High-Dimensional Descriptor Selection and Computational QSAR Modeling for Antitumor Activity of ARC-111 Analogues Based on Support Vector Regression (SVR)

Abstract
:1. Introduction
2. Results and Discussion
2.1. Comparative QSAR Modeling with the Low-Dimensional Literature Descriptors Using Stepwise MLR, PLS, ANN and SVR Techniques
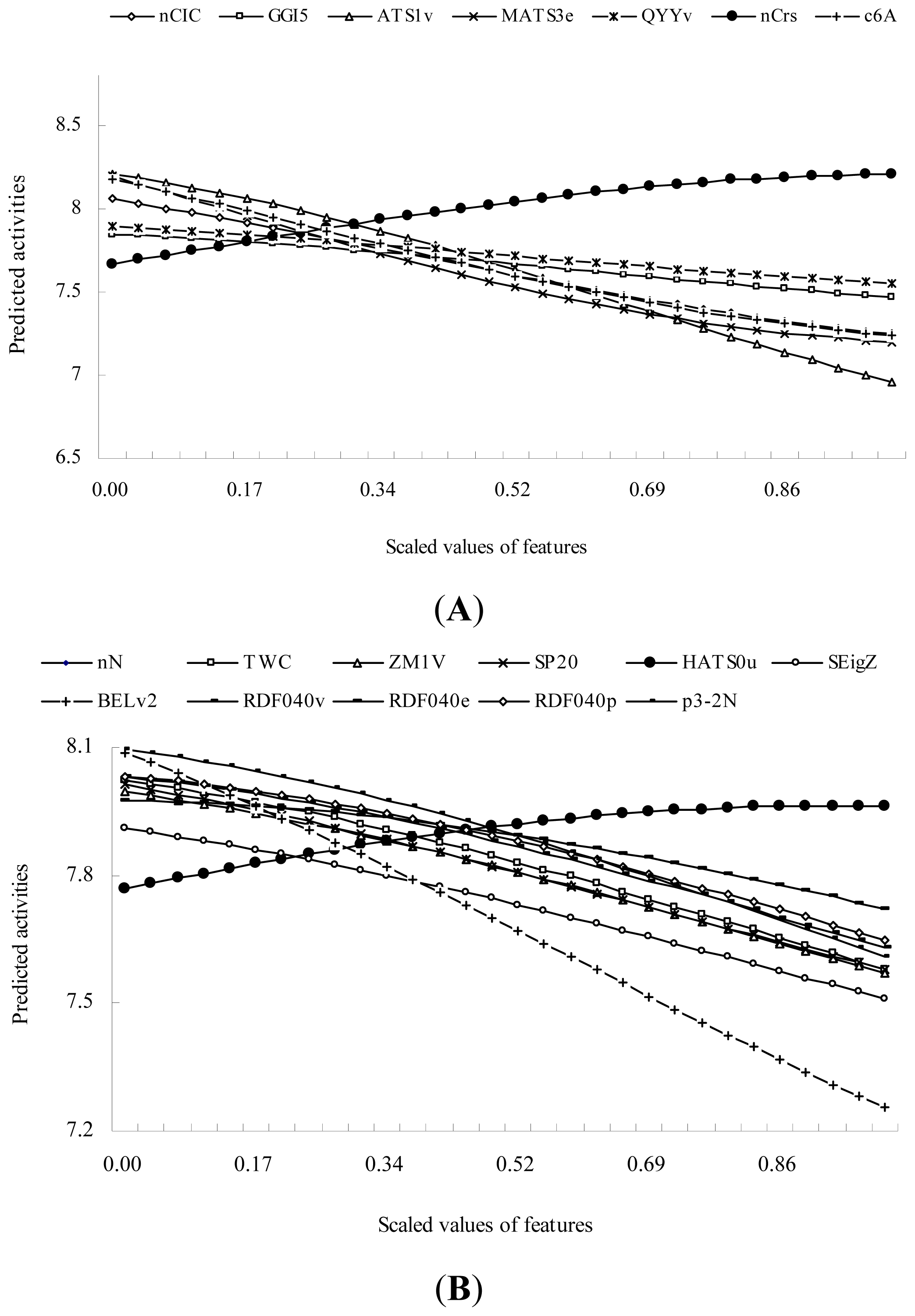
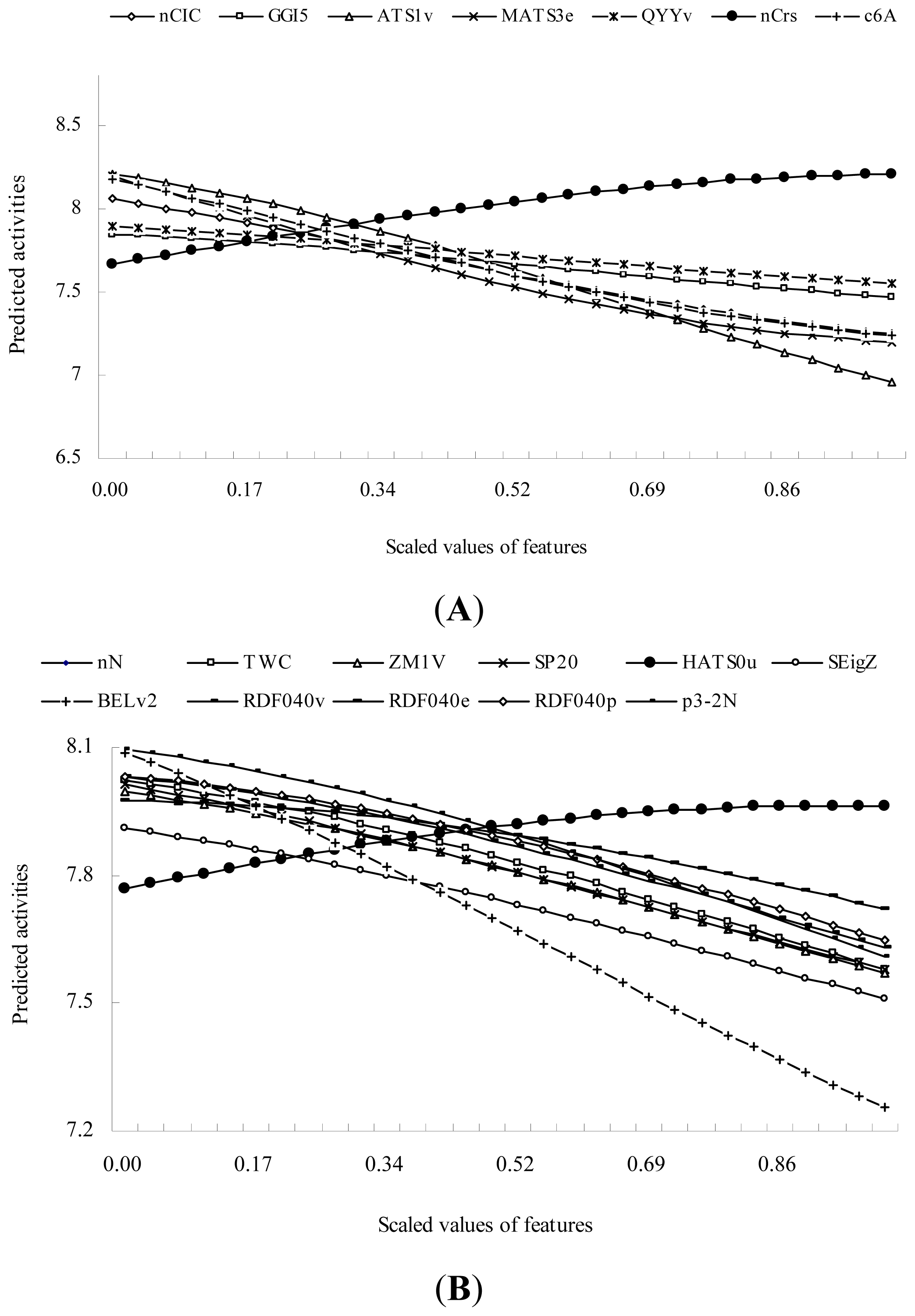
2.2. QSAR Modeling with the High-Dimensional Descriptors Using SVR Technique
3. Materials and Methods
3.1. Structures and Activities
3.2. Calculation of Molecular Descriptors
3.3. Model Development
3.4. Model Evaluation
4. Conclusions
Acknowledgments
References
- Rasheed, Z.A.; Rubin, E.H. Mechanisms of resistance to topoisomerase I-targeting drugs. Oncogene 2003, 22, 7296–7304. [Google Scholar]
- Li, T.K.; Houghton, P.J.; Desai, S.D.; Daroui, P.; Liu, A.A.; Hars, E.S.; Ruchelman, A.L.; LaVoie, E.J.; Liu, L.F. Characterization of ARC-111 as a novel Topoisomerase I-targeting anticancer drug. Cancer Res. 2003, 63, 8400–8407. [Google Scholar]
- Ruchelman, A.L.; Houghton, P.J.; Zhou, N.; Liu, A.; Liu, L.F.; LaVoie, E.J. 5-(2-Aminoethyl)dibenzo[c,h][1,6]naphthyridin-6-ones: variation of N-alkyl substituents modulates sensitivity to efflux transporters associated with multidrug resistance. J. Med. Chem 2005, 48, 792–804. [Google Scholar]
- Yu, Y.J.; Su, R.X.; Wang, L.B.; Qi, W.; He, Z.M. Comparative QSAR modeling of antitumor activity of ARC-111 analogues using stepwise MLR, PLS, and ANN techniques. Med. Chem. Res 2010, 19, 1233–1244. [Google Scholar]
- Tetko, I.V.; Gasteiger, J.; Todeschini, R.; Mauri, A.; Livingstone, D.; Ertl, P.; Palyulin, V.A.; Radchenko, E.V.; Zefirov, N.S.; Makarenko, A.S.; et al. Virtual computational chemistry laboratory - design and description. J. Comput. Aid. Mol. Des 2005, 19, 453–463. [Google Scholar]
- Cristianini, N.; Shawe-Taylor, J. An Introduction to Support Vector Machines and Other Kernel-Based Learning Methods; Cambridge University Press: Cambridge, UK, 2000. [Google Scholar]
- Tan, X.S.; Yuan, Z.M.; Zhou, T.J.; Wang, C.J.; Xiong, J.Y. Multi-KNN-SVR combinatorial forecast and its application to QSAR of fluorine-containing compounds. Chem. J. Chin. Univ 2008, 29, 95–99. [Google Scholar]
- Dai, Z.J.; Zhou, W.; Yuan, Z.M. A novel method of nonlinear rapid feature selection for high-dimensional features and its application in peptide QSAR modeling based on support vector machine. Acta Phys. Chim. Sin 2011, 27, 1654–1660. [Google Scholar]
- Wang, L.F.; Tan, X.S.; Bai, L.Y.; Yuan, Z.M. Establishing an interpretability system for support vector regression and its application in QSAR of organophosphorus insecticide. Asian J. Chem 2011, 24. in press. [Google Scholar]
- Geerts, T.; Vander Heyden, Y. In-silico predictions of ADME-Tox properties: Drug absorption. Comb. Chem. High Throughput Screen 2011, 14, 339–361. [Google Scholar]
- Marković, V.; Erić, S.; Juranić, Z.D.; Stanojković, T.; Joksović, L.; Ranković, B.; Kosanić, M.; Joksović, M.D. Synthesis, antitumor activity and QSAR studies of some 4-aminomethylidene derivatives of edaravone. Bioorg. Chem 2011, 39, 18–27. [Google Scholar]
- Prabhakar, Y.S.; Rawal, R.K.; Gupta, M.K.; Solomon, V.R.; Katti, S.B. Topological descriptors in modeling the HIV inhibitory activity of 2-Aryl-3-pyridyl-thiazolidin-4-ones. Comb. Chem. High Throughput Screen 2005, 8, 431–437. [Google Scholar]
- Riahi, S.; Ganjali, M.R.; Pourbasheer, E.; Norouzi, P. QSRR study of GC retention indices of essential-oil compounds by multiple linear regression with a genetic algorithm. Chromatographia 2008, 67, 917–922. [Google Scholar]
- Goudarzia, N.; Fatemib, M.H.; Samadi-Maybodib, A. Quantitative structure-properties relationship study of the 29Si-NMR chemical shifts of some silicate species. Spectrosc. Lett 2009, 42, 186–193. [Google Scholar]
- Cao, D.S.; Xu, Q.S.; Liang, Y.Z.; Chen, X.; Li, H.D. Prediction of aqueous solubility of druglike organic compounds using partial least squares, back-propagation network and support vector machine. J. Chemometr 2010, 24, 584–595. [Google Scholar]
- Gozalbes, R.; Pineda-Lucena, A. QSAR-based solubility model for drug-like compounds. Bioorgan. Med. Chem 2010, 18, 7078–7084. [Google Scholar]
- Lapinsh, M.; Veiksina, S.; Uhlén, S.; Petrovska, R.; Mutule, I.; Mutulis, F.; Yahorava, S.; Prusis, P.; Wikberg, J.E.S. Proteochemometric mapping of the interaction of organic compounds with melanocortin receptor subtypes. Mol. Pharmacol 2005, 67, 50–59. [Google Scholar]
- Liang, G.Z.; Yang, S.B.; Zhou, Y.; Zhou, P.; Li, Z.L. Using scores of amino acid topological descriptors for quantitative sequence-mobility modeling of peptides based on support vector machine. Chin. Sci. Bull 2006, 51, 2700–2705. [Google Scholar]
- Bansal, R.; Karthikeyan, C.; Moorthy, N.S.H.N.; Trivedi, P. QSAR analysis of some phthalimide analogues based inhibitors of HIV-1 integrase. Arkivoc 2007, XV, 66–81. [Google Scholar]
- Malik, A.; Singh, H.; Andrabi, M.; Husain, S.A.; Ahmad, S. Databases and QSAR for cancer research. Cancer Inform 2006, 2, 99–111. [Google Scholar]
- Lapins, M.; Wikberg, J.E. Proteochemometric modeling of drug resistance over the mutational space for multiple HIV protease variants and multiple protease inhibitors. J. Chem. Inf. Model 2009, 49, 1202–1210. [Google Scholar]
- Tanabe, K.; Lučić, B.; Amić, D.; Kurita, T.; Kaihara, M.; Onodera, N.; Suzuki, T. Prediction of carcinogenicity for diverse chemicals based on substructure grouping and SVM modeling. Mol. Divers 2010, 14, 789–802. [Google Scholar]
- Gharagheizi, F. An accurate model for prediction of autoignition temperature of pure compounds. J. Hazard. Mater 2011, 189, 211–221. [Google Scholar]
- Fedorowicz, A.; Zheng, L.; Singh, H.; Demchuk, E. QSAR study of skin sensitization using local lymph node assay data. Int. J. Mol. Sci 2004, 5, 56–66. [Google Scholar]
- González, M.P.; Terán, C.; Teijeira, M.; Besada, P.; González-Moa, M.J. BCUT descriptors for predicting affinity toward A3 adenosine receptors. Bioorg. Med. Chem. Lett 2005, 15, 3491–3495. [Google Scholar]
- Pasha, F.A.; Srivastava, H.K.; Srivastava, A.; Singh, P.P. QSTR study of small organic molecules against Tetrahymena pyriformis. QSAR Comb. Sci 2007, 26, 69–84. [Google Scholar]
- Putz, M.V.; Putz, A.-M.; Lazea, M.; Ienciu, L.; Chiriac, A. Quantum-SAR extension of the spectral-SAR algorithm. Application to polyphenolic anticancer bioactivity. Int. J. Mol. Sci 2009, 10, 1193–1214. [Google Scholar]
- Putz, M.V.; Lacrămă, A.-M. Introducing spectral structure activity relationship (S-SAR) analysis. Application to ecotoxicology. Int. J. Mol. Sci 2007, 8, 363–391. [Google Scholar]
- Lacrămă, A.-M.; Putz, M.V.; Ostafe, V. A spectral-SAR model for the anionic-cationic interaction in ionic liquids: application to vibrio fischeri ecotoxicity. Int. J. Mol. Sci 2007, 8, 842–863. [Google Scholar]
- Chicu, S.A.; Putz, M.V. Köln-Timişoara molecular activity combined models toward interspecies toxicity assessment. Int. J. Mol. Sci 2009, 10, 4474–4497. [Google Scholar]
- Chen, Y.; Yuan, Z.M.; Zhou, W.; Xiong, X.Y. A novel QSAR model based on geostatistics and support vector regression. Acta Phys. Chim. Sin 2009, 25, 1587–1592. [Google Scholar]
- Zhang, S.X.; Wei, L.Y.; Bastow, K.; Zheng, W.F.; Brossi, A.; Lee, K.H.; Tropsha, A. Antitumor agents 252. Application of validated QSAR models to database mining: discovery of novel tylophorine derivatives as potential anticancer agents. J. Comput. Aid. Mol. Des 2007, 21, 97–112. [Google Scholar]

{kind=link}
{kind=link}
| Stepwise MLR | PLS | ANN | SVR1 | SVR2 | |
|---|---|---|---|---|---|
| Number of descriptors | 5 | 7 | 9 | 9 | 6 |
| MSE | 0.201 | 0.167 | 0.050 | 0.141 | 0.061 |
| R2 | 0.910 | 0.890 | 0.962 | 0.937 | 0.950 |
| R2 | 0.730 | 0.775 | 0.933 | 0.811 | 0.918 |
| Stepwise MLR | PLS | ANN | SVR3 | SVR4 | |
|---|---|---|---|---|---|
| Number of descriptors | 5 | 7 | 9 | 7 | 11 |
| MSE | 0.201 | 0.167 | 0.050 | 0.032 | 0.028 |
| R2 | 0.910 | 0.890 | 0.962 | 0.964 | 0.971 |
| Rpred2 | 0.730 | 0.775 | 0.933 | 0.957 | 0.962 |
| Model | Group name | Descriptor name | F-value |
|---|---|---|---|
| SVR3 | GSFRAG | c6A: Number of fragments Cyc6[A] | 26.555 ** |
| 2D autocorrelations | ATS1v: Broto-Moreau autocorrelation of a topological structure - lag 1 / weighted by atomic van der Waals volumes | 25.175 ** | |
| Constitutional descriptors | nCIC: Number of rings | 12.210 ** | |
| 2D autocorrelations | MATS3e: Moran autocorrelation - lag 3 / weighted by atomic Sanderson electronegativities | 12.114 ** | |
| Functional group counts | nCrs: Number of ring secondary C(sp3) | 5.898 * | |
| Topological charge indices | GGI5: Topological charge index of order 5 | 3.687 | |
| Geometrical descriptors | QYYv: Qyy COMMA2 value / weighted by atomic van der Waals volumes | 2.387 | |
| SVR4 | BCUT descriptors | BELv2: Lowest eigenvalue n. 2 of Burden matrix / weighted by atomic van der Waals volumes | 11.382 * |
| GSFRAG-L | p3-2N: Number of fragments Path3 with label N on atom 2 | 3.771 | |
| Randic molecular profiles | SP20: Shape profile no. 20 | 3.511 | |
| Eigenvalue-based indices | SEigZ: Eigenvalue sum from Z weighted distance matrix (Barysz matrix) | 2.456 | |
| Constitutional descriptors | nN: Number of Nitrogen atoms | 2.456 | |
| RDF descriptors | RDF040v: Radial distribution function - 4.0 / weighted by atomic van der Waals volumes | 2.435 | |
| Walk and path counts | TWC: Total walk count | 2.425 | |
| RDF descriptors | RDF040p: Radial distribution function - 4.0 / weighted by atomic polarizabilities | 2.398 | |
| Topological descriptors | ZM1V: first Zagreb index by valence vertex degrees | 2.084 | |
| RDF descriptors | RDF040e: Radial distribution function - 4.0 / weighted by atomic Sanderson electronegativities | 1.304 | |
| GETAWAY descriptors | HATS0u: Leverage-weighted autocorrelation of lag 0 / unweighted | 0.599 |
| Experimental drugs | Theoretical drugs | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Compound | Type | Substituent | pIC50 (expt.) | Compound | Type | Substituent | pIC50 (pred.) b | ||||||
| R1 | R2 | R3 | R4 | R1 | R2 | R3 | R4 | ||||||
| 1 | I | Me | Me | 8.699 | 1 | I | Me | Et | 8.651 | ||||
| 2 | Me | Bn | 7.276 | 2 | Me | t-Bu | 8.172 | ||||||
| 3 | Et | Bn | 7.114 | 3 | Et | t-Bu | 7.876 | ||||||
| 4 | i-Pr | Bn | 6.523 | 4 | t-Bu | t-Bu | 7.388 | ||||||
| 5 | t-Bu | Bn | 6.071 | 5 | t-Bu | i-Pr | 6.908 | ||||||
| 6 | Bn | Bn | 6.420 | 6 | III | Bn | 6.668 | ||||||
| 7 | Et | Et | 8.222 | 7 | Et | 7.208 | |||||||
| 8 | i-Pr | i-Pr | 8.097a | 8 | t-Bu | 6.904 | |||||||
| 9 | H | Me | 9.523 | 9 | i-Pr | 6.617 | |||||||
| 10 | H | Et | 8.699a | 10 | IV | Et | 6.248 | ||||||
| 11 | H | i-Pr | 8.523 | 11 | t-Bu | 6.102 | |||||||
| 12 | H | t-Bu | 8.699 | 12 | i-Pr | 6.100 | |||||||
| 13 | H | Bn | 7.796 | ||||||||||
| 14 | H | H | 8.398 | ||||||||||
| 15 | Me | i-Pr | 8.097a | ||||||||||
| 16 | Et | i-Pr | 8.301 | ||||||||||
| 17 | II | 8.523 | |||||||||||
| 18 | III | H | 8.155 | ||||||||||
| 19 | Me | 7.523 | |||||||||||
| 20 | IV | Bn | 6.398a | ||||||||||
| 21 | H | 7.046 | |||||||||||
| 22 | Me | 6.523 | |||||||||||
| Group No. | Group of descriptors | Count | Group No. | Group of descriptors | Count |
|---|---|---|---|---|---|
| 1 | Constitutional descriptors | 48 | 13 | RDF descriptors | 150 |
| 2 | Topological descriptors | 119 | 14 | 3D-MoRSE descriptors | 160 |
| 3 | Walk and path counts | 47 | 15 | WHIM descriptors | 99 |
| 4 | Connectivity indices | 33 | 16 | GETAWAY descriptors | 197 |
| 5 | Information indices | 47 | 17 | Functional group counts | 121 |
| 6 | 2D autocorrelations | 96 | 18 | Atom-centered fragments | 120 |
| 7 | Edge adjacency indices | 107 | 19 | Charge descriptors | 14 |
| 8 | BCUT descriptors | 64 | 20 | Molecular properties | 28 |
| 9 | Topological charge indices | 21 | 21 | ET-state Indices | >300 |
| 10 | Eigenvalue-based indices | 44 | 22 | ET-state Properties * | 3 |
| 11 | Randic molecular profiles | 41 | 23 | GSFRAG Descriptor | 307 |
| 12 | Geometrical descriptors | 74 | 24 | GSFRAG-L Descriptor | 886 |
| Total: | >3000 | ||||
© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Zhou, W.; Dai, Z.; Chen, Y.; Wang, H.; Yuan, Z. High-Dimensional Descriptor Selection and Computational QSAR Modeling for Antitumor Activity of ARC-111 Analogues Based on Support Vector Regression (SVR). Int. J. Mol. Sci. 2012, 13, 1161-1172. https://doi.org/10.3390/ijms13011161
Zhou W, Dai Z, Chen Y, Wang H, Yuan Z. High-Dimensional Descriptor Selection and Computational QSAR Modeling for Antitumor Activity of ARC-111 Analogues Based on Support Vector Regression (SVR). International Journal of Molecular Sciences. 2012; 13(1):1161-1172. https://doi.org/10.3390/ijms13011161
Chicago/Turabian StyleZhou, Wei, Zhijun Dai, Yuan Chen, Haiyan Wang, and Zheming Yuan. 2012. "High-Dimensional Descriptor Selection and Computational QSAR Modeling for Antitumor Activity of ARC-111 Analogues Based on Support Vector Regression (SVR)" International Journal of Molecular Sciences 13, no. 1: 1161-1172. https://doi.org/10.3390/ijms13011161
APA StyleZhou, W., Dai, Z., Chen, Y., Wang, H., & Yuan, Z. (2012). High-Dimensional Descriptor Selection and Computational QSAR Modeling for Antitumor Activity of ARC-111 Analogues Based on Support Vector Regression (SVR). International Journal of Molecular Sciences, 13(1), 1161-1172. https://doi.org/10.3390/ijms13011161