Human Gene Control by Vital Oncogenes: Revisiting a Theoretical Model and Its Implications for Targeted Cancer Therapy

{kind=link}
{kind=link}
{kind=link}
Abstract
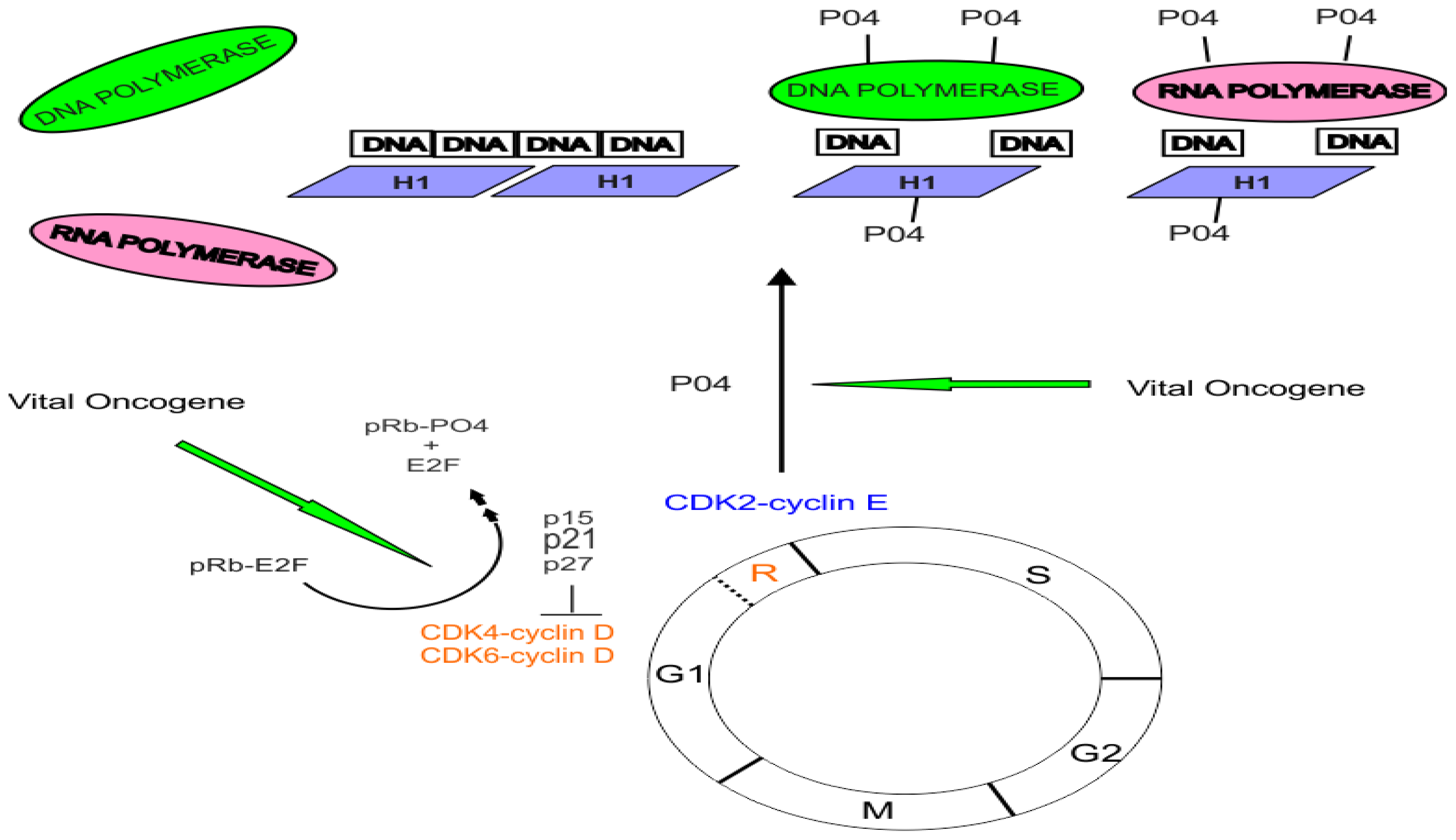
:1. Introduction
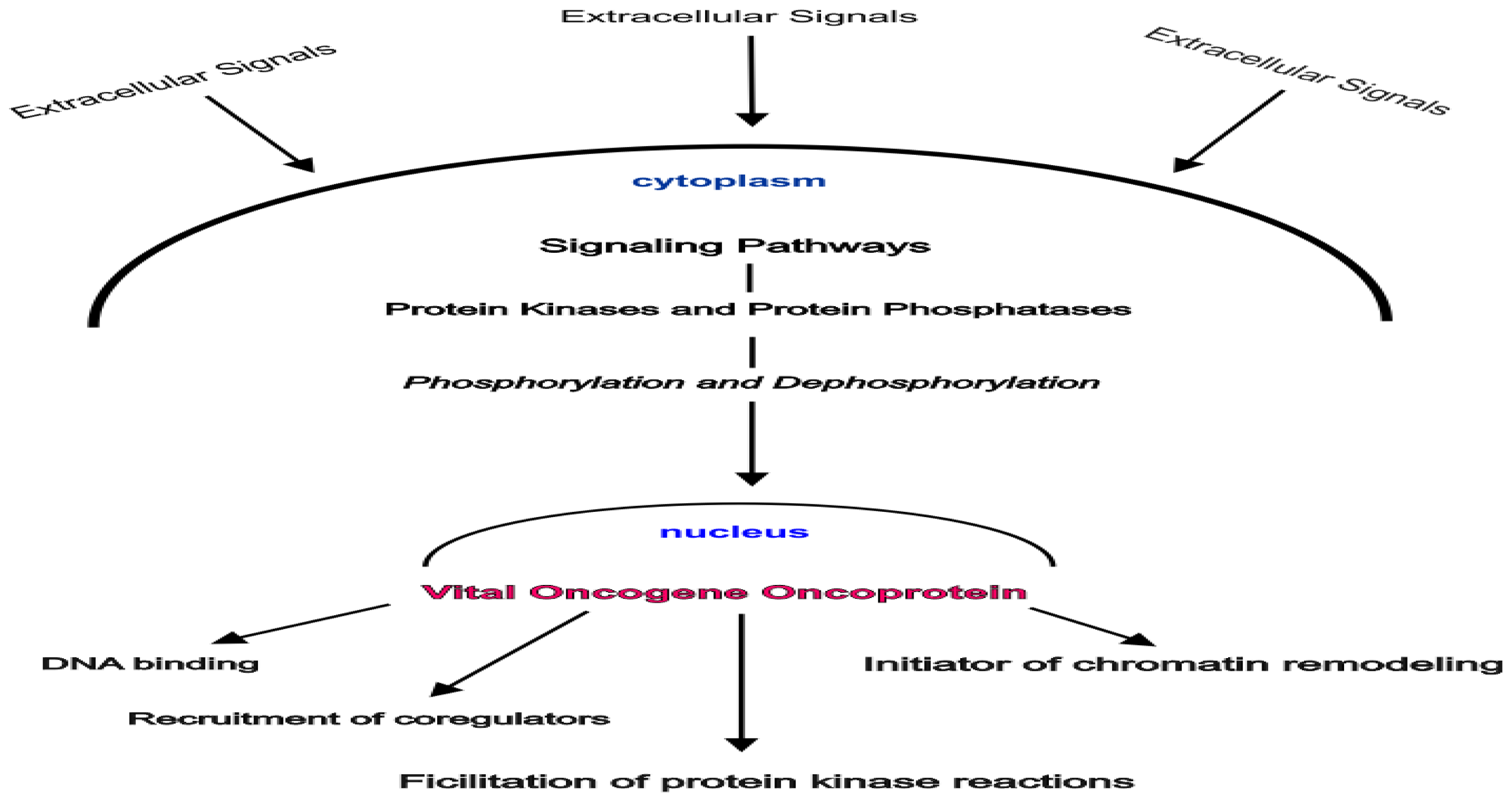
2. Gene Control by Phosphoproteins: A Basic Theory
- Nuclear proto-oncogene protein kinases are activated by phosphorylation.
- These phosphorylated activated protein kinases bind to recognition sites in the major groove of the DNA helix as it winds around the nucleosome core histones.
- A series of phosphorylation reactions mediated by these nuclear phosphoproteins and facilitated by histone H1 phosphorylation result in the displacement of histone H1 away from the site of gene activation eventually allowing access by the transcription initiation complex.
- The exposed DNA sequences are essentially promoter sites.
- The phosphorylation reaction cascade includes recruitment and phosphorylation of either DNA polymerase or RNA polymerase.
- The intact histone octamer that makes up the nucleosome core complex functions as a scaffold during gene activity. The model assigned a central role to nuclear phosphoproteins.
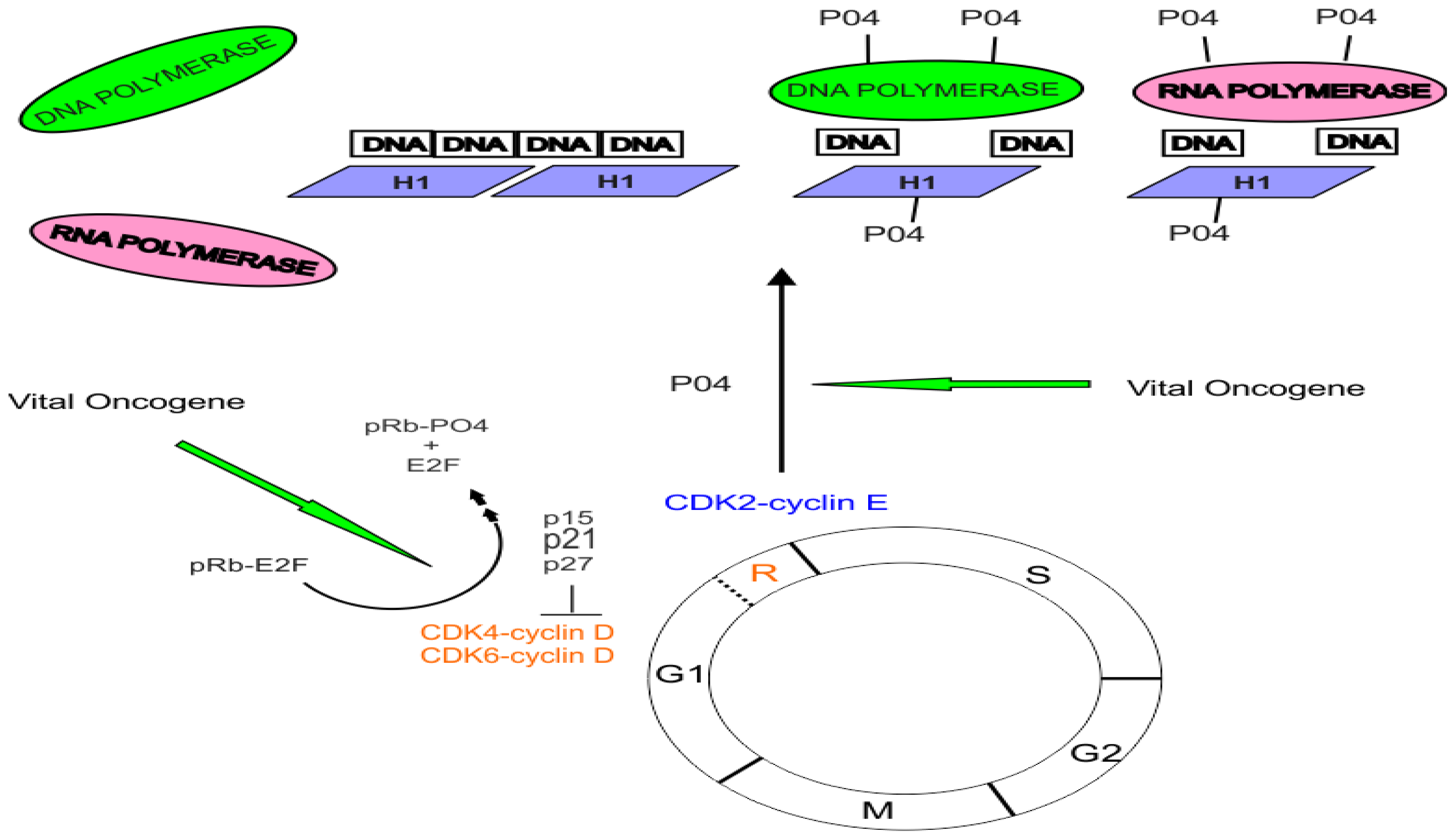
3. The Role of Cyclin-Dependent Kinases and the pRB Protein in Cell Cycle Control
4. Genomic Structure and Epigenetic Regulation
5. c-Myc a Prototype Vital Oncogene
6. Conclusion
Supplementary Material
ijms-13-00316-s001.pdfReferences
- Britten, R.J.; Davidson, E.H. Gene regulation for higher cells: A theory. Science 1969, 165, 349–357. [Google Scholar]
- Venters, B.J.; Pugh, B.F. How eukaryotic genes are transcribed. Crit. Rev. Biochem. Mol. Biol 2009, 44, 117–141. [Google Scholar]
- Rous, P. Transmission of a malignant new growth by means of a cell-free filtrate. J. Am. Med. Assoc 1983, 250, 1445–1449. [Google Scholar]
- Bishop, J.M. Cellular oncogenes and retroviruses. Annu. Rev. Biochem 1983, 52, 301–354. [Google Scholar]
- Panganiban, A.T. Retroviral DNA integration. Cell 1985, 42, 5–6. [Google Scholar]
- Bishop, J.M. Viral oncogenes. Cell 1985, 42, 23–38. [Google Scholar]
- Marx, J.L. Tumor viruses and the kinase connection. Science 1981, 211, 1336–1338. [Google Scholar]
- Erikson, R.I.; Collett, M.S.; Erikson, E.; Purchio, A.F.; Brugge, J.S. Protein phosphorylation mediated by partially purified avian sarcoma virus transforming-gene product. Cold Spring Harbor Symp. Quant. Biol 1980, 44, 907–917. [Google Scholar]
- Rubin, C.S.; Rosen, O.M. Protein phosphorylation. Annu. Rev. Biochem 1975, 44, 831–887. [Google Scholar]
- Krebs, E.G.; Beavo, J.A. Phosphorylation-dephosphorylation of enzymes. Annu. Rev. Biochem 1979, 48, 923–959. [Google Scholar]
- Barford, D.; Das, A.K.; Egloff, M.P. The structure and mechanism of protein phosphatases: Insights into catalysis and regulation. Annu. Rev. Biophys. Biomol. Struct 1998, 27, 133–164. [Google Scholar]
- Willis, R.E. Gene control by phosphoproteins: A theoretical model for eukaryotic DNA regulation. Med. Hypotheses 1984, 13, 407–437. [Google Scholar]
- Hunter, T.; Karin, M. The regulation of transcription by phosphorylation. Cell 1992, 70, 375–387. [Google Scholar]
- Sahyoun, N.; LeVine, H.; McConnell, R.; Bronson, D.; Cuatrecasas, P. A specific phosphoprotein phosphatase acts on histone H1 phosphorylated by protein kinase C. Proc. Natl. Acad. Sci. USA 1983, 80, 6760–6764. [Google Scholar]
- Cho, H.; Kim, T.K.; Mancebo, H.; Lane, W.S.; Flores, O.; Reinberg, D. A protein phosphatase functions to recycle RNA polymerase II. Genes Dev 1999, 13, 1540–1552. [Google Scholar]
- Cheng, A.; Ross, K.E.; Kaldis, P.; Solomon, M.J. Dephosphorylation of cyclin-dependent kinases by type 2C protein phosphatases. Genes Dev 1999, 13, 2946–2957. [Google Scholar]
- Haspel, R.L.; Darnell, J.E. A nuclear protein tyrosine phosphatase is required for the inactivation of Stat1. Proc. Natl. Acad. Sci. USA 1999, 96, 10188–10193. [Google Scholar]
- Camps, M.; Nichols, A.; Arkinstall, S. Dual specificity phosphatases: A gene family for control of MAP kinase functions. FASEB J 2000, 14, 6–16. [Google Scholar]
- Janssens, V.; Goris, J. Protein phosphatase 2A: A highly regulated family of serine/threonine phosphatases implicated in cell growth and signaling. Biochem. J 2001, 353, 417–439. [Google Scholar]
- Ostman, A.; Hellberg, C.; Bohmer, F.D. Protein-tyrosine phosphatases and cancer. Nat. Rev. Cancer 2006, 6, 307–320. [Google Scholar]
- Owens, D.M.; Keyse, S.M. Differential regulation of MAP kinase signaling by dual-specificity protein phosphatases. Oncogene 2007, 26, 3203–3213. [Google Scholar]
- Chitale, D.; Gong, Y.; Taylor, B.S.; Broderick, S.; Brennan, C.; Somwar, R.; Golas, B.; Wang, L.; Motoi, N.; Szoke, J. An integrated genomic analysis of lung cancer reveals loss of DUSP4 in EGFR-mutant tumors. Oncogene 2009, 28, 2773–2783. [Google Scholar]
- Novakofski, J. Role of proto-oncogenes in normal growth and development. J. Anim Sci 1991, 69, 56–73. [Google Scholar]
- Klampfer, L. Signal transducers and activators of transcription (STATs): Novel targets of chemopreventive and chemotherapeutic drugs. Curr. Cancer Drug Targets 2006, 6, 107–121. [Google Scholar]
- Karin, M.; Hunter, T. Transcriptional control by protein phosphorylation: Signal transmission from the cell surface to the nucleus. Curr. Biol 1995, 5, 747–757. [Google Scholar]
- Futreal, P.A.; Coin, L.; Marshall, M.; Down, T.; Hubbard, T.; Wooster, R.; Rahman, N.; Stratton, M.R. A census of human cancer genes. Nat. Rev. Cancer 2004, 4, 177–183. [Google Scholar]
- Collins, K.; Jacks, T.; Pavletich, N.P. The cell cycle and cancer. Proc. Natl. Acad. Sci. USA 1997, 94, 2776–2778. [Google Scholar]
- Sherr, C.J. Cancer cell cycles. Science 1996, 274, 1672–1677. [Google Scholar]
- Park, M.T.; Lee, S.J. Cell cycle and cancer. J. Biochem. Mol. Biol 2003, 36, 60–65. [Google Scholar]
- Weinstein, I.B.; Joe, A.K. Mechanisms of disease: Oncogene addiction—A rationale for molecular targeting in cancer therapy. Nat. Clin. Pract. Oncol 2006, 3, 448–457. [Google Scholar]
- Weinstein, I.B.; Joe, A. Oncogene addiction. Cancer Res 2008, 68, 3077–3080. [Google Scholar]
- Sharma, S.V.; Settleman, J. Oncogene addiction: Setting the stage for molecularly targeted cancer therapy. Genes Dev 2007, 21, 3214–3231. [Google Scholar]
- Guccione, E.; Martinato, F.; Finocchiaro, G.; Luzi, L.; Tizzoni, L.; Dall’ Olio, V.; Zardo, G.; Nervi, C.; Bernard, L.; Amati, B. Myc-binding-site recognition in the human genome is determined by chromatin context. Nat. Cell Biol 2006, 8, 764–770. [Google Scholar]
- Worcel, A.; Benyajati, C. Higher order coiling of DNA in chromatin. Cell 1977, 12, 83–100. [Google Scholar]
- Felsenfeld, G. Chromatin. Nature 1978, 271, 115–122. [Google Scholar]
- Cartwright, I.L.; Abmayr, S.M.; Fleischmann, G.; Lowenhaupt, K.; Elgin, S.C.; Keene, M.A.; Howard, G.C. Chromatin structure and gene activity: The role of nonhistone chromosomal proteins. CRC Crit. Rev. Biochem 1982, 13, 1–86. [Google Scholar]
- Thoma, F.; Koller, T. Unravelled nucleosomes, nucleosome beads and higher order structures of chromatin: Influence of non-histone components and histone H1. J. Mol. Biol 1981, 149, 709–373. [Google Scholar]
- Thoma, F.; Koller, T. Influence of histone H1 on chromatin structure. Cell 1977, 12, 101–107. [Google Scholar]
- Berger, S.L. Histone modifications in transcriptional regulation. Curr. Opin. Genet. Dev 2002, 12, 142–148. [Google Scholar]
- Berger, S.L. The complex language of chromatin regulation during transcription. Nature 2007, 447, 407–412. [Google Scholar]
- Dou, Y.; Mizzen, C.A.; Abrams, M.; Allis, C.D.; Gorovsky, M.A. Phosphorylation of linker histone H1 regulates gene expression in vivo by mimicking H1 removal. Mol. Cell 1999, 4, 641–347. [Google Scholar]
- Roque, A.; Ponte, I.; Arrondo, J.L.; Suau, P. Phosphorylation of the carboxy-terminal domain of histone H1: Effects on secondary structure and DNA condensation. Nucleic Acids Res 2008, 36, 4719–4726. [Google Scholar]
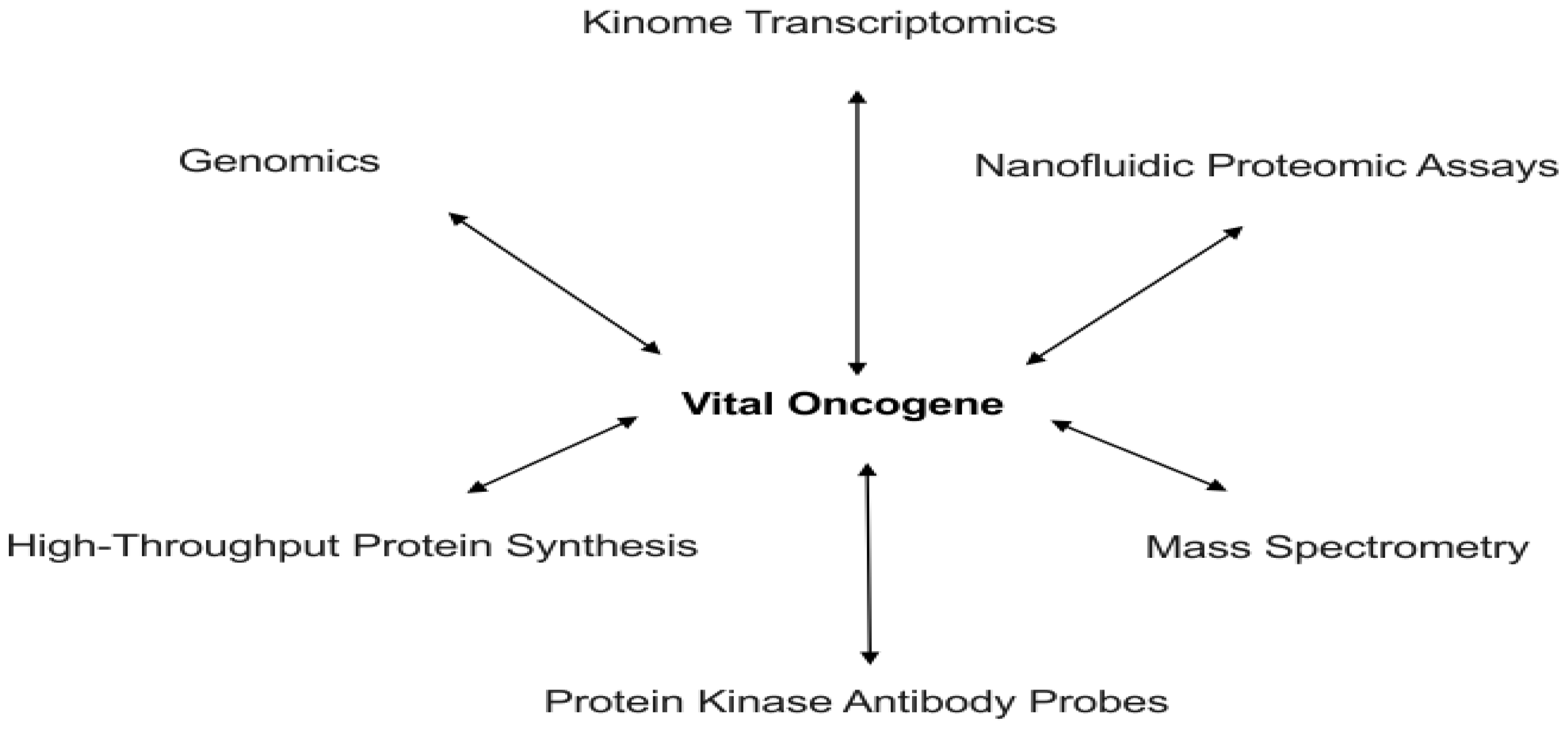
- Baak, J.P.; Janssen, E.A.; Soreide, K.; Heikkilae, R. Genomics and proteomics—The way forward. Ann. Oncol 2005, 16, ii30–ii44. [Google Scholar]
- Duggan, D.J.; Bittner, M.; Chen, Y.; Meltzer, P.; Trent, J.M. Expression profiling using cDNA microarrays. Nat. Genet 1999, 21, 10–14. [Google Scholar]
- Trevino, V.; Falciani, F.; Barrera-Saldana, H.A. DNA microarrays: A powerful genomic tool for biomedical and clinical research. Mol. Med 2007, 13, 527–541. [Google Scholar]
- Schuldiner, O.; Benvenisty, N. A DNA microarray screen for genes involved in c-MYC and N-MYC oncogenesis in human tumors. Oncogene 2001, 20, 4984–4994. [Google Scholar]
- Tao, S.C.; Chen, C.S.; Zhu, H. Applications of protein microarray technology. Comb. Chem. High Througput Screen 2007, 10, 706–718. [Google Scholar]
- Amit, I.; Citri, A.; Shay, T.; Lu, Y.; Katz, M.; Zhang, F.; Tarcic, G.; Siwak, D.; Lahad, J.; Jacob-Hirsch, J. A module of negative feedback regulators defines growth factor signaling. Nat. Genet 2007, 39, 503–512. [Google Scholar]
- Afjehi-Sadat, L.; Engidawork, E.; Felizardo-Cabatic, M.; Slavc, I.; Lubec, G. Proteomic profiling of signaling proteins in ten different tumor cell lines. Cancer Genomics 2004, 1, 427–454. [Google Scholar]
- Manning, G.; Plowman, G.D.; Hunter, T.; Sudarsanam, S. Evolution of protein kinase signaling from yeast to man. Trends Biochem. Sci 2002, 27, 514–520. [Google Scholar]
- Hubbard, M.J.; Cohen, P. On target with a new mechanism for the regulation of protein phosphorylation. Trends Biochem. Sci 1993, 18, 172–177. [Google Scholar]
- Mann, M.; Ong, S.; Gronborg, M.; Steen, H.; Jensen, O.; Pandey, A. Analysis of protein phosphorylation using mass spectrometry: Deciphering the phosphoproteome. Trends Biotechnol 2002, 20, 261–268. [Google Scholar]
- Yu, L.-R.; Issaq, H.J.; Veenstra, T.D. Phosphoproteomics for the discovery of kinases as cancer biomarkers and drug targets. Proteomics 2007, 1, 1042–1057. [Google Scholar]
- Brennan, D.J.; O’Connor, D.P.; Rexhepaj, E.; Ponten, F.; Gallagher, W.M. Antibody-based proteomics: Fast-tracking molecular diagnostics in oncology. Nat. Rev. Cancer 2010, 10, 605–617. [Google Scholar]
- Willis, R.E. Hypothesis: Is viral transformation mediated by alterations in chromosomal proteins? Med. Hypotheses 1982, 9, 517–528. [Google Scholar]
- Whitmarsh, A.J.; Davis, R.J. Regulation of transcription factor function by phosphorylation. Cell. Mol. Life Sci 2000, 57, 1172–1183. [Google Scholar]
- Barberis, A.; Petrascheck, M. Transcription Activation in Eukaryotic Cells; John Wiley & Sons, Ltd: Hoboken, NJ, USA, eLS, 2001. [Google Scholar]
- Jones, P.A.; Baylin, S.B. The fundamental role of epigenetic events in cancer. Nat. Rev. Genet 2002, 3, 415–428. [Google Scholar]
- Choi, J.K.; Howe, L.J. Histone acetylation: Truth of consequences? Biochem. Cell Biol 2009, 87, 139–150. [Google Scholar]
- Chadee, D.N.; Taylor, W.R.; Hurta, R.A.; Allis, C.D.; Wright, J.A.; Davie, J.R. Increased phosphorylation of histone H1 in mouse fibroblasts transformed with oncogenes or constitutively active mitogen-activated protein kinase kinase. J. Biol. Chem 1995, 270, 20098–20105. [Google Scholar]
- Taylor, W.R.; Chadee, D.N.; Allis, C.D.; Wright, J.A.; Davie, J.R. Fibroblasts transformed by combinations of ras, myc and mutant p53 exhibit increased phosphorylation of histone H1 that is independent of metastatic potential. FEBS Lett 1995, 377, 51–53. [Google Scholar]
- Chadee, D.N.; Allis, C.D.; Wright, J.A.; Davie, J.R. Histone H1b phosphorylation is dependent upon ongoing transcription and replication in normal and ras-transformed mouse fibroblasts. J.Biol. Chem 1997, 272, 8113–8116. [Google Scholar]
- Dahia, P.L. PTEN, a unique tumor suppressor gene. Endocr. Related Cancer 2000, 7, 115–129. [Google Scholar]
- Ahuja, D.; Saenz-Robles, M.T.; Pipas, J.M. SV40 large T antigen targets multiple cellular pathways to elicit cellular transformation. Oncogene 2005, 24, 7729–7745. [Google Scholar]
- Sotillo, E.; Garriga, J.; Kurimchak, A.; Grana, X. Cyclin E and SV40 small T antigen cooperate to bypass quiescence and contribute to transformation by activating CDK2 in human fibroblasts. J. Biol. Chem 2008, 283, 11280–11292. [Google Scholar]
- Watanabe, G.; Howe, A.; Lee, R.J.; Albanese, C.; Shu, I.W.; Karnezis, A.N.; Zon, L.; Kyriakis, J.; Rundell, K.; Pestell, R.G. Induction of cyclin D1 by simian virus 40 small tumor antigen. Proc. Natl. Acad. Sci. USA 1996, 93, 12861–12866. [Google Scholar]
- Aranda, A.; Pascual, A. Nuclear hormone receptors and gene expression. Physiol. Rev 2001, 81, 1269–1304. [Google Scholar]
- Weigel, N.L.; Moore, N.L. Steroid receptor phosphorylation: A key modulator of multiple receptor functions. Mol. Endocrinol 2007, 21, 2311–2319. [Google Scholar]
- Likhite, V.S.; Stossi, F.; Kim, K.; Katzenellenbogen, B.S.; Katzenellenbogen, J.A. Kinase-specific phosphorylation of the estrogen receptor changes receptor interactions with ligand, deoxyribonucleic acid, coregulators associated with alterations in estrogen and tamoxifen activity. Mol. Endocrinol 2006, 20, 3120–3132. [Google Scholar]
- Bunone, G.; Briand, P.A.; Miksicek, R.J.; Picard, D. Activation of the unliganded estrogen receptor by EGF involves the MAP kinase pathway and direct phosphorylation. EMBO J 1996, 15, 2174–2183. [Google Scholar]
- Ikeda, K.; Ogawa, S.; Tsukui, T.; Horie-Inoue, K.; Ouchi, Y.; Kato, S.; Muramatsu, M.; Inoue, S. Protein phosphatase 5 is a negative regulator of estrogen receptor-mediated transcription. Mol. Endocrinol 2004, 18, 1131–1143. [Google Scholar]
- Narayanan, R.; Adigun, A.A.; Edwards, D.P.; Weigel, N.L. Cyclin-dependent kinase activity is required for progesterone receptor function: Novel role for cyclin A/Cdk2 as a progesterone receptor coactivator. Mol. Cell. Biol 2005, 25, 264–277. [Google Scholar]
- Hawking, S.W. A Brief History of Time: From the Big Bang to Black Holes; Bantam Dell Publishing Group: New York, NY, USA, 1988; Volume 10. [Google Scholar]
- Garrett, D. Cell cycle control and cancer. Curr. Sci 2001, 81, 515–521. [Google Scholar]
- Nurse, P. A long twentieth century of the cell cycle and beyond. Cell 2000, 100, 71–78. [Google Scholar]
- Johnson, D.G.; Walker, C.L. Cyclins and cell cycle checkpoints. Annu. Rev. Pharmacol. Toxicol 1999, 39, 295–312. [Google Scholar]
- Malumbres, M.; Barbacid, M. Mammalian cyclin-dependent kinases. Trends Biochem. Sci 2005, 30, 630–641. [Google Scholar]
- Morgan, D.O. Cyclin-dependent kinases: Engines, clocks, and microprocessors. Annu. Rev. Cell Dev. Biol 1997, 13, 261–291. [Google Scholar]
- Morgan, D.O. Principles of CDK regulation. Nature 1995, 374, 131–134. [Google Scholar]
- Kaldis, P. The cdk-activating kinase (CAK): From yeast to mammals. Cell. Mol. Life Sci 1999, 55, 284–296. [Google Scholar]
- Kaldis, P.; Solomon, M.J. Analysis of CAK activities from human cells. Eur. J. Biochem. FEBS 2000, 267, 4213–4221. [Google Scholar]
- Harbour, J.W.; Luo, R.X.; Dei Santi, A.; Postigo, A.A.; Dean, D.C. Cdk phosphorylation triggers sequential intramolecular interactions that progressively block Rb functions as cells move through G1. Cell 1999, 98, 859–869. [Google Scholar]
- Cánepa, E.T.; Scassa, M.E.; Ceruti, J.M.; Marazita, M.C.; Carcagno, A.L.; Sirkin, P.F.; Ogara, M.F. INK4 proteins, a family of mammalian CDK inhibitors with novel biological functions. IUBMB Life 2007, 59, 419–426. [Google Scholar]
- Massague, J. G1 cell-cycle control and cancer. Nature 2004, 432, 298–306. [Google Scholar]
- Stamatakos, M.; Palla, V.; Karaiskos, I.; Xiromeritis, K.; Alexiou, I.; Pateras, I.; Kontzoglou, K. Cell cyclins: Triggering elements of cancer or not. World J. surg. Oncol 2010, 8, 111:1–111:8. [Google Scholar]
- Santamaria, D.; Ortega, S. Cyclins and CDKS in development and cancer: Lessons from genetically modified mice. Front. Biosci 2006, 11, 1164–1188. [Google Scholar]
- Mermelshtein, A.; Gerson, A.; Walfisch, S.; Delgado, B.; Shechter-Maor, G.; Delgado, J.; Fich, A.; Gheber, L. Expression of D-type cyclins in colon cancer and in cell lines from colon carcinomas. Br. J. Cancer 2005, 93, 338–345. [Google Scholar]
- Deshpande, A.; Sicinski, P.; Hinds, P.W. Cyclins and cdks in development and cancer: A perspective. Oncogene 2005, 24, 2909–2915. [Google Scholar]
- Tajara, E.H. Oral cancer and cyclins. Int. J. Oral Maxillofac. Surg 2004, 33, 518, author reply 519. [Google Scholar]
- Sutherland, R.L.; Musgrove, E.A. Cyclins and breast cancer. J. Mammary Gland Biol. Neoplasia 2004, 9, 95–104. [Google Scholar]
- Miehlke, S.; Yu, J.; Ebert, M.; Szokodi, D.; Vieth, M.; Kuhlisch, E.; Buchcik, R.; Schimmin, W.; Wehrmann, U.; Malfertheiner, P. Expression of G1 phase cyclins in human gastric cancer and gastric mucosa of first-degree relatives. Dig. Dis. Sci 2002, 47, 1248–1256. [Google Scholar]
- Bartkova, J.; Rajpert-de Meyts, E.; Skakkebaek, N.E.; Bartek, J. D-type cyclins in adult human testis and testicular cancer: Relation to cell type, proliferation, differentiation, and malignancy. J. Pathol 1999, 187, 573–581. [Google Scholar]
- Pines, J. Cyclins, CDKs and cancer. Semin. Cancer Biol 1995, 6, 63–72. [Google Scholar]
- Weinberg, R.A. The retinoblastoma protein and cell cycle control. Cell 1995, 81, 323–330. [Google Scholar]
- Grana, X.; Garriga, J.; Mayol, X. Role of the retinoblastoma protein family, pRB, p107 and p130 in the negative control of cell growth. Oncogene 1998, 17, 3365–3383. [Google Scholar]
- Takemura, M.; Yamamoto, T.; Kitagawa, M.; Taya, Y.; Akiyama, T.; Asahara, H.; Linn, S.; Suzuki, S.; Tamai, K.; Yoshida, S. Stimulation of DNA Polymerase [alpha] Activity by Cdk2-Phosphorylated Rb Protein. Biochem. Biophys. Res. Commun 2001, 282, 984–990. [Google Scholar]
- Akiyama, T.; Ohuchi, T.; Sumida, S.; Matsumoto, K.; Toyoshima, K. Phosphorylation of the retinoblastoma protein by cdk2. Proc. Natl. Acad. Sci. USA 1992, 89, 7900–7904. [Google Scholar]
- Ewen, M.E.; Sluss, H.K.; Sherr, C.J.; Matsushime, H.; Kato, J.; Livingston, D.M. Functional interactions of the retinoblastoma protein with mammalian D-type cyclins. Cell 1993, 73, 487–497. [Google Scholar]
- Giacinti, C.; Giordano, A. RB and cell cycle progression. Oncogene 2006, 25, 5220–5227. [Google Scholar]
- Chambeyron, S.; Bickmore, W.A. Chromatin Decondensation and Nuclear Reorganization of the HoxB Locus upon Induction of Transcription; Cold Spring Harbor Laboratory Press: New York, NY, USA, 2004. [Google Scholar]
- Gilbert, N.; Thomson, I.; Boyle, S.; Allan, J.; Ramsahoye, B.; Bickmore, W.A. DNA methylation affects nuclear organization, histone modifications, and linker histone binding but not chromatin compaction. J. Cell. Biol 2007, 177, 401–411. [Google Scholar]
- Zheng, Y.; John, S.; Pesavento, J.J.; Schultz-Norton, J.R.; Schiltz, R.L.; Baek, S.; Nardulli, A.M.; Hager, G.L.; Kelleher, N.L.; Mizzen, C.A. Histone H1 phosphorylation is associated with transcription by RNA polymerases I and II. J. Cell. Biol 2010, 189, 407–415. [Google Scholar]
- Horn, P.J.; Carruthers, L.M.; Logie, C.; Hill, D.A.; Solomon, M.J.; Wade, P.A.; Imbalzano, A.N.; Hansen, J.C.; Peterson, C.L. Phosphorylation of linker histones regulates ATP-dependent chromatin remodeling enzymes. Nat. Struct. Biol 2002, 9, 263–267. [Google Scholar]
- Contreras, A.; Hale, T.K.; Stenoien, D.L.; Rosen, J.M.; Mancini, M.A.; Herrera, R.E. The dynamic mobility of histone H1 is regulated by cyclin/CDK phosphorylation. Mol. Cell. Biol 2003, 23, 8626–8636. [Google Scholar]
- Nesbit, C.; Tersak, J.; Prochownik, V. Myc oncogenes and human neoplastic disease. Oncogene 1999, 18, 3004–3016. [Google Scholar]
- Adhikary, S.; Eilers, M. Transcriptional regulation and transformation by Myc proteins. Nat. Rev. Mol. Cell Biol 2005, 6, 635–645. [Google Scholar]
- Meyer, N.; Penn, L.Z. Reflecting on 25 years with MYC. Nat. Rev. Cancer 2008, 8, 976–990. [Google Scholar]
- Holzel, M.; Kohlhuber, F.; Schlosser, I.; Holzel, D.; Luscher, B.; Eick, D. Myc/max/mad regulate the frequency but not the duration of productive cell cycles. EMBO Rep 2001, 21, 1125–1132. [Google Scholar]
- Obaya, A.J.; Mateyak, M.; Sedivy, J. Mysterious liaisons: The relationship between c-myc and the cell cycle. Oncogene 1999, 18, 2934–2941. [Google Scholar]
- Berns, K.; Hijmanns, E.; Bernards, R. Repression of c-Myc responsive genes in cycling cells causes G1 arrest through reduction of cyclin E/CDK2 kinase activity. Oncogene 1997, 15, 1347–56. [Google Scholar]
- Fernandez, P.C.; Frank, S.R.; Wang, L.; Schroeder, M.; Liu, S.; Greene, J.; Cocito, A.; Amati, B. Genomic targets of the human c-Myc protein. Genes Dev 2003, 17, 1115–1129. [Google Scholar]
- Perez-Roger, I.; Solomon, D.L.; Sewing, A.; Land, H. Myc activation of cyclin E/Cdk2 kinase involves induction of cyclin E gene transcription and inhibition of p27(Kip1) binding to newly formed complexes. Oncogene 1997, 14, 2373–2381. [Google Scholar]
- Bouchard, C.; Thieke, K.; Maier, A.; Saffrich, R.; Hanley-Hyde, J.; Ansorge, W.; Reed, S.; Sicinski, P.; Bartek, J.; Eilers, M. Direct induction of cyclin D2 by Myc contributes to cell cycle progression and sequestration of p27. EMBO J 1999, 18, 5321–5333. [Google Scholar]
- Jansen-Durr, P.; Meichle, A.; Steiner, P.; Pagano, M.; Finke, K.; Botz, J.; Wessbecher, J.; Draetta, G.; Eilers, M. Differential modulation of cyclin gene expression by MYC. Proc. Natl. Acad. Sci. USA 1993, 90, 3685–3689. [Google Scholar]
- Hermeking, H.; Rago, C.; Schuhmacher, M.; Li, Q.; Barrett, J.F.; Obaya, A.J.; O’Connell, B.C.; Mateyak, M.K.; Tam, W.; Kohlhuber, F. Identification of CDK4 as a target of c-MYC. Proc. Natl. Acad. Sci. USA 2000, 97, 2229–2234. [Google Scholar]
- Rustgi, A.K.; Dyson, N.; Hill, D.; Bernards, R. The c-myc oncoprotein forms a specific complex with the product of the retinoblastoma gene. Cold Spring Harbor Symp. Quant. Biol 1991, 56, 163–167. [Google Scholar]
- Brown, J.R.; Nigh, E.; Lee, R.J.; Ye, H.; Thompson, M.A.; Saudou, F.; Pestell, R.G.; Greenberg, M.E. Fos family members induce cell cycle entry by activating cyclin D1. Mol. Cell. Biol 1998, 18, 5609–5619. [Google Scholar]
- Wisdom, R.; Johnson, R.S.; Moore, C. c-Jun regulates cell cycle progression and apoptosis by distinct mechanisms. EMBO J 1999, 18, 188–197. [Google Scholar]
- Haller, K.; Wu, Y.; Derow, E.; Schmitt, I.; Jeang, K.; Grassman, R. Physical interaction of human T-cell leukemia virus type 1 tax with cyclin-dependent kinase 4 stimulates the phosphorylation of retinoblastoma protein. Mol. Cell. Biol 2002, 22, 3327–3338. [Google Scholar]
- Martin, N.G.; McAndrew, P.C.; Eve, P.D.; Garrett, M.D. Phosphorylation of cyclin dependent kinase 4 on tyrosine 17 is mediated by Src family kinases. FEBS J 2008, 275, 3099–3109. [Google Scholar]
- Cohen, S.; Levi-Montalcini, R. Purification and properties of a nerve growth-promoting factor isolated from mouse sarcoma 180. Cancer Res 1957, 17, 15–20. [Google Scholar]
- Gschwind, A.; Fischer, O.M.; Ullrich, A. The discovery of receptor tyrosine kinases: Targets for cancer therapy. Nat. Rev. Cancer 2004, 4, 361–370. [Google Scholar]
- Ross, J.S.; Schenkein, D.P.; Pietrusko, R.; Rolfe, M.; Linette, G.P.; Stec, J.; Stagliano, N.E.; Ginsburg, G.S.; Symmans, W.F.; Pusztai, L. Targeted therapies for cancer 2004. Am. J. Clin. Pathol 2004, 122, 598–609. [Google Scholar]
- Arora, A.; Scholar, E.M. Role of tyrosine kinase inhibitors in cancer therapy. J. Pharmacol. Exp. Ther 2005, 315, 971–979. [Google Scholar]
- Krause, D.S.; van Etten, R.A. Tyrosine kinases as targets for cancer therapy. N. Eng. J. Med 2005, 353, 172–187. [Google Scholar]
- Kwak, E.L.; Bang, Y.J.; Camidge, D.R.; Shaw, A.T.; Solomon, B.; Maki, R.G.; Ou, S.H.I.; Dezube, B.J.; Jänne, P.A.; Costa, D.B. Anaplastic lymphoma kinase inhibition in non-small-cell lung cancer. N. Eng. J. Med 2010, 363, 1693–1703. [Google Scholar]
- Flaherty, K.T.; Puzanov, I.; Kim, K.B.; Ribas, A.; McArthur, G.A.; Sosman, J.A.; O’Dwyer, P.J.; Lee, R.J.; Grippo, J.F.; Nolop, K. Inhibition of mutated, activated BRAF in metastatic melanoma. N. Eng. J. Med 2010, 363, 809–819. [Google Scholar]
- Fojo, T.; Parkinson, D.R. Biologically targeted cancer therapy and marginal benefits: Are we making too much of too little or are we achieving too little by giving too much? Clin. Cancer Res 2010, 16, 5972–5980. [Google Scholar]
- Darnell, J.E. Transcription factors as targets for cancer therapy. Nat. Rev. Cancer 2002, 2, 740–749. [Google Scholar]
- Stadler, Z.K.; Vijai, J.; Thom, P.; Kirchhoff, T.; Hansen, N.A.; Kauff, N.D.; Robson, M.; Offit, K. Genome-wide association studies of cancer. J. Clin. Oncol 2010, 28, 4255–4267. [Google Scholar]
- Pusztai, L.; Ayers, M.; Stec, J.; Hortobagyi, G.N. Clinical application of cDNA microarrays in oncology. Oncologist 2003, 8, 252–258. [Google Scholar]
- LaTulippe, E.; Satagopan, J.; Smith, A.; Scher, H.; Scardino, P.; Reuter, V.; Gerald, W.L. Comprehensive gene expression analysis of prostate cancer reveals distinct transcriptional programs associated with metastatic disease. Cancer Res 2002, 62, 4499–4506. [Google Scholar]
- Harada, T.; Chelala, C.; Crnogorac-Jurcevic, T.; Lemoine, N.R. Genome-wide analysis of pancreatic cancer using microarray-based techniques. Pancreatology 2009, 9, 13–24. [Google Scholar]
- Welsh, J.B.; Sapinoso, L.M.; Su, A.I.; Kern, S.G.; Wang-Rodriguez, J.; Moskaluk, C.A.; Frierson, H.F.; Hampton, G.M. Analysis of gene expression identifies candidate markers and pharmacological targets in prostate cancer. Cancer Res 2001, 61, 5974–8. [Google Scholar]
- Chen, X.; Cheung, S.T.; So, S.; Fan, S.T.; Barry, C.; Higgins, J.; Lai, K.M.; Ji, J.; Dudoit, S.; Ng, I.O.L. Gene expression patterns in human liver cancers. Mol. Biol. Cell 2002, 13, 1929–39. [Google Scholar]
- Fehrmann, R.S.; Li, X.Y.; van der Zee, A.G.; de Jong, S.; Te Meerman, G.J.; de Vries, E.G.; Crijns, A.P. Profiling studies in ovarian cancer: A review. Oncologist 2007, 12, 960–966. [Google Scholar]
- Sotiriou, C.; Pusztai, L. Gene-expression signatures in breast cancer. N. Eng. J. Med 2009, 360, 790–800. [Google Scholar]
- Chung, C.H.; Seeley, E.H.; Roder, H.; Grigorieva, J.; Tsypin, M.; Roder, J.; Burtness, B.A.; Argiris, A.; Forastiere, A.A.; Gilbert, J. Detection of tumor epidermal growth factor receptor pathway dependence by serum mass spectrometry in cancer patients. Cancer Epidemiol. Biomark. Prev 2010, 19, 358–365. [Google Scholar]
- Chen, H.B.; Pan, K.; Tang, M.K.; Chui, Y.L.; Chen, L.; Su, Z.J.; Shen, Z.Y.; Li, E.M.; Xie, W.; Lee, K.K. Comparative proteomic analysis reveals differentially expressed proteins regulated by a potential tumor promoter, BRE, in human esophageal carcinoma cells. Biochem. Cell Biol 2008, 86, 302–311. [Google Scholar]
- Rush, J.; Moritz, A.; Lee, K.A.; Guo, A.; Goss, V.L.; Spek, E.J.; Zhang, H.; Zha, X.; Polakiewicz, R.D.; Comb, M.J. Immunoaffinity profiling of tyrosine phosphorylation in cancer cells. Nat. Biotech 2005, 23, 94–101. [Google Scholar]
- Fan, A.C.; Deb-Basu, D.; Orban, M.W.; Gotlib, J.R.; Natkunam, Y.; O’Neill, R.; Padua, R.A.; Xu, L.; Taketa, D.; Shirer, A.E. Nanofluidic proteomic assay for serial analysis of oncoprotein activation in clinical specimens. Nat. Med 2009, 15, 566–571. [Google Scholar]

© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Willis, R.E. Human Gene Control by Vital Oncogenes: Revisiting a Theoretical Model and Its Implications for Targeted Cancer Therapy. Int. J. Mol. Sci. 2012, 13, 316-335. https://doi.org/10.3390/ijms13010316
Willis RE. Human Gene Control by Vital Oncogenes: Revisiting a Theoretical Model and Its Implications for Targeted Cancer Therapy. International Journal of Molecular Sciences. 2012; 13(1):316-335. https://doi.org/10.3390/ijms13010316
Chicago/Turabian StyleWillis, Rudolph E. 2012. "Human Gene Control by Vital Oncogenes: Revisiting a Theoretical Model and Its Implications for Targeted Cancer Therapy" International Journal of Molecular Sciences 13, no. 1: 316-335. https://doi.org/10.3390/ijms13010316