Determination of Phenolic Acids and Flavonoids in Taraxacum formosanum Kitam by Liquid Chromatography-Tandem Mass Spectrometry Coupled with a Post-Column Derivatization Technique




Abstract
:1. Introduction
2. Results and Discussion
2.1. Comparison of Extraction Solvents
2.2. Comparison of Various Mobile Phases in HPLC
2.3. Comparison of Various Elution Volumes in SPE
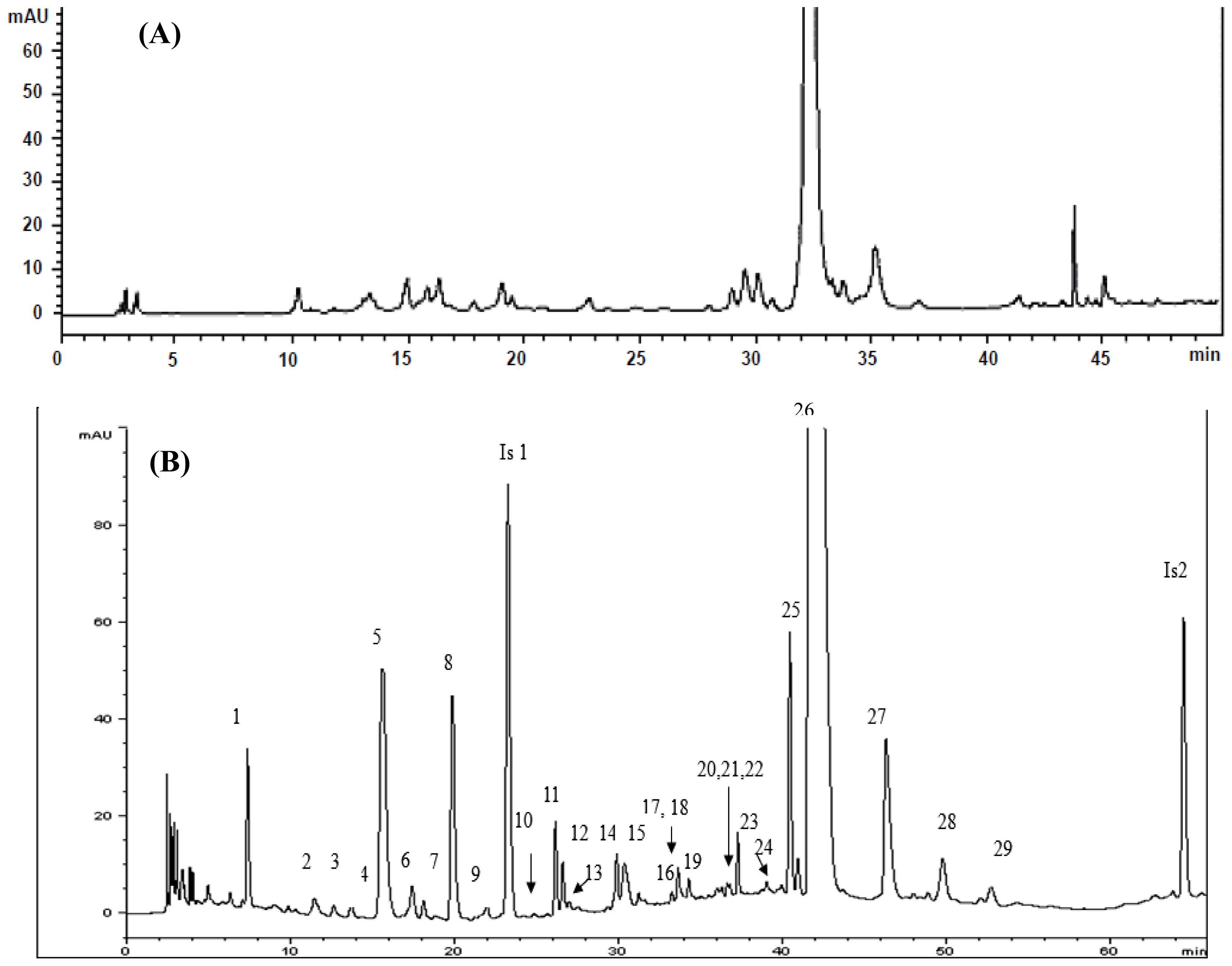
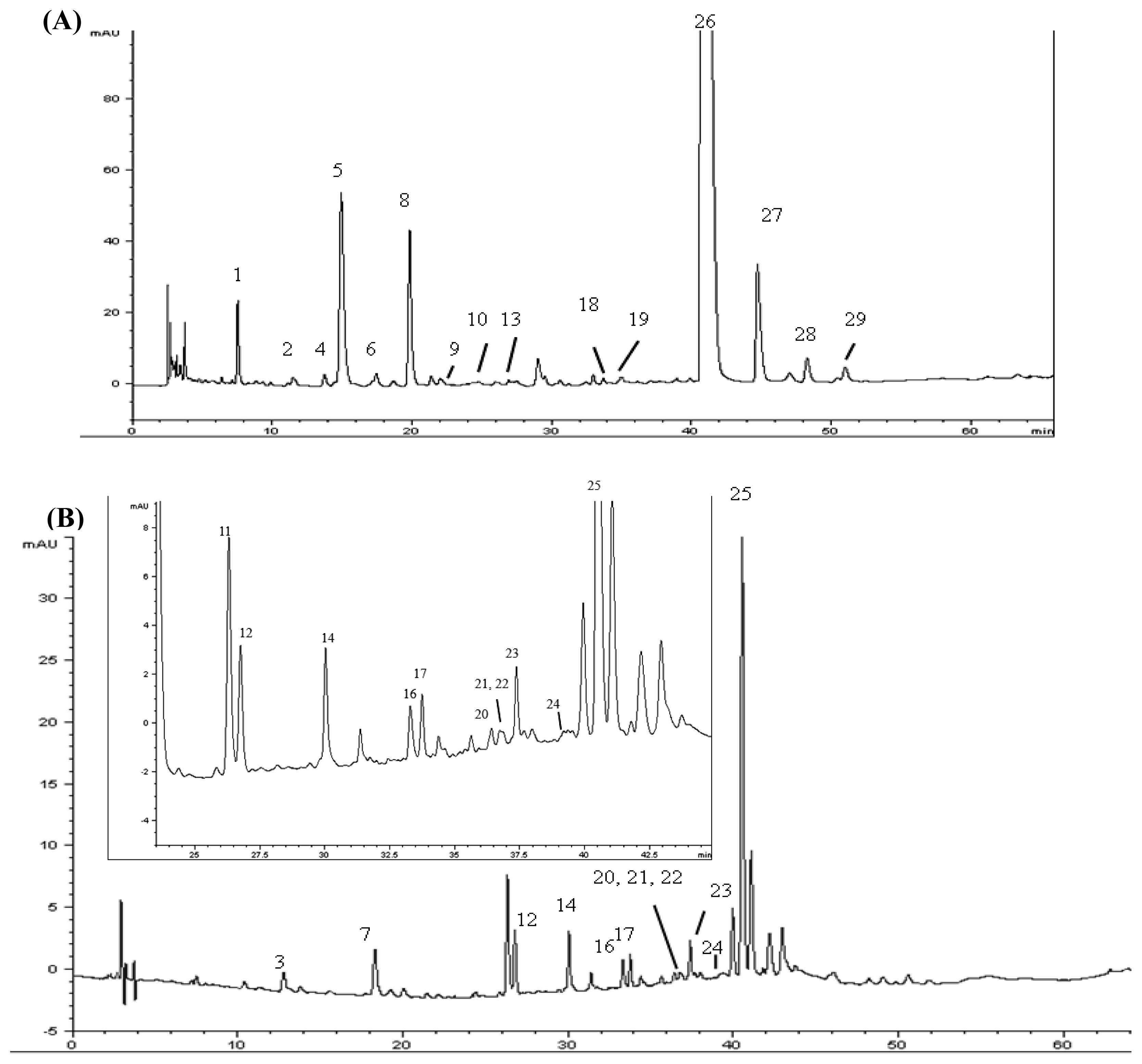
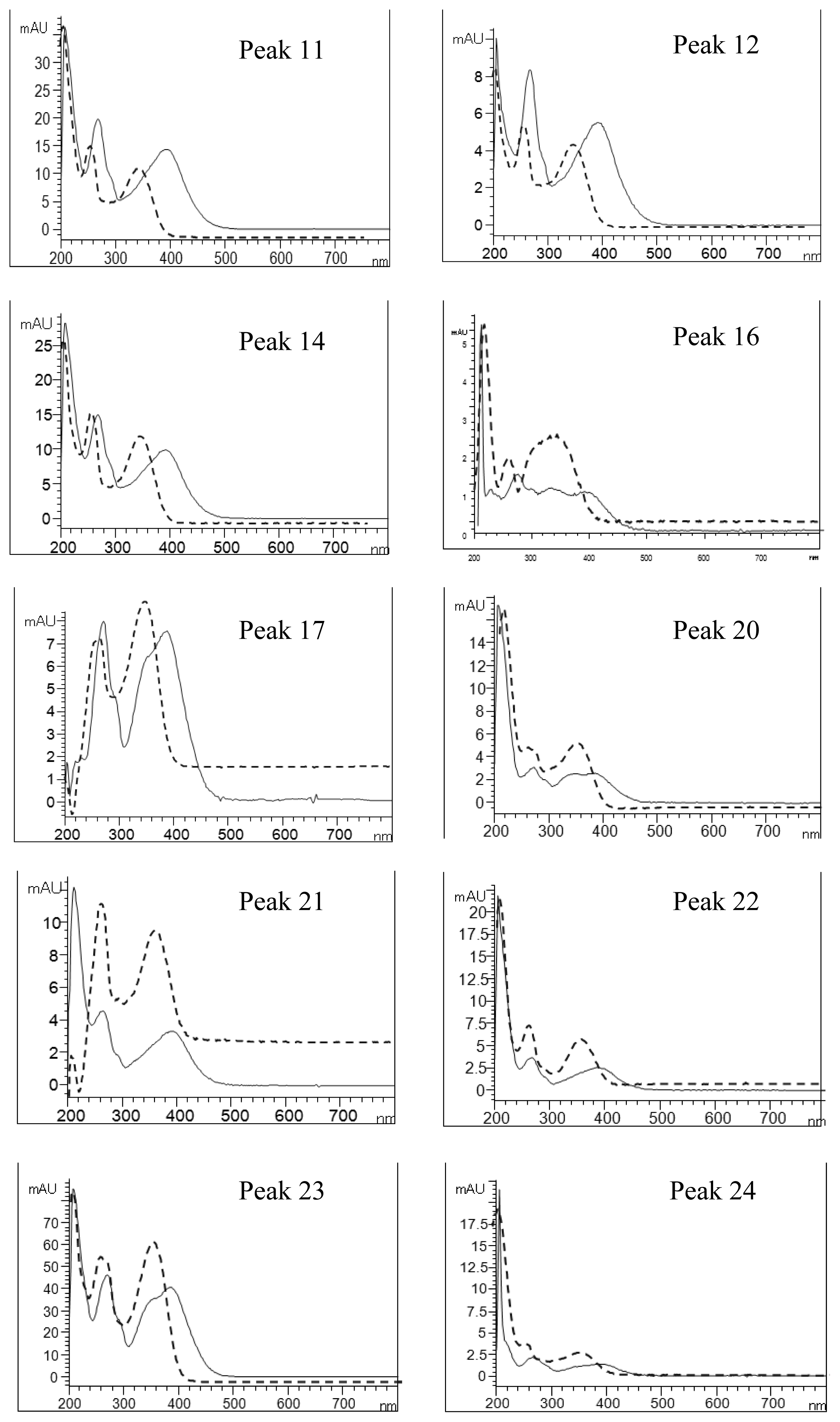
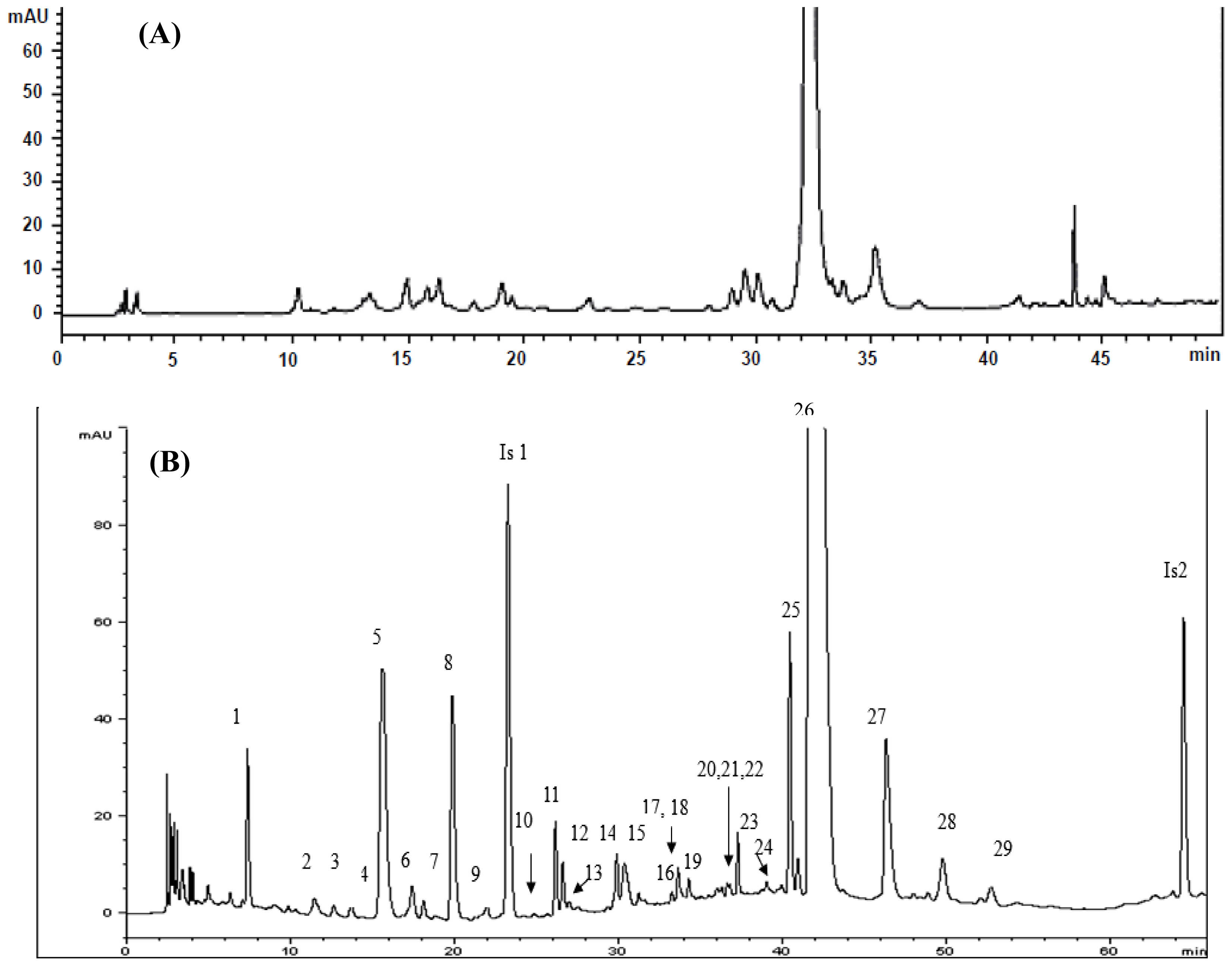
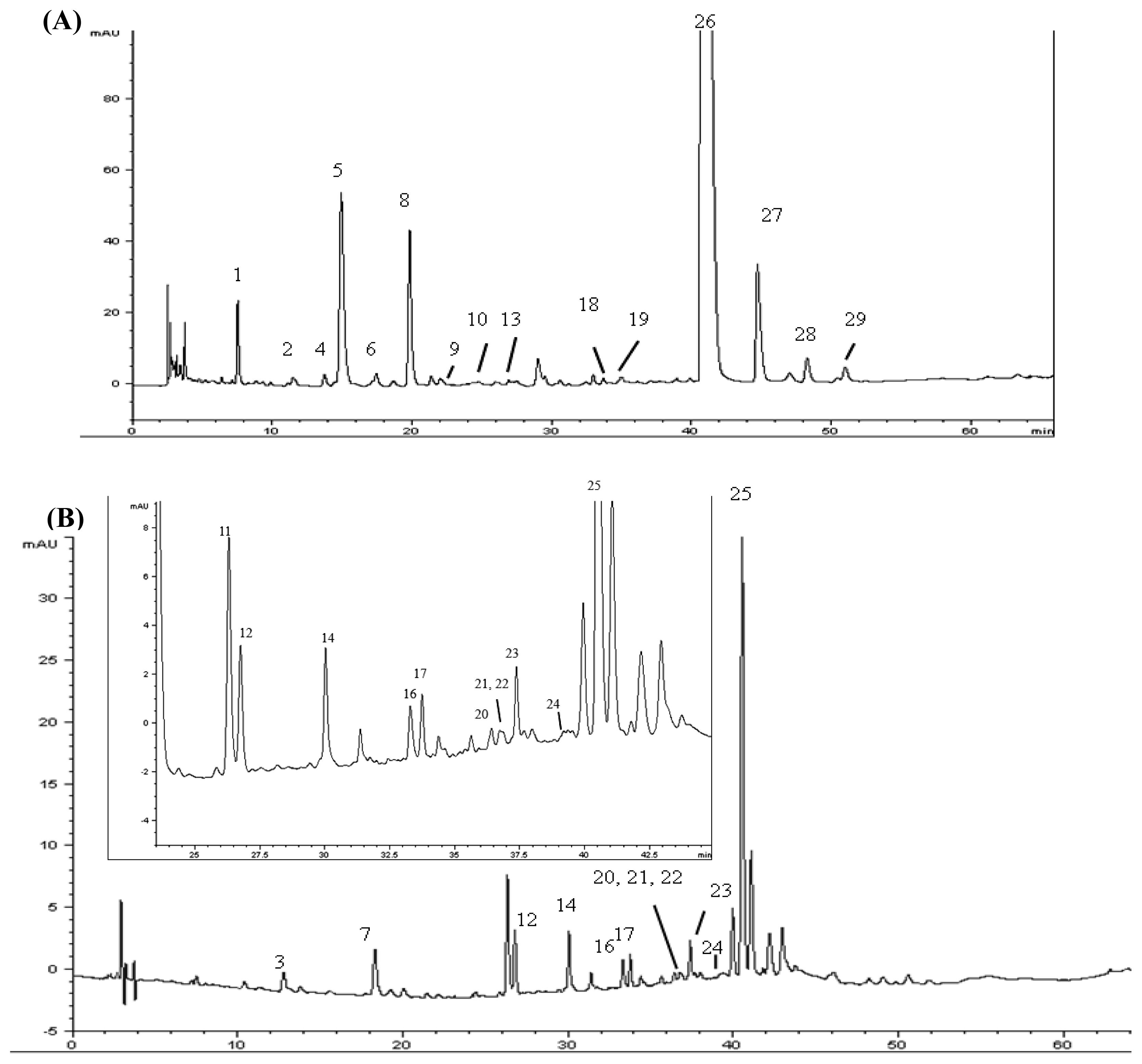
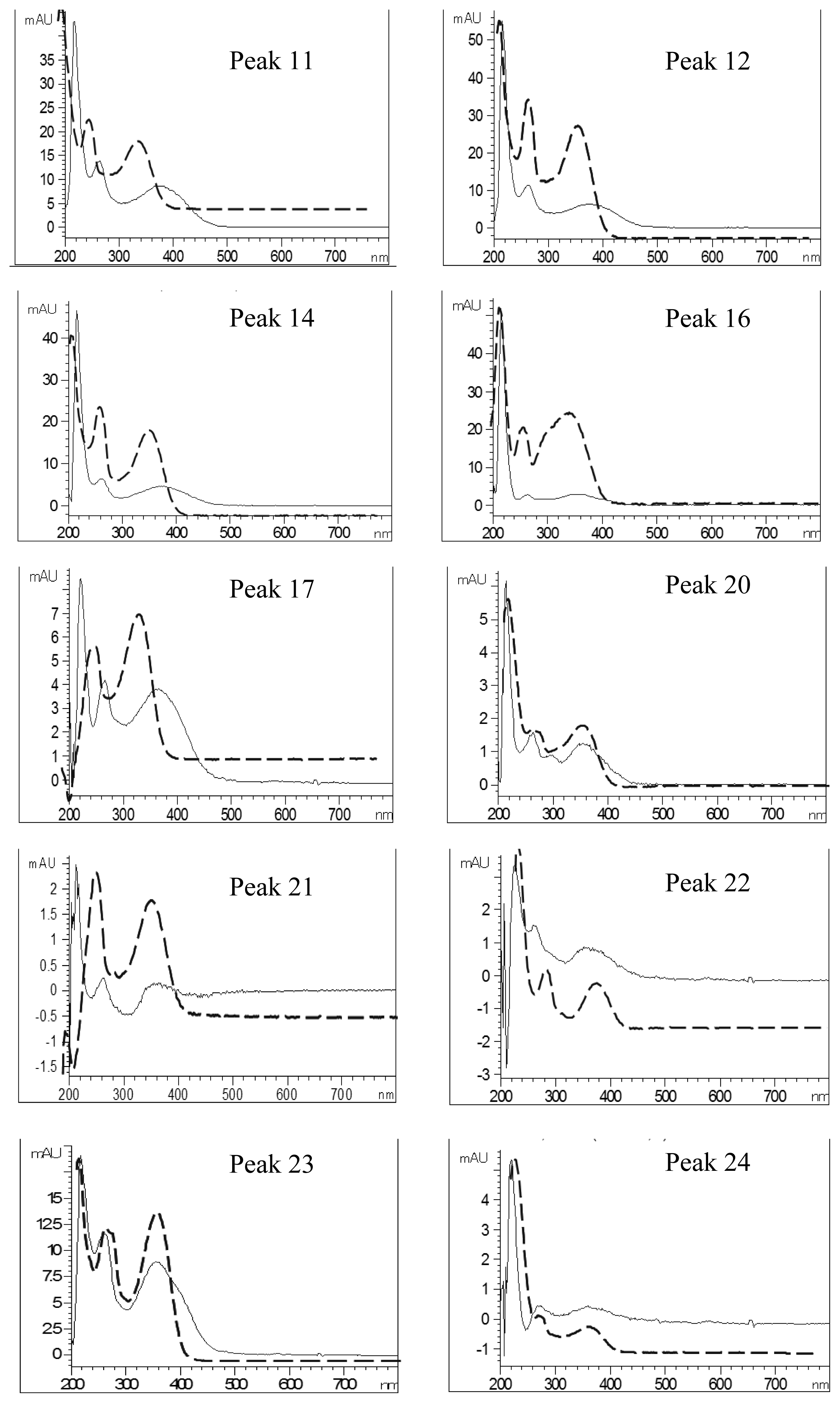
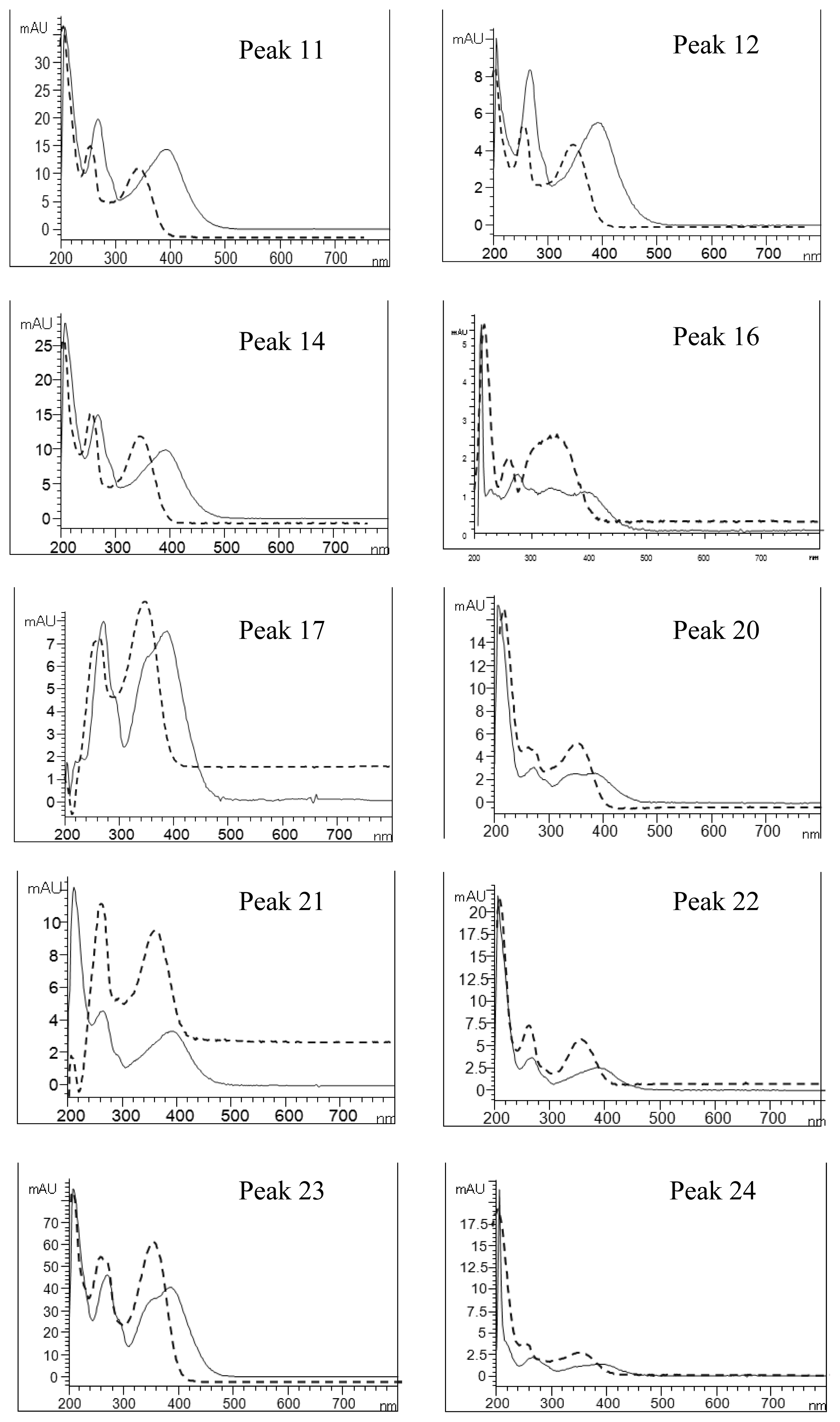
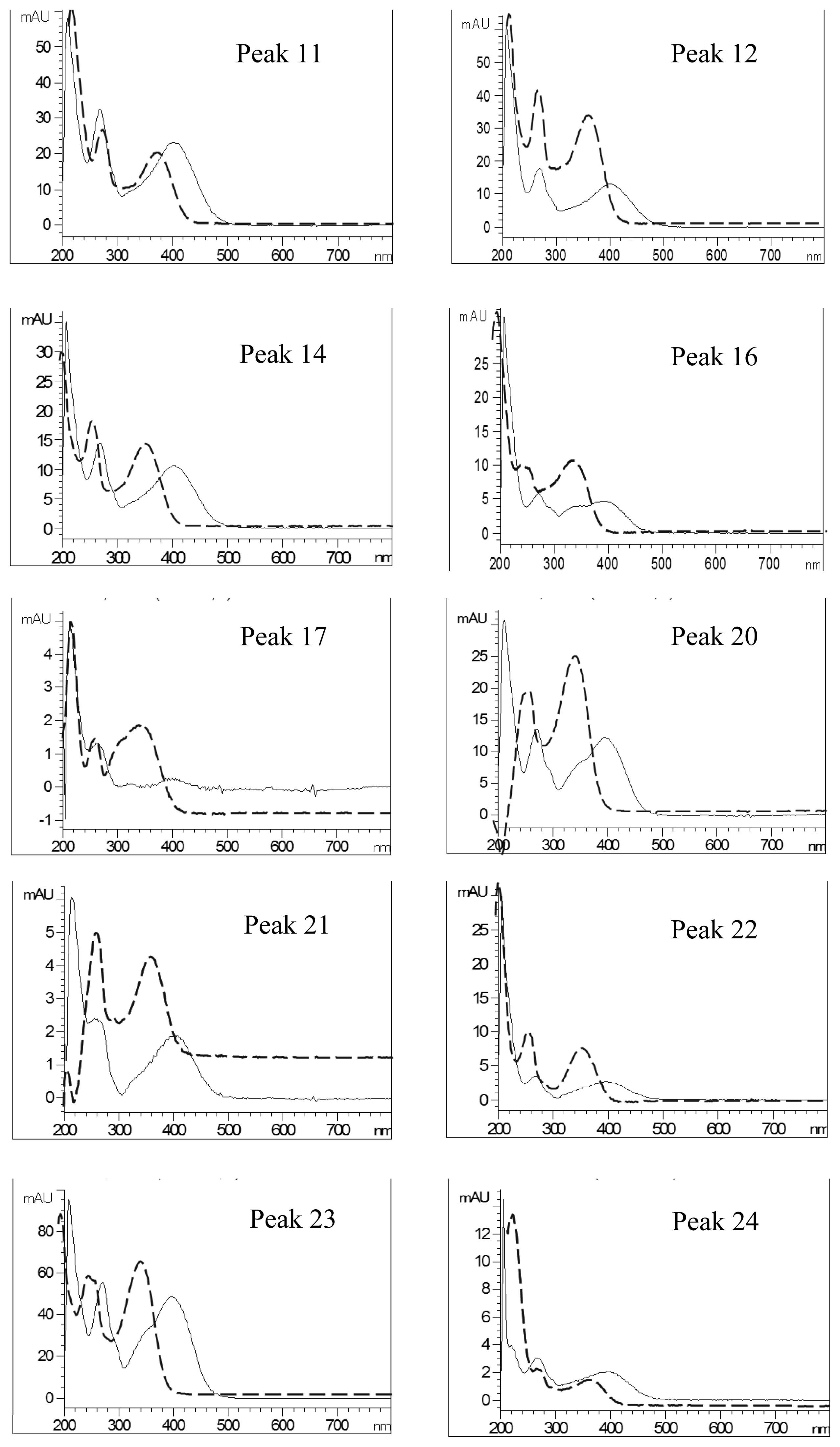
2.4. Identification of Various Phenolic Acids and Flavonoids in Taraxacum formosanum
2.5. Quality Control
3. Experimental Section
3.1. Materials
3.2. Instrumentation
3.3. Extraction and Purification
3.4. HPLC Separation
3.5. Identification
3.6. Quantitation
3.7. Quality Control
3.8. Statistical Analysis
4. Conclusions
References
- Koo, H.N.; Hong, S.H.; Song, B.K.; Kim, C.H.; Yoo, Y.H.; Kim, H.M. Taraxacum officinale induces cytotoxicity through TNF-α and IL-1α secretion n HepG2 cells. Life Sci 2004, 74, 1149–1157. [Google Scholar]
- Rana, A.C.; Sumanth, M. In vivo anti-oxidant activity of hydroalcoholic acid of Taraxacum officinale root in rats. Indian J. Physiol. Pharmacol 2006, 38, 54–55. [Google Scholar]
- Seo, S.W.; Koo, H.N.; An, H.J.; Kwon, K.B.; Lim, B.C.; Seo, E.A.; Ryu, D.G.; Moon, G.; Kim, H.Y.; Kim, H.M.; et al. Taraxacum officinale protects against cholecystokinin-induced acute pancreatitis in rats. World J. Gastroenterol 2005, 11, 597–599. [Google Scholar]
- Hu, C.; Kitts, D.D. Antioxidant, proxidant, and cytotoxic activities of solvent-fractionated dandelion (Taraxacum officinale) flower extracts in vitro. J. Agric. Food Chem 2003, 51, 301–310. [Google Scholar]
- Rocha-Guzmán, N.E.; Gallegos-Infante, J.A.; González-Laredo, R.F.; Reynoso-Camacho, R.; Ramos-Gómez, M.; Garcia-Gasca, T.; Rodríguez-Muñoz, M.E.; Guzmán-Maldonado, S.H.; Medina-Torres, L.; Lujan-García, B.A. Antioxidant activity and genotoxic effect on HeLa cells of phenolic compounds from infusions of Quercus resinosa leaves. Food Chem 2009, 115, 1320–1325. [Google Scholar]
- Ayaz, F.A.; Hayırlıoglu-Ayaz, S.; Alpay-Karaoglu, S.; Grúz, K.; Valentová, K.; Ulrichová, J.; Strnad, M. Phenolic acid contents of kale (Brassica oleraceae L. var. acephala DC) extracts and their antioxidant and antibacterial activities. Food Chem 2008, 107, 19–25. [Google Scholar]
- Campos, F.M.; Couto, J.A.; Figueiredo, A.R.; Tóth, I.V.; Rangel, A.O.S.S.; Hogg, T.A. Cell membrane damage induced by phenolic acids on wine lactic acid bacteria. Int. J. Food Microbiol 2009, 135, 144–151. [Google Scholar]
- Caridi, D.; Trenerry, C.; Rochfort, S.; Duong, S.; Laugher, D.; Jones, R. Profiling and quantities of quercetin glucosides in onion (Allium cepa L.) varieties using capillary zone electrophoresis and high performance liquid chromatography. Food Chem 2007, 105, 691–699. [Google Scholar]
- Tsai, Y.C.; Lin, C.L.; Chen, B.H. Preparative chromatography of flavonoids and saponins in Gynostemma pentaphyllum and their antiproliferation effect on hepatoma cell. Phytomedicine 2010, 15, 2–10. [Google Scholar]
- Kao, T.H.; Wu, W.M.; Hong, C.F.; Wu, W.B.; Chen, B.H. Anti-inflammatory effect of isoflavone powder produced from soybean cake. J. Agric. Food Chem 2007, 55, 11068–11079. [Google Scholar]
- Robbins, R.J. Phenolic acids in foods. J. Agric. Food Chem 2003, 51, 2866–2887. [Google Scholar]
- Qian, J.Y.; Liu, D.; Huang, A.G. The efficiency of flavonoids in polar extracts of Lycium Chinese Mill fruit as free radical scavenger. Food Chem 2004, 87, 283–288. [Google Scholar]
- Hu, C.; Kitts, D.D. Luteolin and luteolin-7-O-glucoside from dandelion flower suppress iNOS and COX-2 in RAW264.7 cells. Mol. Cell. Biochem 2004, 265, 107–113. [Google Scholar]
- Rohn, S.; Buchner, N.; Driemel, G.; Rauser, M.; Kroh, L.W. Thermal degradation of onion quercetin glucosides under roasting conditions. J. Agric. Food Chem 2007, 55, 1568–1573. [Google Scholar]
- Wills, R.B.H.; Stuart, D.L. Effect of handling and storage on alkamides and chicoric acid in Echinacea purpurea. J. Sci. Food Agric 2000, 80, 1402–1406. [Google Scholar]
- Yu, J.; Vasanthan, T.; Temelli, F. Analysis of phenolic acids in barley by high-performance liquid chromatography. J. Agric. Food Chem 2001, 49, 4352–4358. [Google Scholar]
- Inbaraj, B.S.; Lu, H.; Kao, T.H.; Chen, B.H. Simultaneous determination of phenolic acids and flavonoids in Lycium barbarum Linnaeus by HPLC-DAD-ESI-MS. J. Pharm. Biomed. Anal 2010, 51, 549–556. [Google Scholar]
- Zheng, W.; Clifford, M.N. Profiling the chlorogenic acids of sweet potato (Ipomea batatas) from China. Food Chem 2008, 106, 147–152. [Google Scholar]
- Herchi, W.; Sawalha, S.; Arráez-Román, D.; Boukhchina, S.; Segura-Carretero, A.; Kallel, H.; Fernández-Gutierrez, A. Determination of phenolic and other polar compounds in flaxseed oil using liquid chromatography coupled with time-of-flight mass spectrometry. Food Chem 2011, 126, 332–338. [Google Scholar]
- Kao, T.H.; Huang, S.C.; Inbaraj, B.S.; Chen, B.H. Determination of flavonoids and saponins in Gynostemma pentaphyllum (Thunb.) Makino by liquid chromatography-mass spectrometry. Anal. Chim. Acta 2008, 626, 200–211. [Google Scholar]
- Schütz, K.; Kammerer, D.R.; Carle, R.; Schieber, A. Characterization of phenolic acids and flavonoids in dandelion (Taraxacum officinale WEB. Ex WIGG) root and herb by high-performance liquid chromatography/electrospray ionization mass spectrometry. Rapid Commun. Mass Spectrom 2005, 19, 179–186. [Google Scholar]
- Mabry, T.J.; Markham, K.R.; Thomas, M.B. The Systematic Identification of Flavonoids; Springer-Verlag: New York, NY, USA, 1970; p. 354. [Google Scholar]
- Colombo, R.; Yariwake, J.H.; Queiroz, E.F.; Ndjoko, K.; Hostettmann, K. On-line identification of further flavone C- and O-glycosides from sugarcane (Saccharum officinarum L. Gramineae) by HPLC-UV-MS. Phytochem. Anal 2006, 17, 337–343. [Google Scholar]
- Tiberti, L.A.; Yariwake, J.H.; Ndjoko, K.; Hostettmann, K. Identification of flavonols in leaves of Maytenus ilicifolia and M. aquifolium (Celastraceae) by LC/MS/MS analysis. J. Chromatogr. B 2007, 846, 378–384. [Google Scholar]
- Prasain, J.K.; Barnes, S. Metabolism and bioavailability of flavonoids in chemoprevention: current analytical strategies and future prospectus. Mol. Pharm 2007, 4, 846–864. [Google Scholar]
- Fang, B.; Yu, S.; Prior, R.L. LC/MS/MS characterization of phenolic constituents in dried plums. J. Agric. Food Chem 2002, 50, 3579–3585. [Google Scholar]
- Fang, Z.; Zhang, M.; Wang, L. HPLC-DAD-ESIMS analysis of phenolic compounds in bayberries (Myrica rubra Sieb. Et Zucc.). Food Chem 2007, 100, 845–852. [Google Scholar]
- Shakya, R.; Navarre, D.A. Rapid screening of ascorbic acid, glycoalkaloids, and phenolics in potato using high-performance liquid chromatography. J. Agric. Food Chem 2006, 54, 5253–5260. [Google Scholar]
- Atoui, A.K.; Mansouri, A.; Boskou, G.; Kefalas, P. Tea and herbal influsions: Their antioxidant activity and phenolic profile. Food Chem 2005, 89, 27–36. [Google Scholar]
- Arranz, S.; Calixto, F.S. Analysis of polyphenols in cereals may be improved performing acidic hydrolysis: A study in wheat flour and wheat bran and cereals of the diet. J. Cereal Sci 2010, 51, 313–318. [Google Scholar]
- Sakakibara, H.; Honda, Y.; Nakagawa, S.; Ashida, H.; Kanazawa, K. Simultaneous determination of all polyphenols in vegetables, fruits, and teas. J. Agric. Food Chem 2003, 51, 571–581. [Google Scholar]
- Rochfort, S.J.; Imsic, M.; Jones, R.; Trenerry, V.C.; Tomkins, B. Characterization of flavonol conjugates in immature leaves of pak choi [Brassica rapa L. Ssp. chinensis L. (Hanelt.)] by HPLC-DAD and LC-MS/MS. J. Agric. Food Chem 2006, 54, 4855–4860. [Google Scholar]
- Gouveia, S.C.; Castilho, P.C. Characterization of phenolic compounds in Helichrysum melaleucum by high-performance liquid chromatography with on-line ultraviolet and mass spectrometry detection. Rapid Commun. Mass Spectrom 2010, 24, 1851–1868. [Google Scholar]
- Mertz, C.; Cheynier, V.; Günata, Z.; Bra, P. Analysis of phenolic compounds in two blackberry species (Rubus glaucus and Rubus adenotrichus) by high-performance liquid chromatography with diode array detection and electrospray ion trap mass spectrometry. J. Agric. Food Chem 2007, 55, 8618–8624. [Google Scholar]
- Arakawa, R.; Yamaguchi, M.; Hotta, H.; Osakai, T.; Kimoto, T. Product analysis of caffeic acid oxidation by on-line electrochemistry/electrospray ionization mass spectrometry. J. Am. Soc. Mass Spectrom 2004, 15, 1228–1236. [Google Scholar]
- Lorrain, B.; Dangles, O.; Genot, C. Chemical modeling of heme-induced lipid oxidation in gastric conditions and inhibition by dietary polyphenols. J. Agric. Food Chem 2010, 58, 676–683. [Google Scholar]
- Rivera-Pastrana, D.M.; Yahia, E.M.; González-Aquilar, G.A. Phenolic and carotenoid profiles of papaya fruit (Carica papaya L.) and their contents under low temperature storage. J. Sci. Food Agric 2010, 90, 2358–2365. [Google Scholar]
- Niranjan, A.; Barthwal, J.; Lehri, A.; Singh, D.P.; Govindrajan, R.; Rawat, A.K.S.; Amla, D.V. Development and validation of an HPLC-UV-MS-MS method for identification and quantification of polyphenols in Artemisia pallens L. Acta. Chromatogr 2009, 21, 105–116. [Google Scholar]
- Manček, B.; Kreft, S. Determination of chicoric acid content in dried press juice of purple coneflower (Echinacea purpurea) with capillary electrophoresis. Talanta 2005, 66, 1094–1097. [Google Scholar]
- ICH. Q2B Validation of Analytical Procedures: Methodology, Presented at International Conference on Harmonization, Geneva, Switzerland, March 1996.
- Statistical Analysis Software, version 6; SAS Institute Inc: Cary, NC, USA, 2009.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peak No. | Identity | Retention Time (tR, min) | Retention Factor (k) | Separation Factor (α) | Peak Purity (%) |
|---|---|---|---|---|---|
| 1 | Protocatechuic acid hexoside | 7.397 | 2.06 | 1.84 (1,2) a | 99.9 |
| 2 | Caffeoyl hexoside | 11.596 | 3.79 | 1.84 (1,2) a | 72.9 |
| 3 | Caffeoyl-d-glucose | 12.781 | 4.28 | 1.13 (2,3) a | 99.5 |
| 4 | p-Hydroxybenzoic acid | 13.86 | 4.73 | 1.10 (3,4) a | 97.2 |
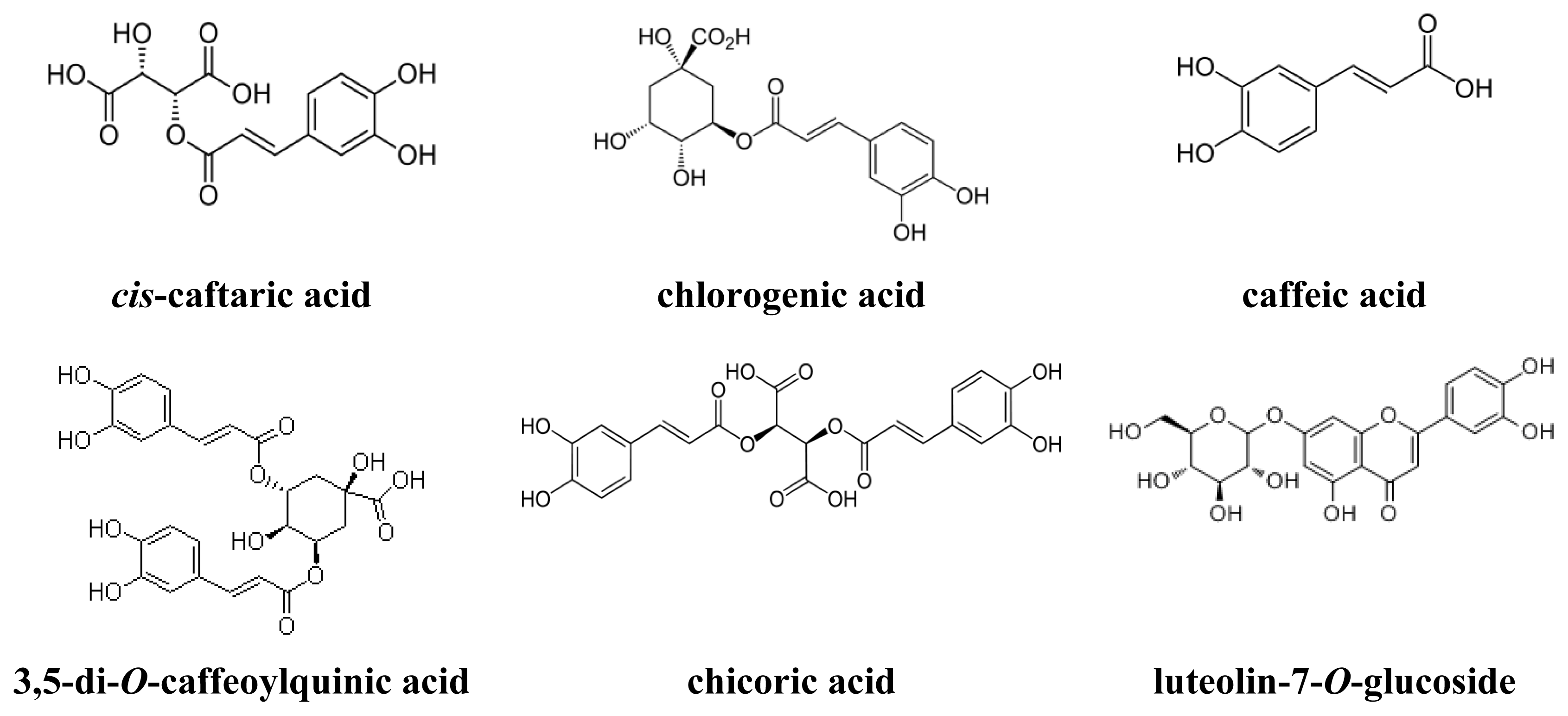
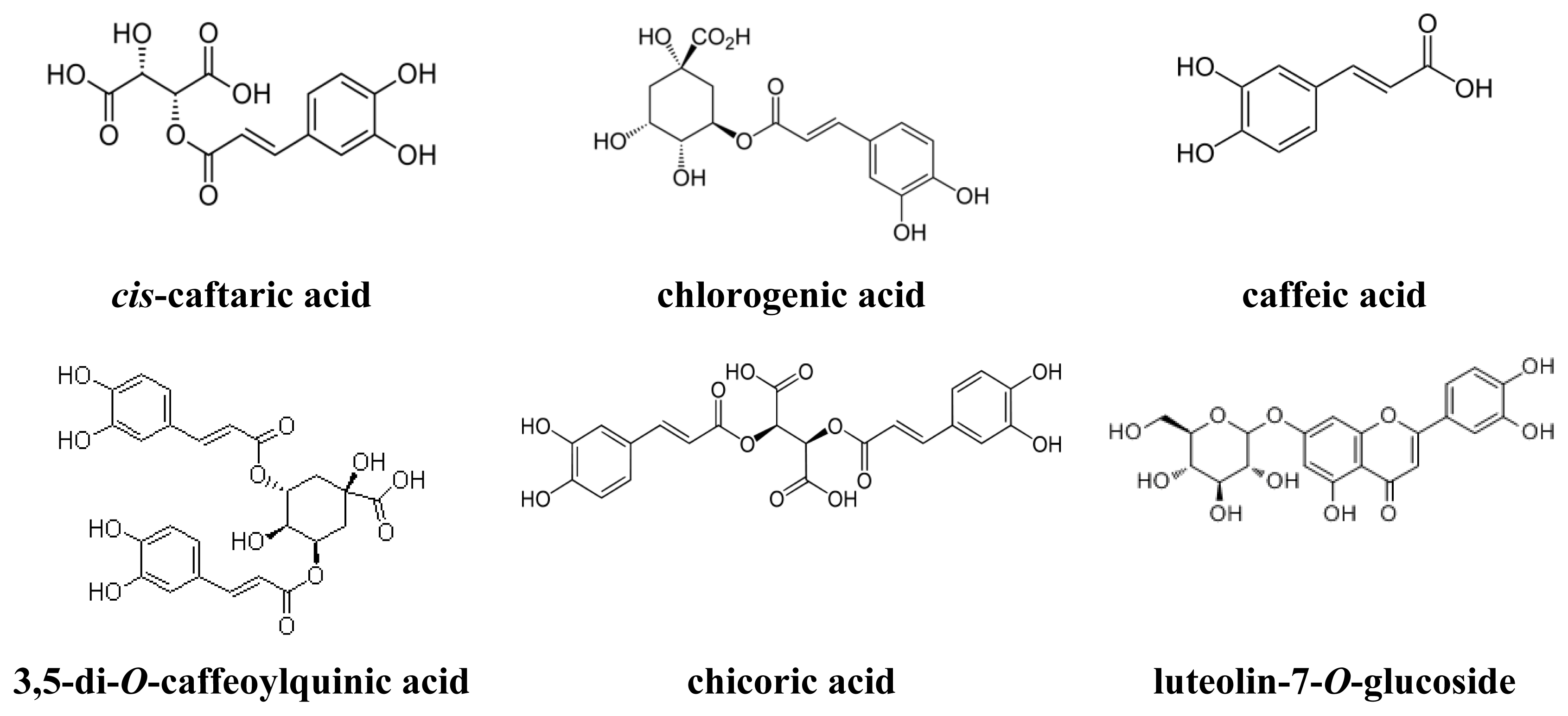
| 5 | cis-Caftaric acid b | 16.11 | 5.66 | 1.20 (4,5) a | 99.9 |
| 6 | Caffeoyl hexoside | 17.58 | 6.26 | 1.11 (5,6) a | 95.5 |
| 7 | Quinic acid derivative | 18.253 | 6.54 | 1.04 (6,7) a | 99.8 |
| 8 | Chlorogenic acid b | 20.098 | 7.30 | 1.12 (7,8) a | 99.9 |
| 9 | Caffeic acid b | 22.151 | 8.15 | 1.12 (8,9) a | 95.0 |
| 10 | Hydroxycinnamic acid derivative | 25.042 | 9.35 | 1.15 (9,10 a | 98.2 |
| 11 | Quercetin-pentoside-hexoside | 26.287 | 9.86 | 1.06 (10,11) a | 99.9 |
| 12 | Quercetin-hexoside-hexoside | 26.729 | 10.05 | 1.02 (11,12) a | 99.9 |
| 13 | Quinic acid derivative | 27.165 | 10.23 | 1.02 (12,13) a | 97.2 |
| 14 | Quercetin-pentoside-hexoside | 30.031 | 11.41 | 1.12 (13,14) a | 90.2 |
| 15 | Caffeoyldihydroxyphenyllactoyltartaric acid | 31.097 | 11.85 | 1.04 (14,15) a | 90.2 |
| 16 | Quercetin-7-O-hexoside-3-O-(malonyl)hexoside | 33.45 | 12.82 | 1.08 (15,16) a | 85.8 |
| 17 | Luteolin hexoside hexoside | 33.754 | 12.95 | 1.01 (16,17) a | 79.8 |
| 18 | Caffeic acid derivative | 34.063 | 13.08 | 1.01 (17,18) a | 98.9 |
| 19 | Chicoric acid derivative | 34.63 | 13.31 | 1.02 (18,19) a | 95.5 |
| 20 | Luteolin-7-O-rutinoside | 36.458 | 14.07 | 1.06 (19,20) a | 99.2 |
| 21 | Quercetin pentoside | 36.745 | 14.18 | 1.01 (20,21) a | 99.2 |
| 22 | Quercetin hexoside | 36.908 | 14.25 | 1.00 (21,22) a | 99.7 |
| 23 | Luteolin-7-O-glucoside b | 37.407 | 14.46 | 1.01 (22,23) a | 99.9 |
| 24 | Quercetin pentoside | 39.198 | 15.20 | 1.05 (23,24) a | 80.9 |
| 25 | 3,5-Di-O-caffeoylquinic acid b | 40.647 | 15.80 | 1.04 (24,25) a | 82.8 |
| 26 | Chicoric acid b | 42.28 | 16.47 | 1.04 (25,26) a | 99.1 |
| 27 | Chicoric acid derivative | 47.19 | 18.50 | 1.12 (26,27) a | 99.8 |
| 28 | Caffeic acid derivative | 50.687 | 19.95 | 1.08 (27,28) a | 93.8 |
| 29 | Caffeoyl hexose-deoxyhexoside | 53.709 | 21.19 | 1.06 (28,29) a | 98.3 |
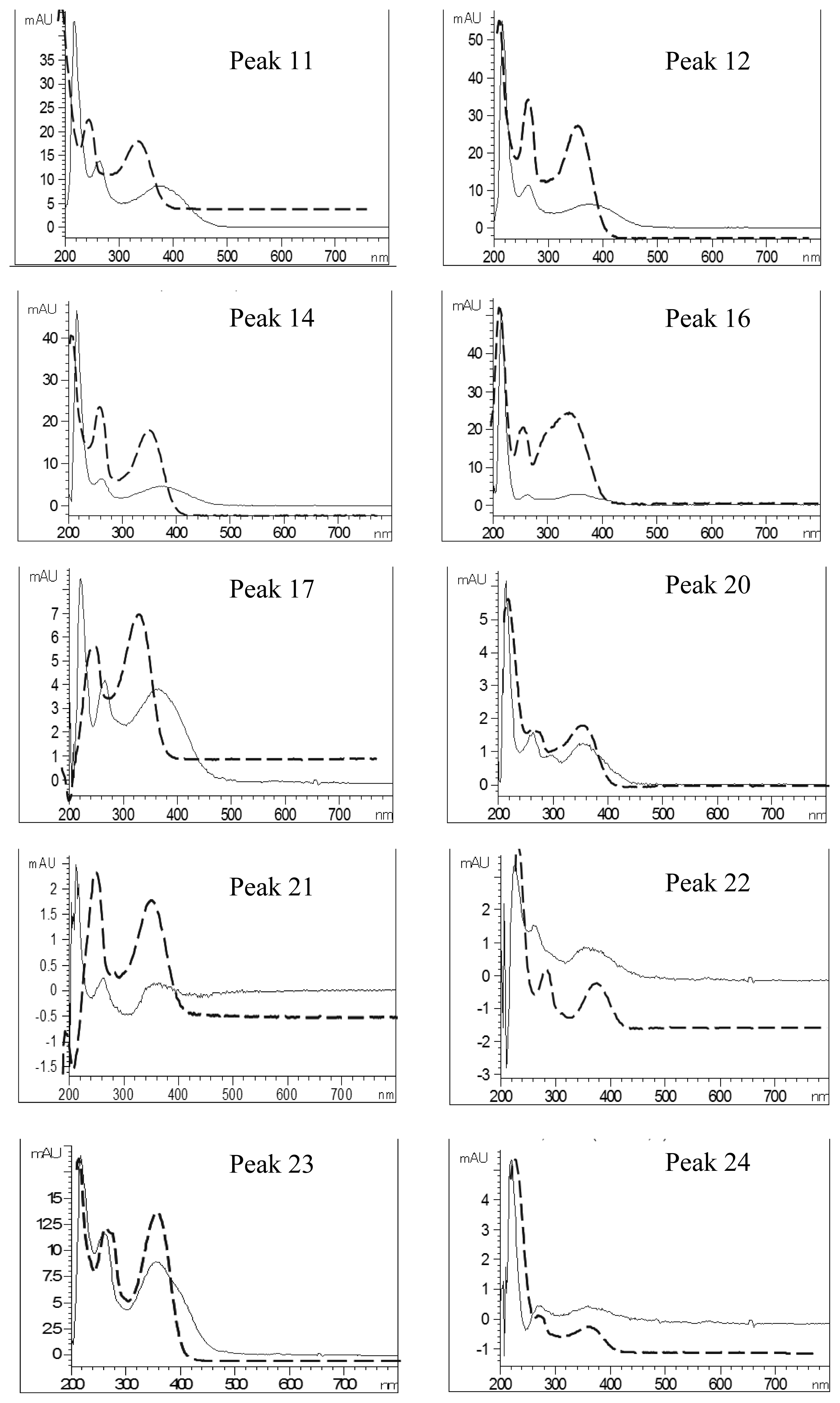
| Peak No. | Identity | Eluent UV Spectra (nm) | NaOAc+NaOH-Shifted UV Spectra (nm) | AlCl3-Shifted UV Spectra (nm) | AlCl3+NaOH-Shifted UV Spectra (nm) | ||||
|---|---|---|---|---|---|---|---|---|---|
| Band I | Band II | Band I | Band II | Band I | Band II | Band I | Band II | ||
| 11 | Quercetin-pentoside-hexoside | 358 | 260 | 376 | 264 | 394 | 268 | 402 | 270 |
| 12 | Quercetin-hexoside-hexoside | 358 | 260 | 378 | 264 | 394 | 268 | 402 | 270 |
| 14 | Quercetin-pentoside-hexoside | 358 | 260 | 374 | 262 | 392 | 268 | 404 | 268 |
| 16 | Quercetin-7-O-hexoside-3-O-(malonyl)hexoside | 354 | 260 | 356 | 264 | 392 | 268 | 398 (328) a | 266 |
| 17 | Luteolin hexoside hexoside | 348 | 260 | 362 | 264 | 386 (356) a | 292 | 400 (348) a | 270 |
| 20 | Luteolin-7-O-rutinoside | 350 | 260 | 354 | 262 | 386 (342) a | 272 | 390(344) a | 272 |
| 21 | Quercetin pentoside | 360 | 258 | 360 | 262 | 392 (286) a | 266 | 402 | 260 |
| 22 | Quercetin hexoside | 356 | 262 | 356 | 262 | 390 (286) a | 268 | 398 | 268 |
| 23 | Luteolin-7-O-glucoside | 348 | 260 | 354 | 260 | 388 (354) a | 270 | 398 (352) a | 270 |
| 24 | Quercetin pentoside | 356 | 258 | 362 | 272 | 390 | 268 | 400 | 268 |
| Peak No. | Retention Time (min) | Identity | [M − H]− (On-Line) (Parent Ion) | Fragment Ions (On-Line, MRM Mode) (Daughter Ion) | [M − H]− (Reported) (Parent Ion) | Fragment Ions (Reported) (Daughter Ion) |
|---|---|---|---|---|---|---|
| 1 | 7.397 | Protocatechuic acid hexoside | 315 b | 153 [M − H − hexose] | 315 b | 153 b |
| 2 | 11.596 | Caffeoyl hexoside | 341 c | 179 [M − H − hexose],135 [M − H − hexose − CO2] | 341 c | 179, 135 c |
| 3 | 12.781 | Caffeoyl-d-glucose | 339 d | - | 339 d | - |
| 4 | 13.86 | p-Hydroxybenzoic acid | 137 e | - | 137 e | - |
| 5 | 16.11 | cis-Caftaric acid a | 311 c | 179 [M − H − tartaric], 149 [M − H − caffeoyl] | 311 c | 149, 179 c |
| 6 | 17.58 | Caffeoyl hexoside | 341 c | 179 [M − H − hexose], 135 [M − H − hexose − CO2] | 341 c | 179, 135 c |
| 7 | 18.253 | Quinic acid derivative | - | - | - | - |
| 8 | 20.098 | Chlorogenic acid a | 353 c | 191 [M − H − caffeoyl], 179 [M − H − quinic] | 353 c | 191, 179 c |
| 9 | 22.151 | Caffeic acid a | 179 c | 135 [M − H − CO2] | 179 c | 135 c |
| 10 | 25.042 | Hydroxycinnamic acid derivative | 421 | - | - | - |
| 11 | 26.287 | Quercetin-pentoside-hexoside | 595 c | 433 [M − H − hexose], 301 [M − H − hexose − pentose] | 595 c | 433, 301 c |
| 12 | 26.729 | Quercetin-hexoside-hexoside | 625 c | 343, 301 [M − H − 2 hexose] | 625 c | 343, 301 c |
| 13 | 27.165 | Quinic acid derivative | 441 c | 279 [M − H − caffeoyl], 235 [M − H − caffeoyl − CO2] | 441 c | 279, 235 c |
| 14 | 30.031 | Quercetin-pentoside-hexoside | 595 c | 433 [M − H − hexose] | 595 c | 433 c |
| 15 | 31.097 | Caffeoyl-dihydroxyphenyllactoyl-tartaric acid | 491 c | 329 [M − H − caffeoyl], 293 [M − H − dihydroxyphenyl lactoyltartaric acid − H2O] | 491 c | 329, 293 c |
| 16 | 33.45 | Quercetin-7-O-hexoside-3-O-(malonyl)hexoside | 711 f | 667 [M − H − CO2], 301 [M − H − hexose − malonyl − hexose] | 711 f | 667, 301 f |
| 17 | 33.754 | Luteolin hexoside hexoside | 609 c | 285 [M − H − 2 hexose] | 609 c | 285 c |
| 18 | 34.063 | Caffeic acid derivative | 635 c | - | 635 c | - |
| 19 | 34.63 | Chicoric acid derivative | 473 c | 311 [M − H − caffeoyl], 293 [M − H − caffeoyl − H2O] | 473 c | 311, 293 c |
| 20 | 36.458 | Luteolin-7-O-rutinoside | 593 c | 285 [M − H − rutinose] | 593 c | 285 c |
| 21 | 36.745 | Quercetin pentoside | 433 c | 301 [M − H − pentose] | 433 c | 301 c |
| 22 | 36.908 | Quercetin hexoside | 463 g | 301 [M − H − hexose] | 463 g | 301 g |
| 23 | 37.407 | Luteolin-7-O-glucoside a | 447 c | 285 [M − H − hexose] | 447 c | 285 c |
| 24 | 39.198 | Quercetin pentoside | 433 c | - | 433 c | - |
| 25 | 40.647 | 3,5-Di-O-caffeoylquinic acid a | 515 c | 353 [M − H − caffeoyl], 173 [M − H − caffeoyl − quinic] | 515 c | 353, 173 c |
| 26 | 42.28 | Chicoric acid a | 473 c | 311 [M − H − caffeoyl] | 473 c | - |
| 27 | 47.19 | Chicoric acid derivative | 473 c | 293 [M − H − caffeoyl − H2O] | 473 c | - |
| 28 | 50.687 | Caffeic acid derivative | 357 h | 179 | 357 h | 179 h |
| 29 | 53.709 | Caffeoyl hexose-deoxyhexoside | 487 i | 308 [M − H − caffeoyl], 179 [M − H − deoxyhexose − hexose] | 487 i | 179 i |
| Peak No. | Identity | λmax (On-Line) | λmax (Reported) | Content (μg/g) |
|---|---|---|---|---|
| 1 | Protocatechuic acid hexoside | 220, 278 | 257, 291 b | 149.1 ± 3.41 |
| 2 | Caffeoyl hexoside | 226, 294, 318 | 234, 288sh, 297 c | 49.2 ± 1.95 |
| 3 | Caffeoyl-d-glucose | 222, 286, 338 | - | 26.3 ± 0.64 |
| 4 | p-Hydroxybenzoic acid | 232, 280, 310 | 278, 310sh d | 26.3 ± 1 |
| 5 | cis-Caftaric acid a | 218, 244, 302, 326 | 232, 277, 321 c | 1227.3 ± 31.71 |
| 6 | Caffeoyl hexoside | 214, 222, 290 | 233, 291 c | 752.4 ± 5.14 |
| 7 | Quinic acid derivative | 222, 264 | 230, 266 c | 204.3 ± 7.63 |
| 8 | Chlorogenic acid a | 218, 240, 298sh, 324 | 236, 303sh, 326 c | 837.2 ± 16.66 |
| 9 | Caffeic acid a | 248, 298, 324 | 241, 305sh, 323 c | 39.1 ± 1.96 |
| 10 | Hydroxycinnamic acid derivative | 236, 314 | 241, 291, 319 e | 14.1 ± 0.25 |
| 11 | Quercetin-pentoside-hexoside | 208, 260, 358 | 231, 260,358 c | 325.8 ± 12.11 |
| 12 | Quercetin-hexoside-hexoside | 208, 260, 358 | 230,261, 358 c | 176.8 ± 11.18 |
| 13 | Quinic acid derivative | 220, 266 | 230, 266 c | 173.3 ± 6.19 |
| 14 | Quercetin-pentoside-hexoside | 212, 260, 358 | 231, 260, 358 c | 192.7 ± 7.96 |
| 15 | Caffeoyl-dihydroxyphenyllactoyl-tartaric acid | 220, 288, 326 | 246, 300sh, 332 c | 135.0 ± 2.17 |
| 16 | Quercetin-7-O-hexoside-3-O-(malonyl)hexoside | 206, 260, 354 | - | 60.0 ± 4.43 |
| 17 | Luteolin hexoside hexoside | 210, 260, 348 | 255, 266sh, 347 c | 31.0 ± 1.84 |
| 18 | Caffeic acid derivative | 216, 246, 328 | 240, 310sh, 325 c | 29.0 ± 1.74 |
| 19 | Chicoric acid derivative | 212, 292, 326 | 242, 305sh, 328 c | 225.4 ± 2.25 |
| 20 | Luteolin-7-O-rutinoside | 206, 260, 350 | 255, 266sh, 348 c | 26.7 ± 1.01 |
| 21 | Quercetin pentoside | 208, 258, 360 | - | 75.6 ± 3.93 |
| 22 | Quercetin hexoside | 210, 262, 356 | 256, 300sh, 354 f | 12.4 ± 4.42 |
| 23 | Luteolin-7-O-glucoside a | 208, 260, 348 | 255, 266sh, 347 c | 175.9 ± 9.44 |
| 24 | Quercetin pentoside | 212, 258, 356 | - | 9.9 ± 0.48 |
| 25 | 3,5-Di-O-caffeoylquinic acid a | 220, 244, 300sh, 326 | 243, 303sh, 327 c | 989.3 ± 22.99 |
| 26 | Chicoric acid a | 220, 244, 304sh, 328 | 242, 305sh, 328 c | 10870.4 ± 150.05 |
| 27 | Chicoric acid derivative | 246, 302, 328 | 242, 305sh, 328 c | 653.4 ± 7.27 |
| 28 | Caffeic acid derivative | 212, 230, 314 | - | 120.5 ± 6.09 |
| 29 | Caffeoyl hexose-deoxyhexoside | 220, 244, 330sh, 328 | 290, 320 g | 51.8 ± 3.51 |
| Peak No. | Phenolic Acid/Flavonoid | Intra-Day Variability a | Inter-Day Variability a | ||
|---|---|---|---|---|---|
| Mean (μg/g) ± SD | RSD (%) | Mean (μg/g) ± SD | RSD (%) | ||
| 1 | Protocatechuic acid hexoside | 149.1 ± 3.4 | 2.3 | 147.8 ± 9.1 | 6.2 |
| 2 | Caffeoyl hexoside | 49.2 ± 2.0 | 4.0 | 51.2 ± 2.8 | 5.5 |
| 3 | Caffeoyl-d-glucose | 26.3 ± 0.6 | 2.4 | 26.9 ± 1.1 | 4.3 |
| 4 | p-Hydroxybenzoic acid | 16.6 ± 1.0 | 6.0 | 15.9 ± 1.2 | 7.5 |
| 5 | cis-Caftaric acid | 1105.3 ± 31.7 | 2.9 | 1096.7 ± 32.0 | 2.9 |
| 6 | Caffeoyl hexoside | 84.9 ± 5.1 | 6.1 | 78.0 ± 5.8 | 7.5 |
| 7 | Quinic acid derivative | 191.5 ± 7.6 | 4.0 | 187.1 ± 3.7 | 2.0 |
| 8 | Chlorogenic acid | 784.9 ± 16.7 | 2.1 | 760.2 ± 18.6 | 2.5 |
| 9 | Caffeic acid | 39.1 ± 2.0 | 5.0 | 38.5 ± 2.6 | 6.8 |
| 10 | Hydroxycinnamic acid derivative | 14.1 ± 0.3 | 1.7 | 14.6 ± 0.8 | 5.7 |
| 11 | Quercetin-pentoside-hexoside | 325.8 ± 12.1 | 3.7 | 329.8 ± 14.7 | 4.4 |
| 12 | Quercetin-hexoside-hexoside | 176.8 ± 11.2 | 6.3 | 175.5 ± 9.9 | 5.7 |
| 13 | Quinic acid derivative | 162.4 ± 6.2 | 3.8 | 164.2 ± 7.3 | 4.4 |
| 14 | Quercetin-pentoside-hexoside | 192.7 ± 8.0 | 4.1 | 188.8 ± 6.8 | 3.6 |
| 15 | Caffeoyldihydroxyphenyllactoyltartaric acid | 135.0 ± 2.2 | 1.6 | 117.6 ± 6.9 | 5.8 |
| 16 | Quercetin-7-O-hexoside-3-O-(malonyl)hexoside | 59.9 ± 4.4 | 7.4 | 56.0 ± 3.9 | 7.0 |
| 17 | Luteolin hexoside hexoside | 26.3 ± 1.8 | 7.0 | 28.4 ± 1.6 | 5.6 |
| 18 | Caffeic acid derivative | 28.9 ± 1.7 | 6.0 | 30.6 ± 2.0 | 6.6 |
| 19 | Chicoric acid derivative | 215.4 ± 2.3 | 1.0 | 219.8 ± 4.3 | 2.0 |
| 20 | Luteolin-7-O-rutinoside | 11.4 ± 0.7 | 6.1 | 14.7 ± 1.1 | 7.1 |
| 21 | Quercetin pentoside | 64.1 ± 3.9 | 6.1 | 70.4 ± 5.1 | 7.3 |
| 22 | Quercetin hexoside | 60.5 ± 4.4 | 7.3 | 60.6 ± 4.9 | 8.1 |
| 23 | Luteolin-7-O-glucoside | 149.3 ± 9.4 | 6.3 | 139.6 ± 2.1 | 1.5 |
| 24 | Quercetin pentoside | 8.4 ± 0.5 | 5.7 | 7.9 ± 0.2 | 2.5 |
| 25 | 3,5-Di-O-caffeoylquinic acid | 890.9 ± 23.0 | 2.6 | 857.3 ± 65.8 | 7.7 |
| 26 | Chicoric acid | 10390.0 ± 150.1 | 1.4 | 10392.4 ± 429.7 | 4.1 |
| 27 | Chicoric acid derivative | 624.5 ± 7.3 | 1.2 | 621.1 ± 25.1 | 4.0 |
| 28 | Caffeic acid derivative | 120.4 ± 6.1 | 5.1 | 110.0 ± 7.5 | 6.9 |
| 29 | Caffeoyl hexose-deoxyhexoside | 51.8 ± 3.5 | 6.8 | 52.4 ± 2.7 | 5.2 |
| Phenolic Acid/Flavonoid | Original (μg) | Spiked (μg) | Found (μg) | Recovery (%) | Mean ± SD (%) | RSD (%) |
|---|---|---|---|---|---|---|
| cis-Caftaric acid | 28.9 | 16.8 | 43.7 | 88.3 | 90.1 ± 2.4 | 2.7 |
| 27.6 | 16.8 | 43.1 | 91.8 | |||
| Chlorogenic acid | 15.3 | 20.7 | 34.5 | 92.7 | 93.8 ± 1.5 | 1.6 |
| 15.3 | 20.7 | 34.9 | 94.8 | |||
| Caffeic acid | 3.0 | 20.3 | 22.0 | 93.6 | 94.1 ± 0.7 | 0.7 |
| 2.7 | 20.3 | 21.9 | 93.7 | |||
| Luteolin-7-O-glucoside | 2.0 | 18.6 | 17.9 | 85.0 | 84.9 ± 0.3 | 0.4 |
| 2.0 | 18.6 | 17.8 | 84.7 | |||
| 3,5-Di-caffeoylquinic acid | 23.1 | 20.9 | 42.8 | 94.5 | 93.1 ± 1.9 | 2.0 |
| 23.6 | 20.9 | 42.8 | 91.8 | |||
| Quercetin | 0 | 20.1 | 18.8 | 93.6 | 95.3 ± 2.5 | 2.6 |
| 0 | 20.1 | 19.5 | 97.0 | |||
| Chicoric acid | 317.1 | 25.8 | 342.2 | 97.5 | 95.6 ± 2.7 | 2.8 |
| 318.0 | 25.8 | 342.2 | 93.7 |
© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Chen, H.-J.; Inbaraj, B.S.; Chen, B.-H. Determination of Phenolic Acids and Flavonoids in Taraxacum formosanum Kitam by Liquid Chromatography-Tandem Mass Spectrometry Coupled with a Post-Column Derivatization Technique. Int. J. Mol. Sci. 2012, 13, 260-285. https://doi.org/10.3390/ijms13010260
Chen H-J, Inbaraj BS, Chen B-H. Determination of Phenolic Acids and Flavonoids in Taraxacum formosanum Kitam by Liquid Chromatography-Tandem Mass Spectrometry Coupled with a Post-Column Derivatization Technique. International Journal of Molecular Sciences. 2012; 13(1):260-285. https://doi.org/10.3390/ijms13010260
Chicago/Turabian StyleChen, Hung-Ju, Baskaran Stephen Inbaraj, and Bing-Huei Chen. 2012. "Determination of Phenolic Acids and Flavonoids in Taraxacum formosanum Kitam by Liquid Chromatography-Tandem Mass Spectrometry Coupled with a Post-Column Derivatization Technique" International Journal of Molecular Sciences 13, no. 1: 260-285. https://doi.org/10.3390/ijms13010260