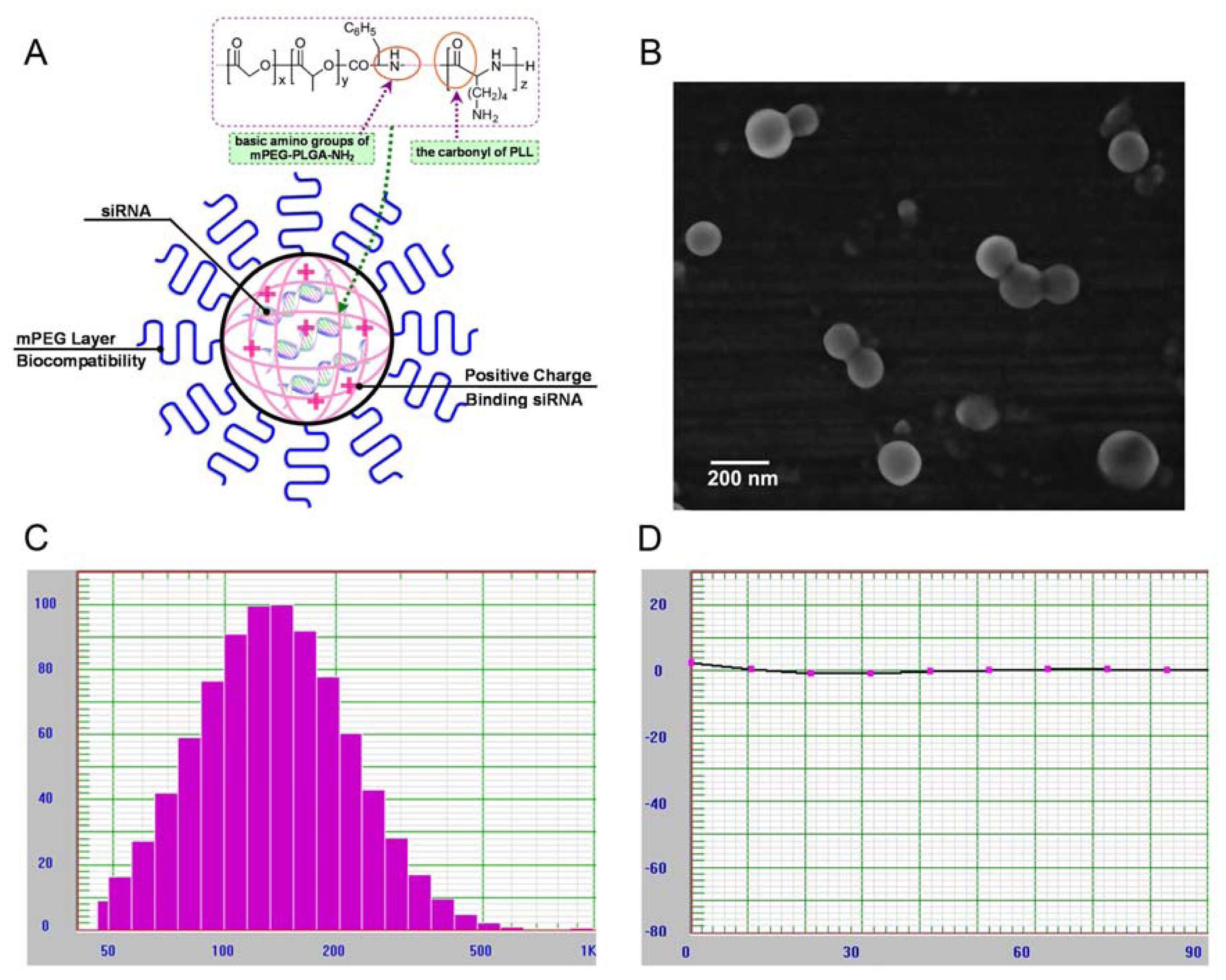
2.1. Characterization of mPEG-PLGA-PLL Triblock Copolymer
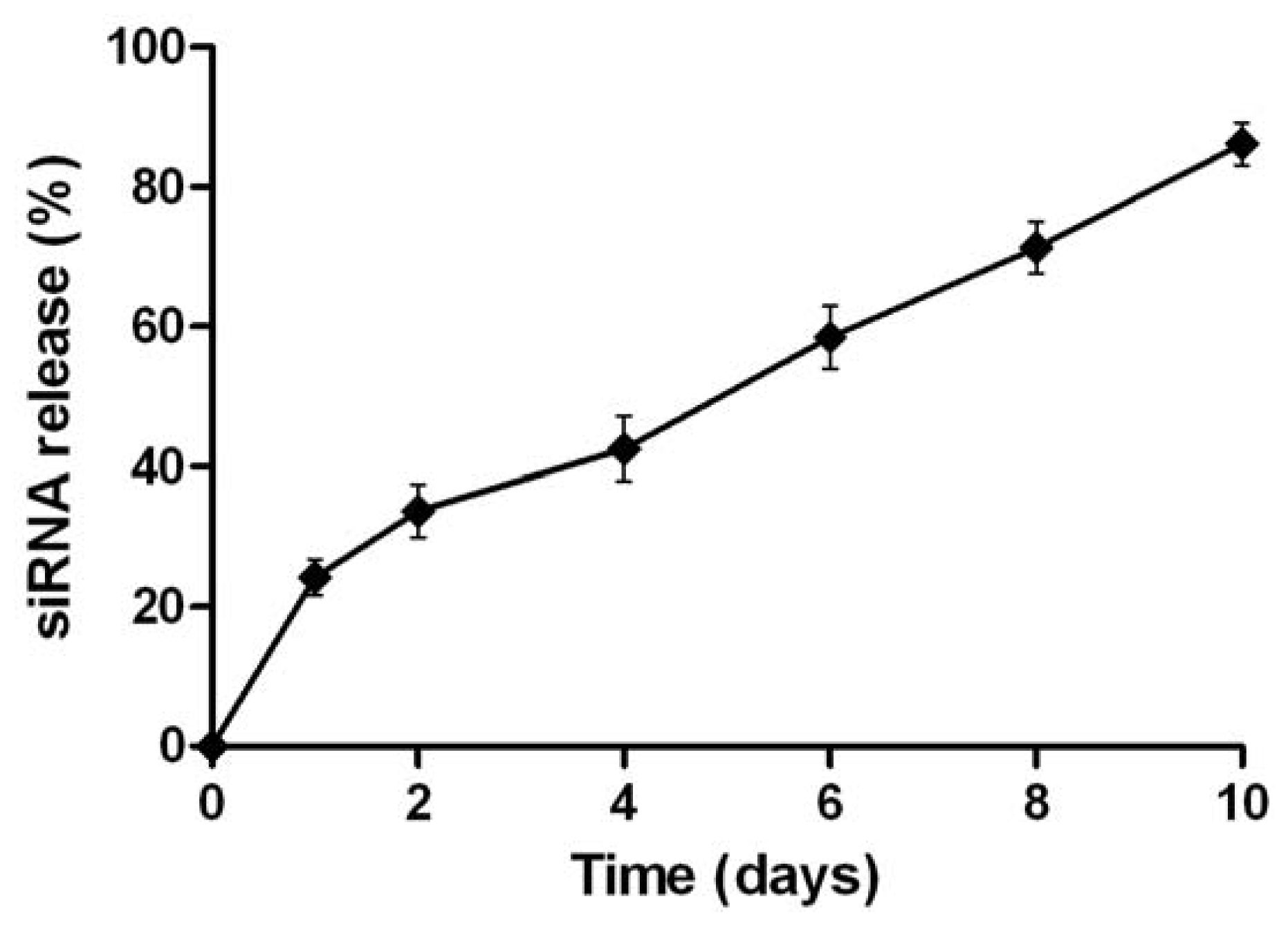
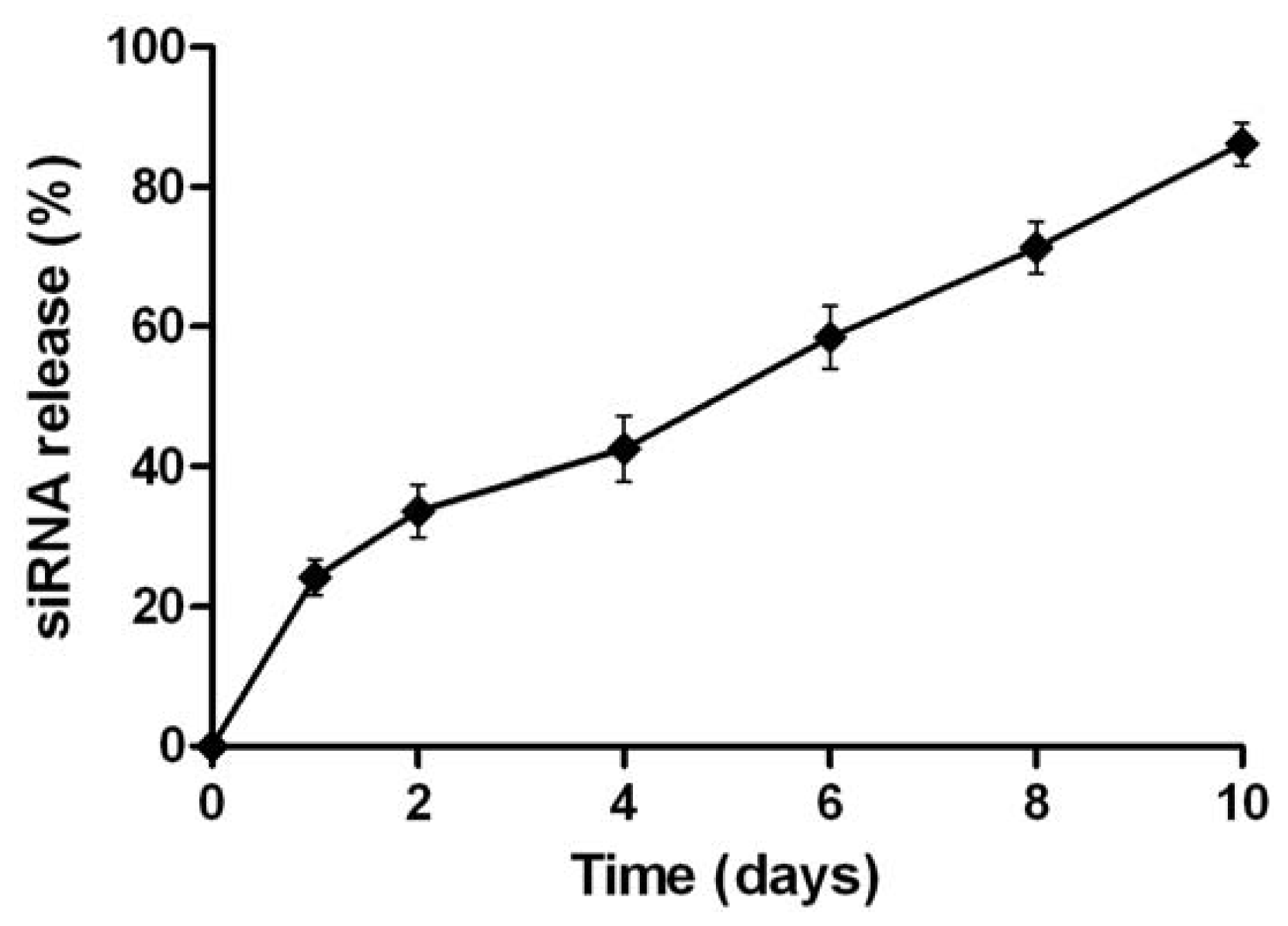
2.2. Physicochemical Characterization of NPs Loading siRNA
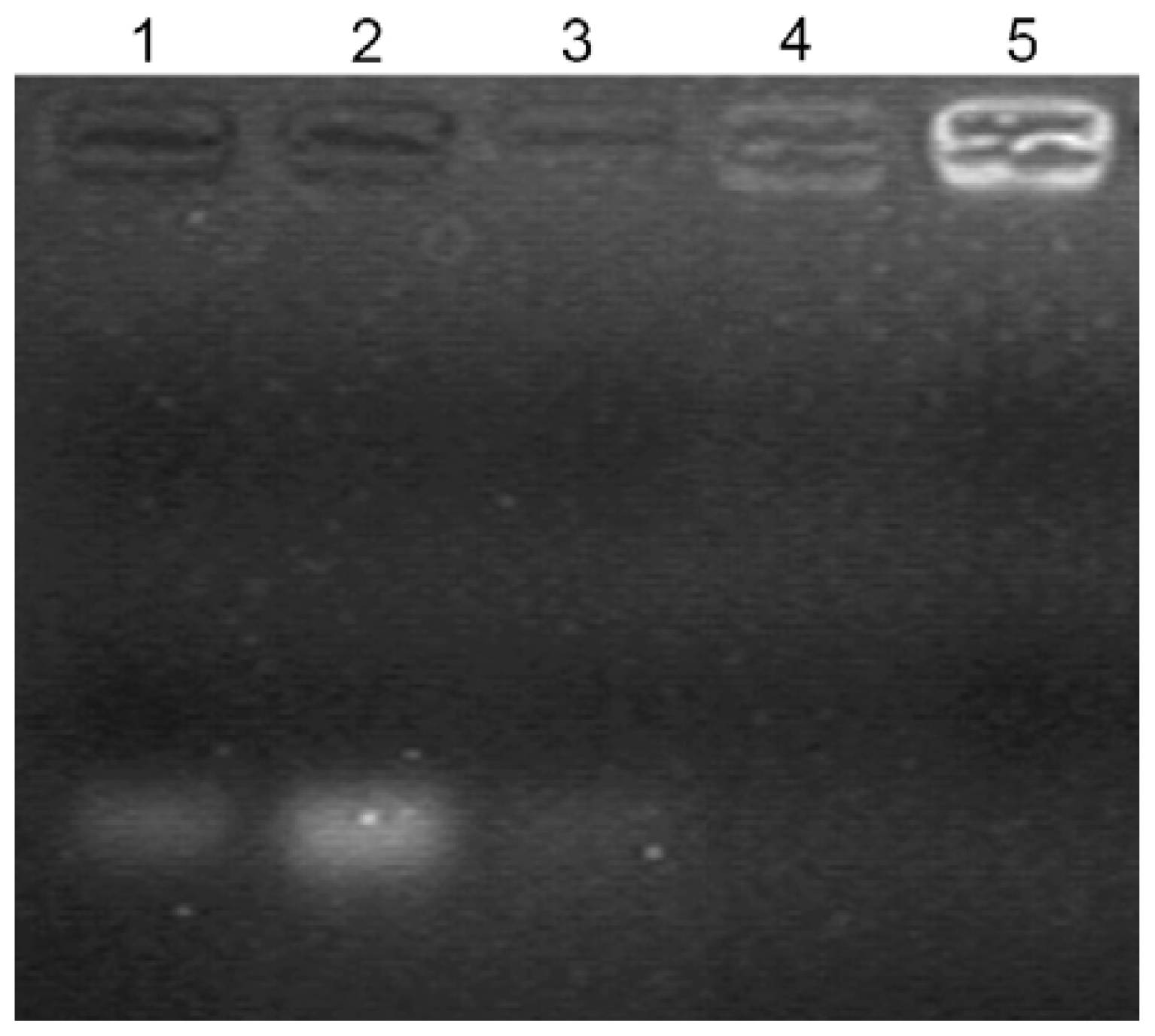
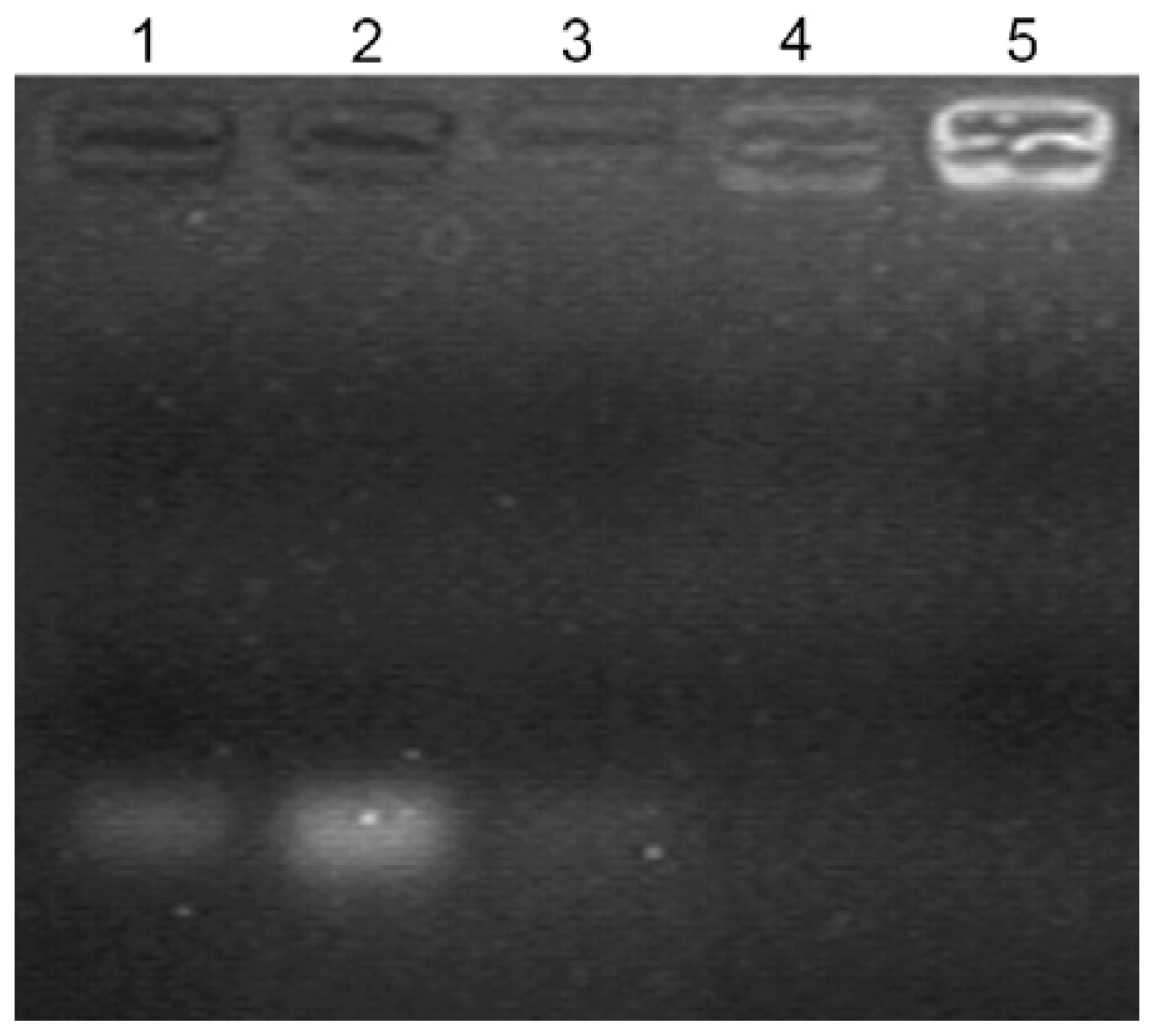
2.3. Gel Retardation Assay
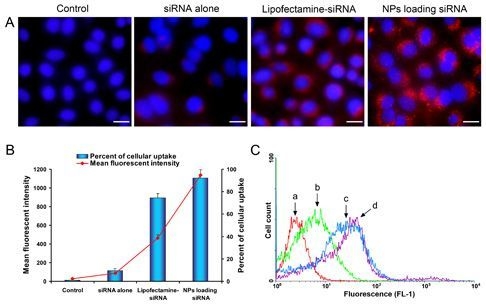
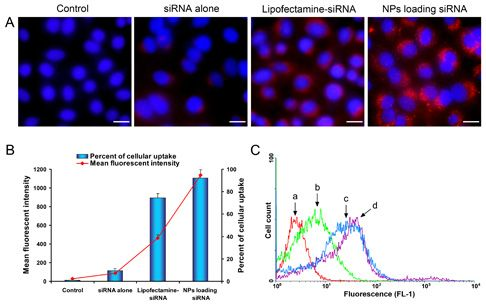
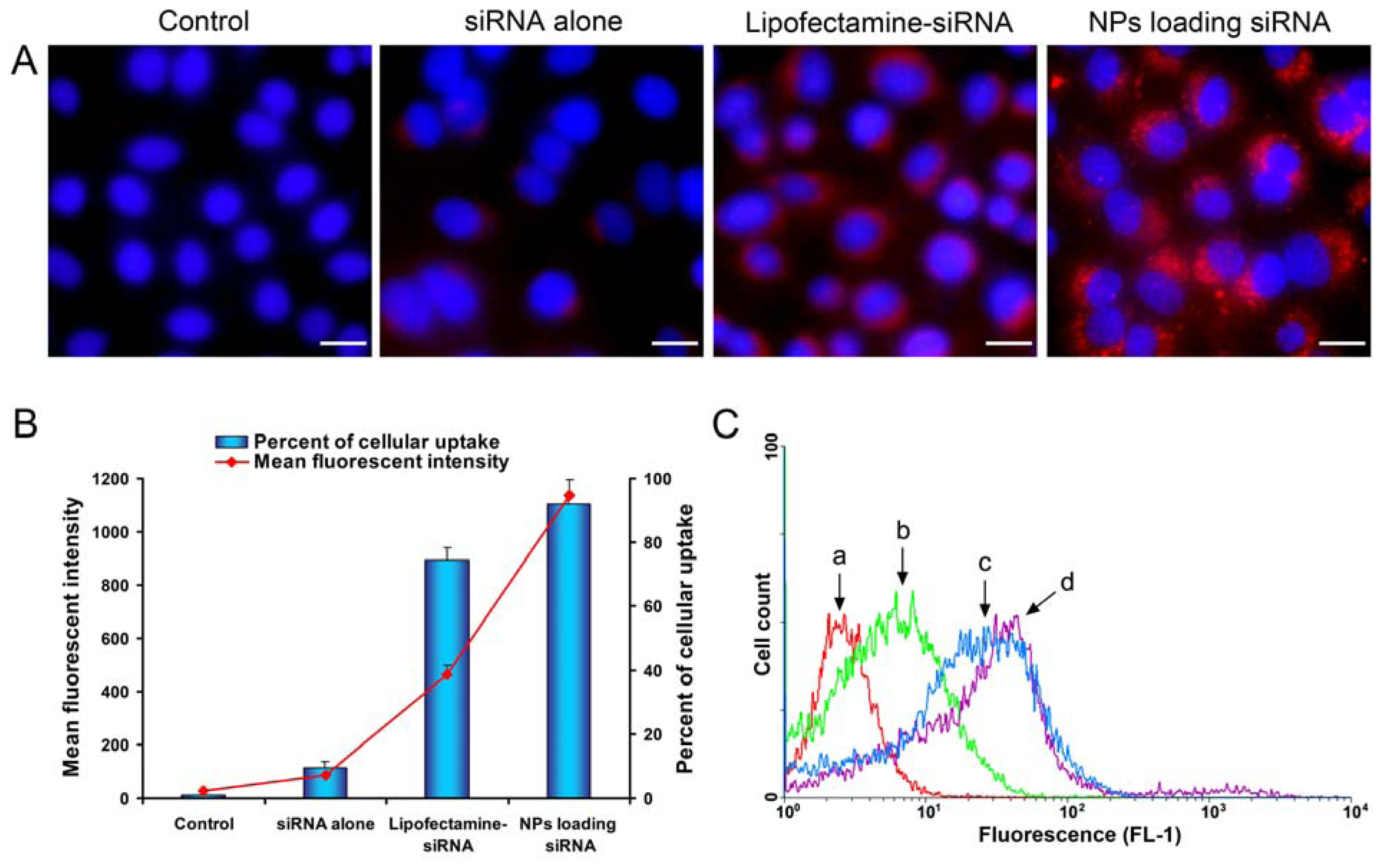
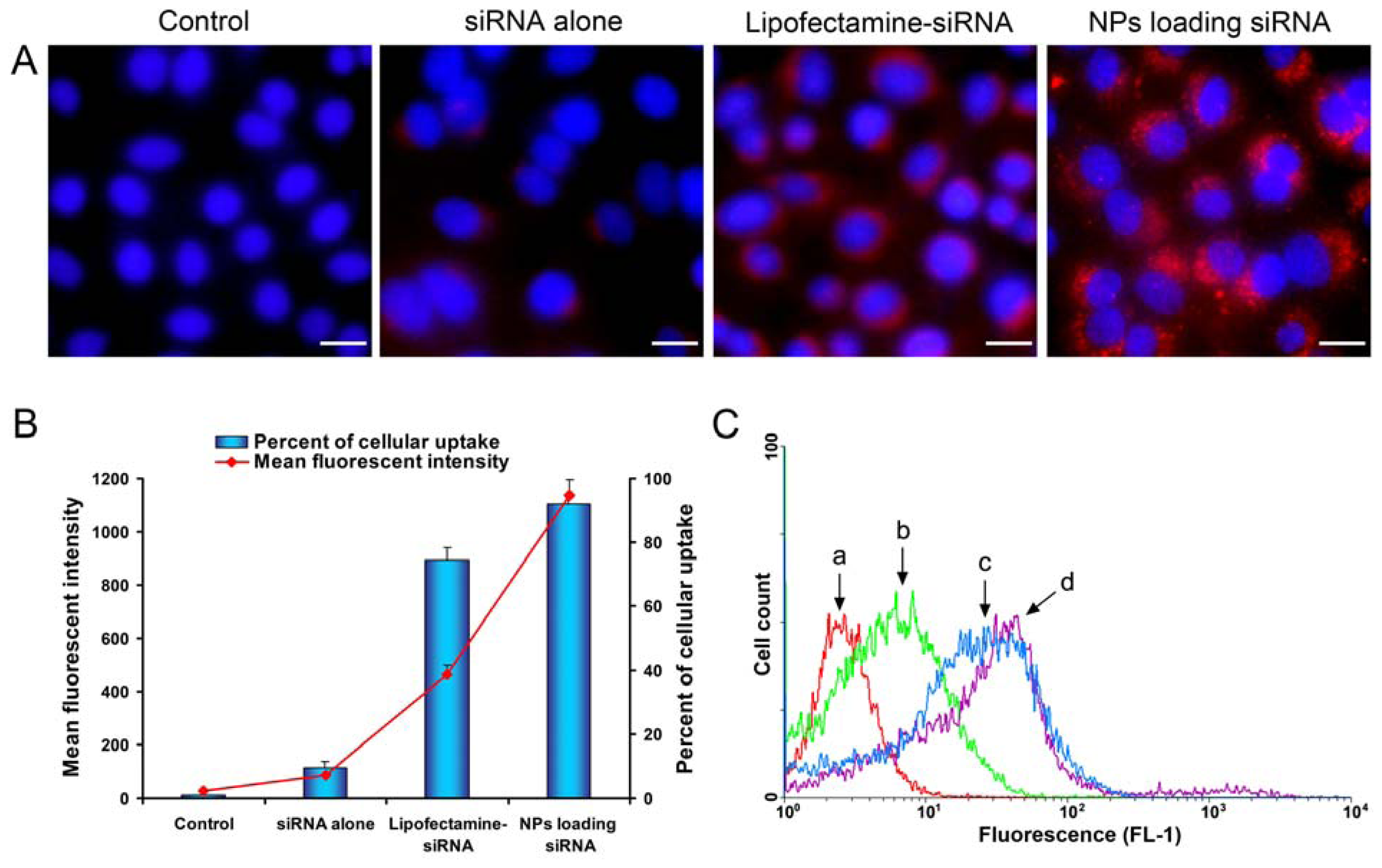
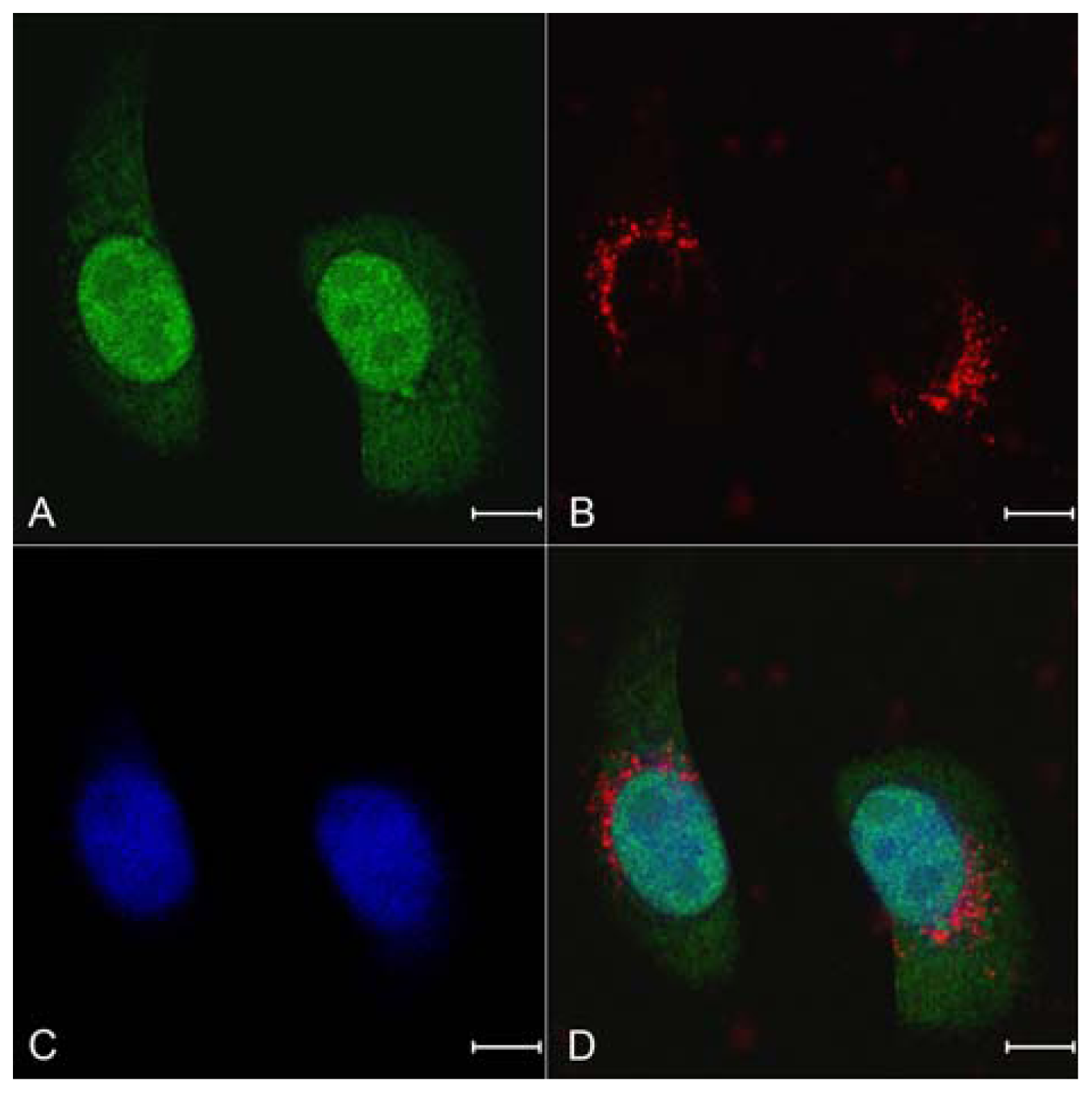
2.4. Celluar Uptake and Location of NPs Loading siRNA
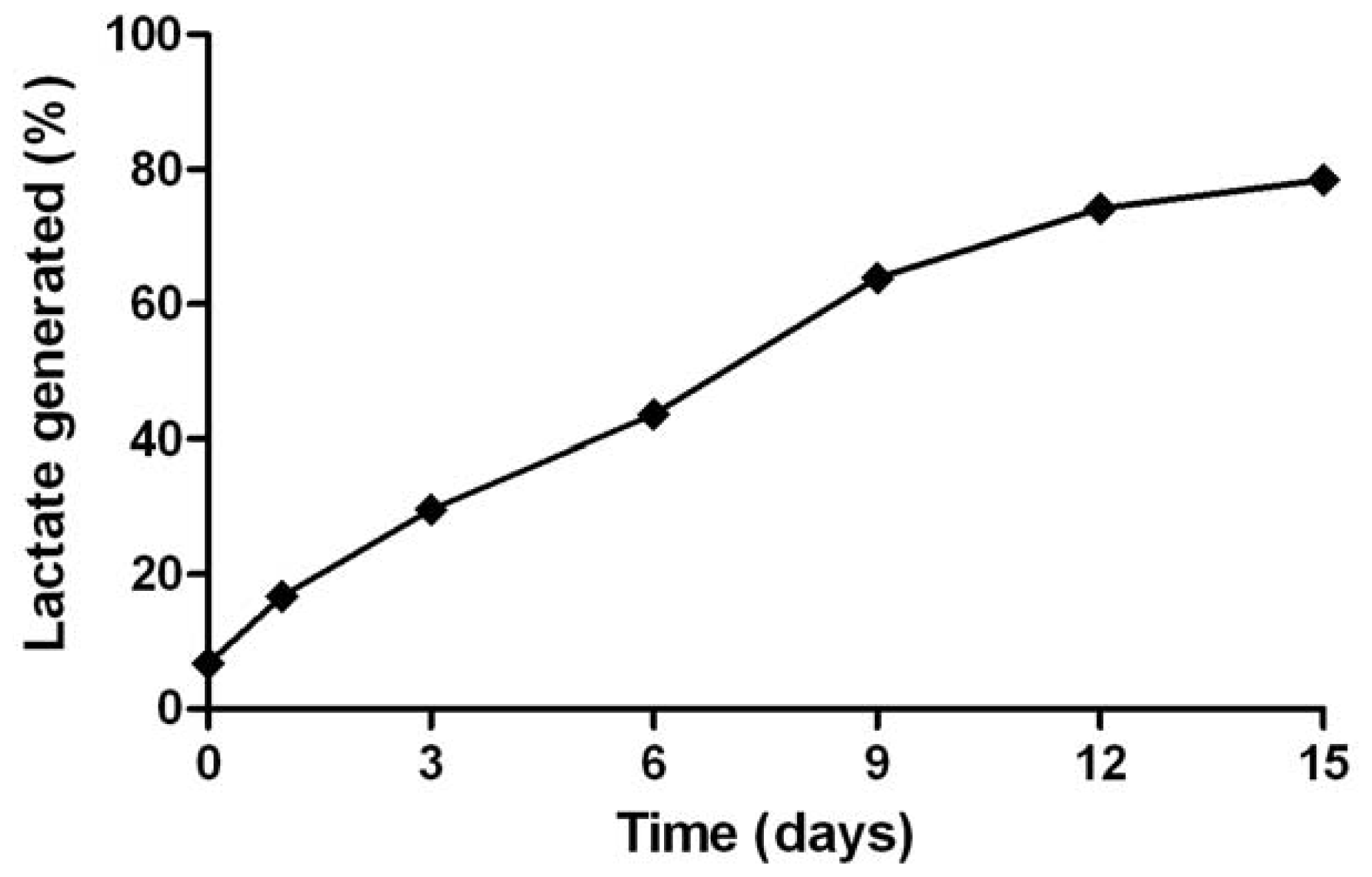
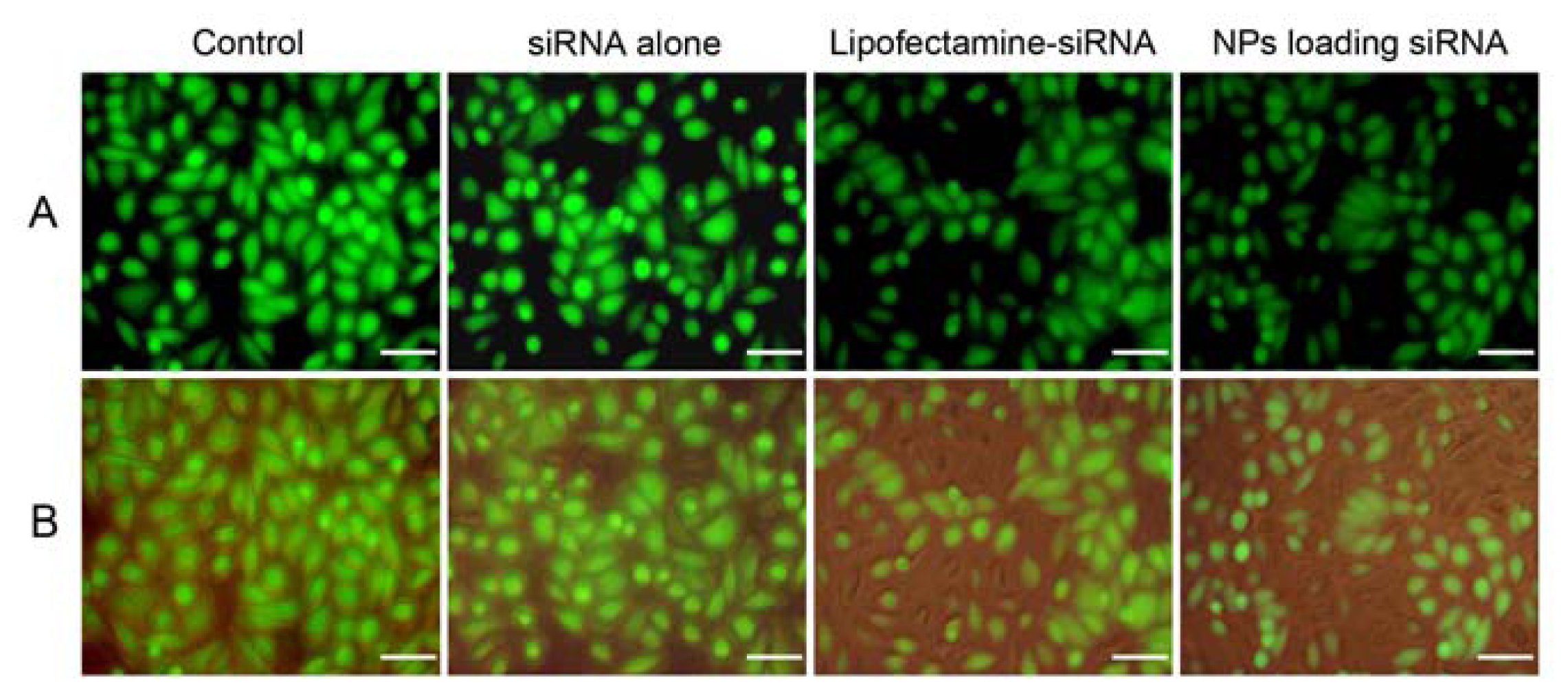
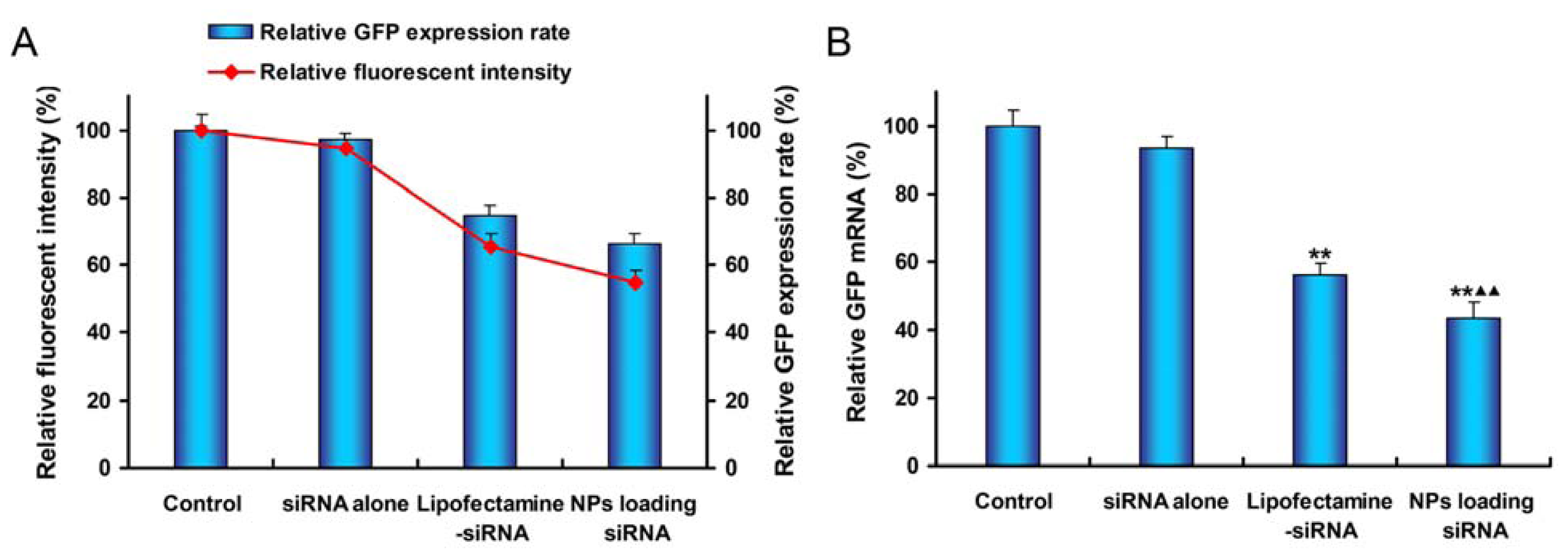
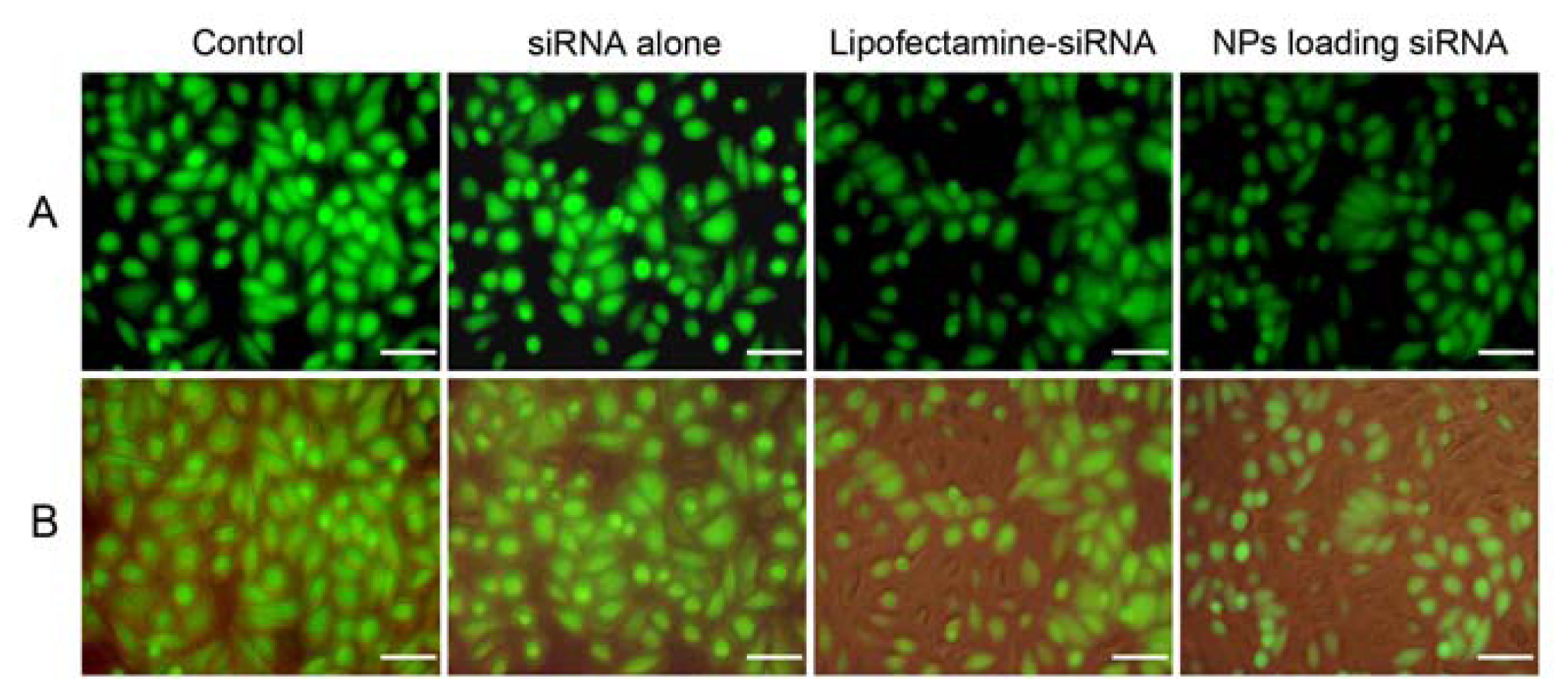
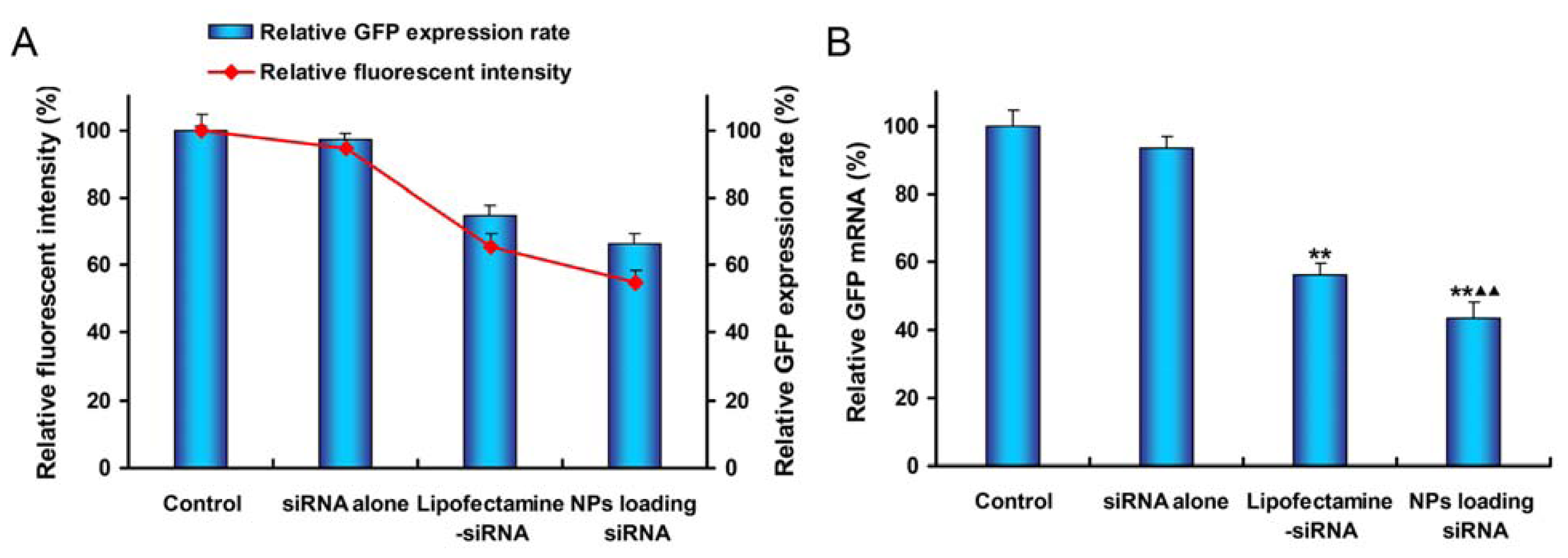
2.5. In Vitro Gene Silencing
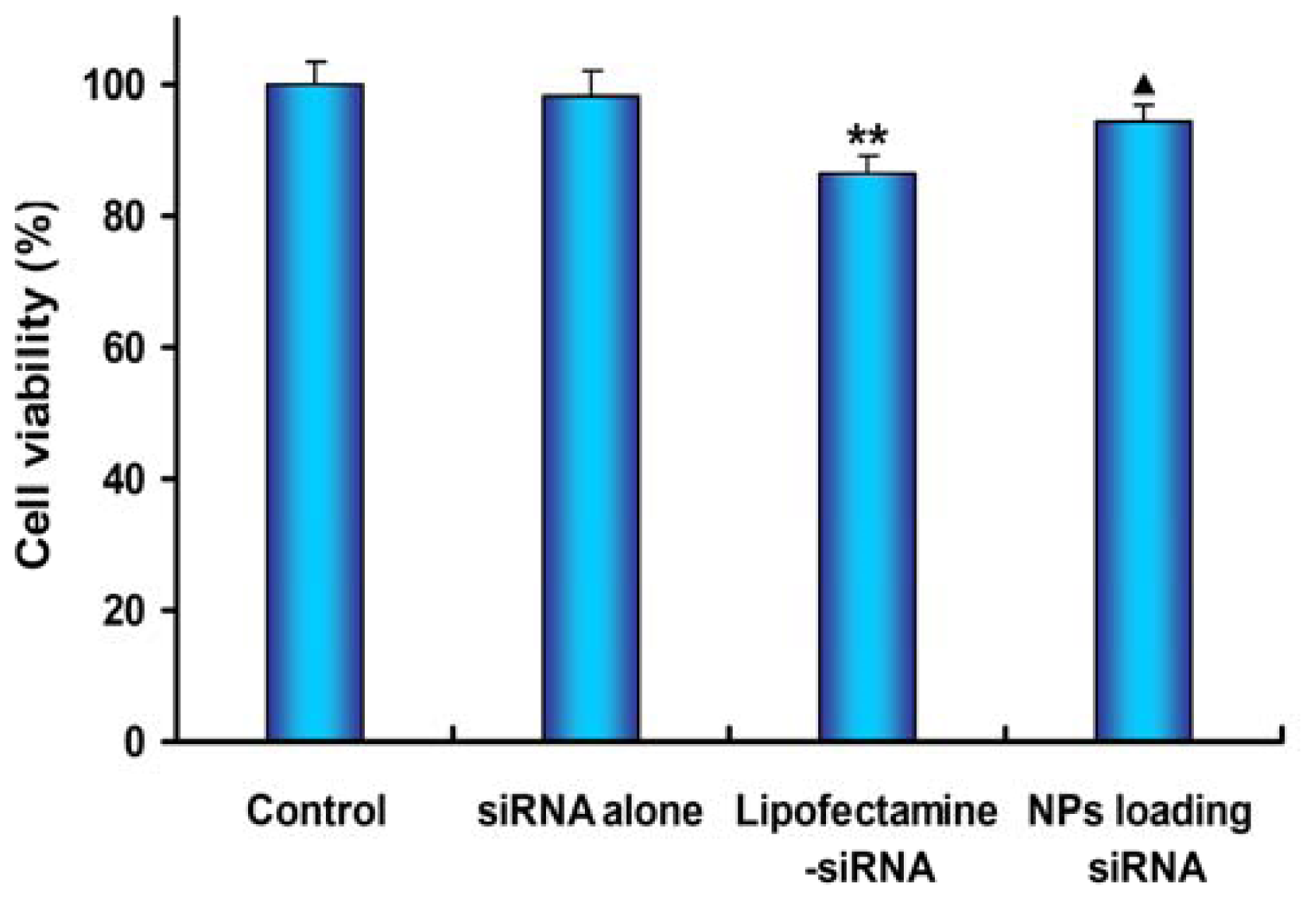
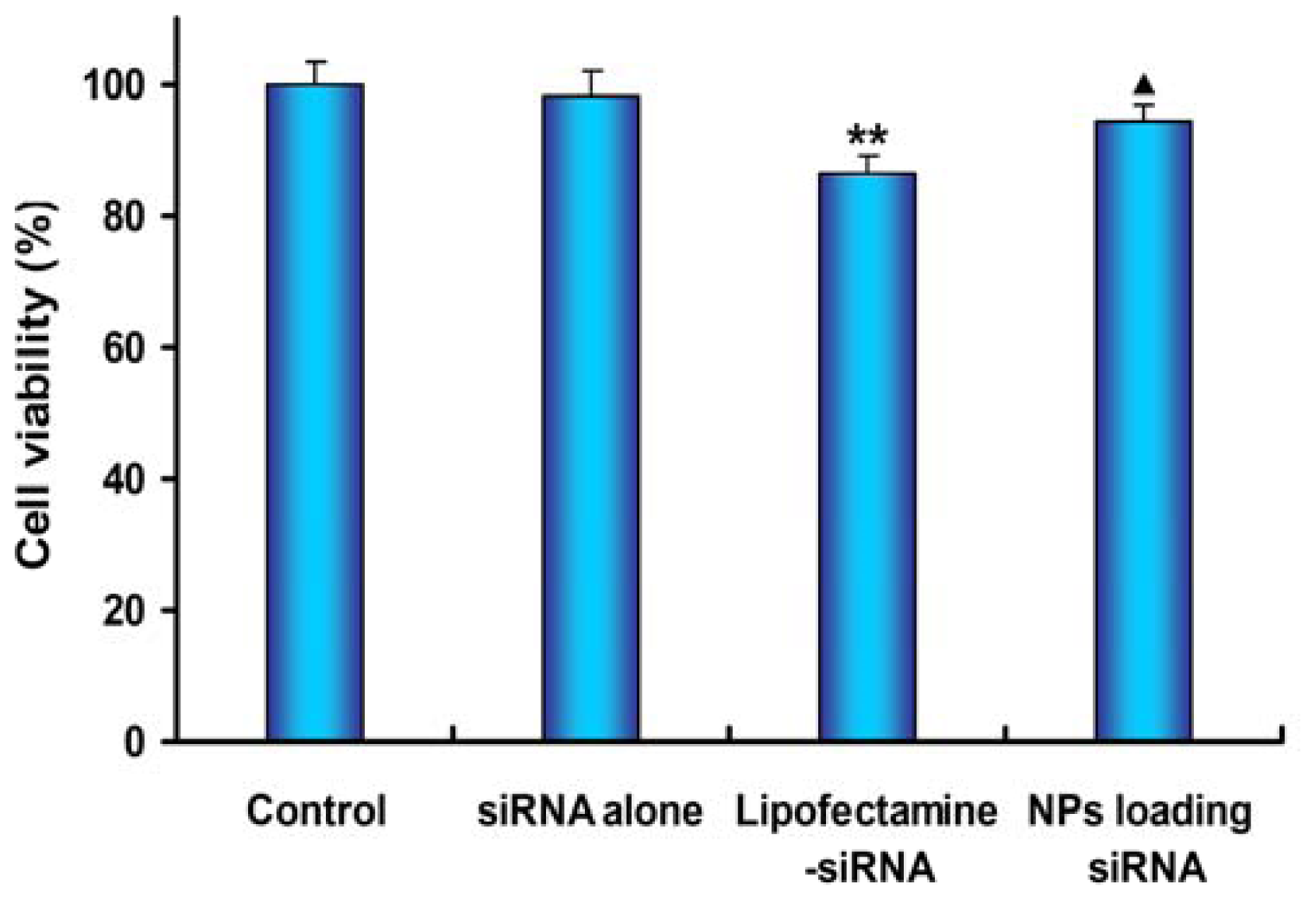
2.6. Cell Viability Assay
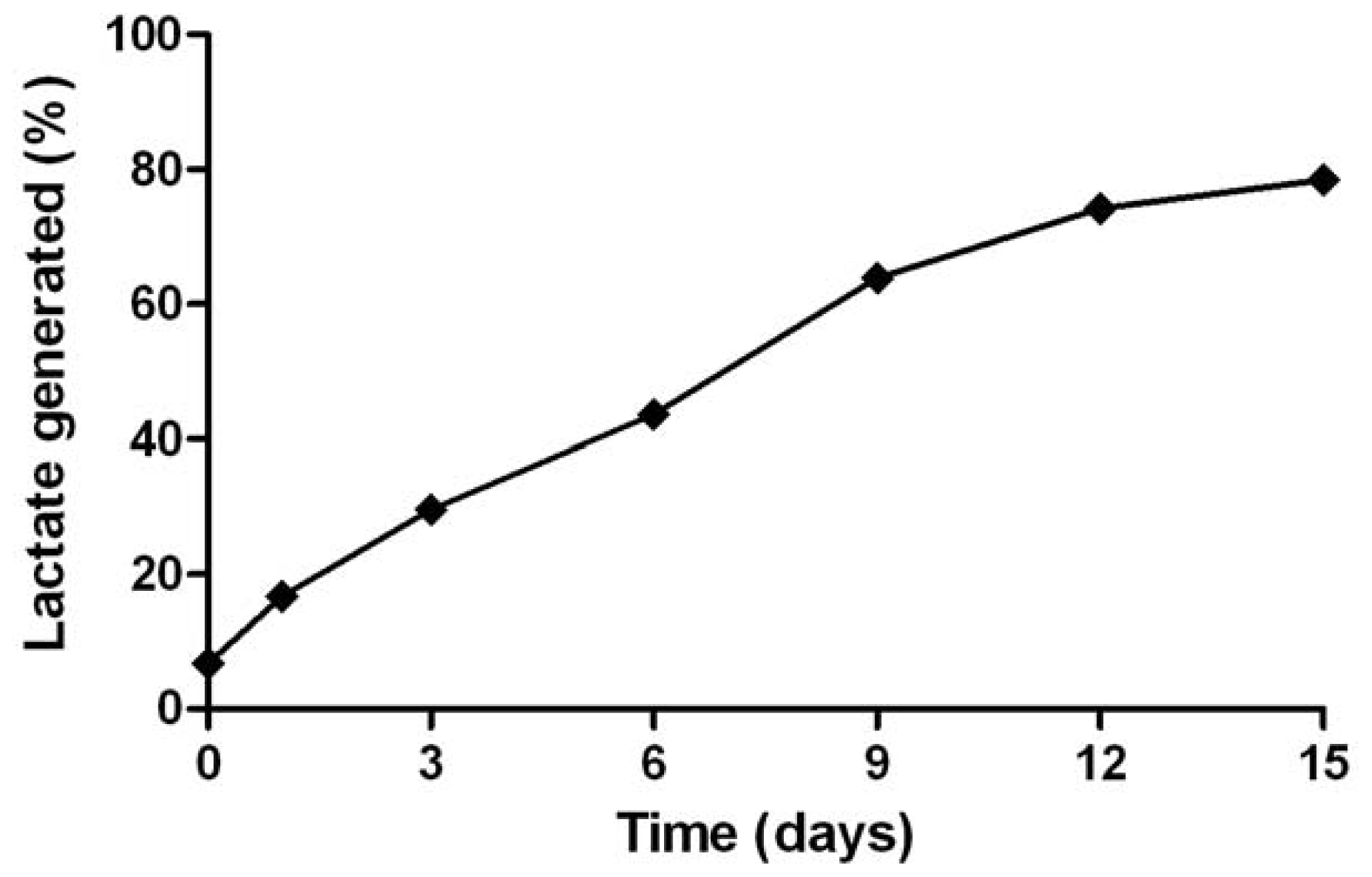
2.7. Discussion
3. Experimental Section
3.1. Materials
3.2. Preparation of mPEG -PLGA-PLL NPs Loading siRNA
3.3. Physicochemical Characterization of NPs Loading siRNA
3.4. Gel Retardation Assay
3.5. Cell Culture
3.6. Celluar Uptake Studies
3.7. Analysis of Gene Silencing
3.8. Cytotoxicity
3.9. Statistical Analysis
4. Conclusions
Acknowledgments
- Conflicts of Interest The authors declare no conflicts of interests related to this manuscript.
References
- Dorsett, Y.; Tuschl, T. siRNAs: Applications in functional genomics and potential as therapeutics. Nat. Rev. Drug Discov 2004, 3, 318–329. [Google Scholar]
- Novina, C.D.; Sharp, P.A. The RNAi revolution. Nature 2004, 430, 161–164. [Google Scholar]
- Dykxhoorn, D.M.; Lieberman, J. The silent revolution: RNA interference as basic biology, research tool, and therapeutic. Annu. Rev. Med 2005, 56, 401–423. [Google Scholar]
- Ryther, R.C.; Flynt, A.S.; Phillips, J.A.; Patton, J.G. siRNA therapeutics: Big potential from small RNAs. Gene Ther 2005, 12, 5–11. [Google Scholar]
- Nimesh, S.; Chandra, R. Polyethylenimine nanoparticles as an efficient in vitro siRNA delivery system. Eur. J. Pharm. Biopharm 2009, 73, 43–49. [Google Scholar]
- Crombez, L.; Charnet, A.; Morris, M.C.; Aldrian-Herrada, G.; Heitz, F.; Divita, G. A non-covalent peptide-based strategy for siRNA delivery. Biochem. Soc. Trans 2007, 35, 44–46. [Google Scholar]
- Hatakeyama, H.; Akita, H.; Ito, E.; Hayashi, Y.; Oishi, M.; Nagasaki, Y.; Danev, R.; Nagayama, K.; Kaji, N.; Kikuchi, H.; et al. Systemic delivery of siRNA to tumors using a lipid nanoparticle containing a tumor-specific cleavable PEG-lipid. Biomaterials 2011, 32, 4306–4316. [Google Scholar]
- Schiffelers, R.M.; Ansari, A.; Xu, J.; Zhou, Q.; Tang, Q.; Storm, G.; Molema, G.; Lu, P.Y.; Scaria, P.V.; Woodle, M.C. Cancer siRNA therapy by tumor selective delivery with ligand-targeted sterically stabilized nanoparticle. Nucleic Acids Res 2004, 32, e149. [Google Scholar]
- Hassani, Z.; Lemkine, G.F.; Erbacher, P.; Palmier, K.; Alfama, G.; Giovannangeli, C.; Behr, J.P.; Demeneix, B.A. Lipid-mediated siRNA delivery down-regulates exogenous gene expression in the mouse brain at picomolar levels. J. Gene Med 2005, 7, 198–207. [Google Scholar]
- Park, T.G.; Jeong, J.H.; Kim, S.W. Current status of polymeric gene delivery systems. Adv. Drug Deliv. Rev 2006, 58, 467–486. [Google Scholar]
- Lee, S.H.; Bae, K.H.; Kim, S.H.; Lee, K.R.; Park, T.G. Amine-functionalized gold nanoparticles as non-cytotoxic and efficient intracellular siRNA delivery carriers. Int. J. Pharm 2008, 364, 94–101. [Google Scholar]
- Prabha, S.; Zhou, W.Z.; Panyam, J.; Labhasetwar, V. Size-dependency of nanoparticle-mediated gene transfection: studies with fractionated nanoparticles. Int. J. Pharm 2002, 244, 105–115. [Google Scholar]
- Nafee, N.; Taetz, S.; Schneider, M.; Schaefer, U.F.; Lehr, C.M. Chitosan-coated PLGA nanoparticles for DNA/RNA delivery: effect of the formulation parameters on complexation and transfection of antisense oligonucleotides. Nanomedicine 2007, 3, 173–183. [Google Scholar]
- Vij, N.; Min, T.; Marasigan, R.; Belcher, C.N.; Mazur, S.; Ding, H.; Yong, K.T.; Roy, I. Development of PEGylated PLGA nanoparticle for controlled and sustained drug delivery in cystic fibrosis. J. Nanobiotechnol 2010, 8, 22. [Google Scholar]
- Prabha, S.; Labhasetwar, V. Critical determinants in PLGA/PLA nanoparticle-mediated gene expression. Pharm. Res 2004, 21, 354–364. [Google Scholar]
- Patil, Y.; Panyam, J. Polymeric nanoparticles for siRNA delivery and gene silencing. Int. J. Pharm 2009, 367, 195–203. [Google Scholar]
- Tahara, K.; Yamamoto, H.; Hirashima, N.; Kawashima, Y. Chitosan-modified poly(d,l-lactideco-glycolide) nanospheres for improving siRNA delivery and gene-silencing effects. Eur. J. Pharm. Biopharm 2010, 74, 421–426. [Google Scholar]
- Sun, X.; Zhang, N. Cationic polymer optimization for efficient gene delivery. Mini Rev. Med. Chem 2010, 10, 108–125. [Google Scholar]
- Patil, M.L.; Zhang, M.; Minko, T. Multifunctional triblock Nanocarrier (PAMAM-PEG-PLL) for the efficient intracellular siRNA delivery and gene silencing. ACS Nano 2011, 5, 1877–1887. [Google Scholar]
- Deng, C.; Chen, X.S.; Yu, H.J.; Sun, J.; Lu, T.C.; Jing, X.B. A biodegradable triblock copolymer poly(ethylene glycol)-b-poly(l-lactide)-b-poly(l-lysine): Synthesis, self-assembly, and RGD peptide modification. Polymer 2007, 48, 139–149. [Google Scholar]
- Fu, C.; Sun, X.L.; Liu, D.H.; Chen, Z.J.; Lu, Z.J.; Zhang, N. Biodegradable tri-block copolymer Poly(lactic acid)-poly(ethylene glycol)-poly(l-lysine)(PLA-PEG-PLL) as a non-viral vector to enhance gene transfection lower gene transfection efficiency. Int. J. Mol. Sci 2011, 12, 1371–1388. [Google Scholar]
- Wolff, J.A.; Rozema, D.B. Breaking the bonds: non-viral vectors become chemically dynamic. Mol. Ther 2008, 16, 8–15. [Google Scholar]
- Niven, R.; Zhang, Y.; Smith, J. Toward development of a non-viral gene therapeutic. Adv. Drug Deliv. Rev 1997, 26, 135–150. [Google Scholar]
- Pişkin, E.; Dinçer, S.; Türk, M. Gene delivery: Intelligent but just at the beginning. J. Biomater. Sci. Polym. Ed 2004, 15, 1181–1202. [Google Scholar]
- Zhang, C.; Tang, N.; Liu, X.; Liang, W.; Xu, W.; Torchilin, V.P. siRNA-containing liposomes modified with polyarginine effectively silence the targeted gene. J. Control. Release 2006, 112, 229–239. [Google Scholar]
- Ishida, T.; Harada, M.; Wang, X.Y.; Ichihara, M.; Irimura, K.; Kiwada, H. Accelerated blood clearance of PEGylated liposomes following preceding liposome injection: Effects of lipid dose and PEG surface-density and chain length of the first-dose liposomes. J. Control. Release 2005, 105, 305–317. [Google Scholar]
- Kim, T.H.; Kim, S.I.; Akaike, T.; Cho, C.S. Synergistic effect of poly(ethylenimine) on the transfection efficiency of galactosylated chitosan/DNA complexes. J. Control. Release 2005, 105, 354–366. [Google Scholar]
- Park, M.R.; Han, K.O.; Han, I.K.; Cho, M.H.; Nah, J.W.; Choi, Y.J.; Cho, C.S. Degradable polyethylenimine-alt-poly(ethylene glycol) copolymers as novel gene carriers. J. Control. Release 2005, 105, 367–380. [Google Scholar]
- Köping-Höggård, M.; Vårum, K.M.; Issa, M.; Danielsen, S.; Christensen, B.E.; Stokke, B.T.; Artursson, P. Improved chitosan-mediated gene delivery based on easily dissociated chitosan polyplexes of highly defined chitosan oligomers. Gene Ther 2004, 11, 1441–1452. [Google Scholar]
- Itaka, K.; Ishii, T.; Hasegawa, Y.; Kataoka, K. Biodegradable polyamino acid-based polycations as safe and effective gene carrier minimizing cumulative toxicity. Biomaterials 2010, 31, 3707–3714. [Google Scholar]
- Jere, D.; Jiang, H.L.; Arote, R.; Kim, Y.K.; Choi, Y.J.; Cho, M.H.; Akaike, T.; Cho, C.S. Degradable polyethylenimines as DNA and small interfering RNA carriers. Expert Opin. Drug Deliv 2009, 6, 827–834. [Google Scholar]
- Nounou, M.I.; Emmanouil, K.; Chung, S.; Pham, T.; Lu, Z.; Bikram, M. Novel reducible linear l-lysine-modified copolymers as efficient nonviral vectors. J. Control Release 2010, 143, 326–334. [Google Scholar]
- Luten, J.; van Nostrum, C.F.; De Smedt, S.C.; Hennink, W.E. Biodegradable polymers as non-viral carriers for plasmid DNA delivery. J. Control. Release 2008, 126, 97–110. [Google Scholar]
- Cun, D.; Foged, C.; Yang, M.; Frøkjaer, S.; Nielsen, H.M. Preparation and characterization of poly(dl-lactide-co-glycolide) nanoparticles for siRNA delivery. Int. J. Pharm 2010, 390, 70–75. [Google Scholar]
- Morille, M.; Passirani, C.; Vonarbourg, A.; Clavreul, A.; Benoit, J.P. Progress in developing cationic vectors for non-viral systemic gene therapy against cancer. Biomaterials 2008, 29, 3477–3496. [Google Scholar] [Green Version]
- Zhang, M.Z.; Kataoka, K. Nano-structured composites based on calcium phosphate for cellular delivery of therapeutic and diagnostic agents. Nano Today 2009, 4, 508–517. [Google Scholar]
- Qi, X.L.; Chen, X.Y.; Sun, Y.; Ma, Z.C.; Guo, X.J.; Lu, W.; Duan, Y.R. Cytotoxicity and cellular uptake evaluation of mitoxantrone-loaded poly (lactic acid-co-lysine) arginine–glycine–aspartic acid nanoparticles. J. Appl. Polym. Sci 2011, 119, 1011–1015. [Google Scholar]
- Deng, C.; Tian, H.; Zhang, P.; Sun, J.; Chen, X.; Jing, X. Synthesis and characterization of poly(ethylene glycol)-b-poly (l-lactide)-b-poly(l-glutamic acid) triblock copolymer. Biomacromolecules 2006, 7, 590–596. [Google Scholar]
- Avgoustakis, K.; Beletsi, A.; Panagi, Z.; Klepetsanis, P.; Karydas, A.G.; Ithakissios, D.S. PLGAmPEG nanoparticles of cisplatin: in vitro nanoparticle degradation, in vitro drug release and in vivo drug residence in blood properties. J. Control. Release 2002, 79, 123–135. [Google Scholar]
- Murata, N.; Takashima, Y.; Toyoshima, K.; Yamamoto, M.; Okada, H. Anti-tumor effects of anti-VEGF siRNA encapsulated with PLGA microspheres in mice. J. Control. Release 2008, 126, 246–254. [Google Scholar]

© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Du, J.; Sun, Y.; Shi, Q.-S.; Liu, P.-F.; Zhu, M.-J.; Wang, C.-H.; Du, L.-F.; Duan, Y.-R. Biodegradable Nanoparticles of mPEG-PLGA-PLL Triblock Copolymers as Novel Non-Viral Vectors for Improving siRNA Delivery and Gene Silencing. Int. J. Mol. Sci. 2012, 13, 516-533. https://doi.org/10.3390/ijms13010516
Du J, Sun Y, Shi Q-S, Liu P-F, Zhu M-J, Wang C-H, Du L-F, Duan Y-R. Biodegradable Nanoparticles of mPEG-PLGA-PLL Triblock Copolymers as Novel Non-Viral Vectors for Improving siRNA Delivery and Gene Silencing. International Journal of Molecular Sciences. 2012; 13(1):516-533. https://doi.org/10.3390/ijms13010516
Chicago/Turabian StyleDu, Jing, Ying Sun, Qiu-Sheng Shi, Pei-Feng Liu, Ming-Jie Zhu, Chun-Hui Wang, Lian-Fang Du, and You-Rong Duan. 2012. "Biodegradable Nanoparticles of mPEG-PLGA-PLL Triblock Copolymers as Novel Non-Viral Vectors for Improving siRNA Delivery and Gene Silencing" International Journal of Molecular Sciences 13, no. 1: 516-533. https://doi.org/10.3390/ijms13010516
APA StyleDu, J., Sun, Y., Shi, Q.-S., Liu, P.-F., Zhu, M.-J., Wang, C.-H., Du, L.-F., & Duan, Y.-R. (2012). Biodegradable Nanoparticles of mPEG-PLGA-PLL Triblock Copolymers as Novel Non-Viral Vectors for Improving siRNA Delivery and Gene Silencing. International Journal of Molecular Sciences, 13(1), 516-533. https://doi.org/10.3390/ijms13010516