Structure–Activity Relationship of Halophenols as a New Class of Protein Tyrosine Kinase Inhibitors



Abstract
:1. Introduction
2. Results and Discussion
2.1. Chemistry
2.2. In vitro PTK Inhibitory Activity
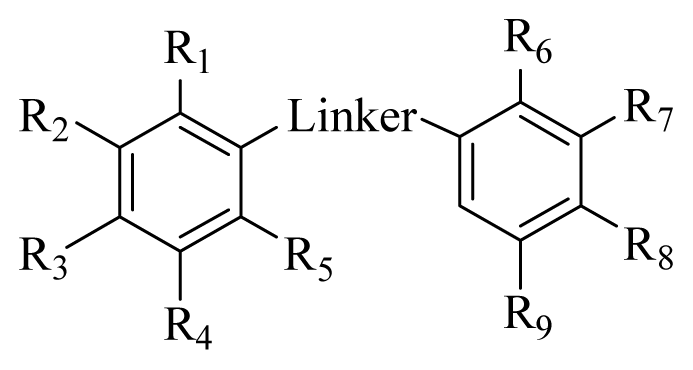
2.3. SAR Analysis
3. Experimental Section
3.1. General
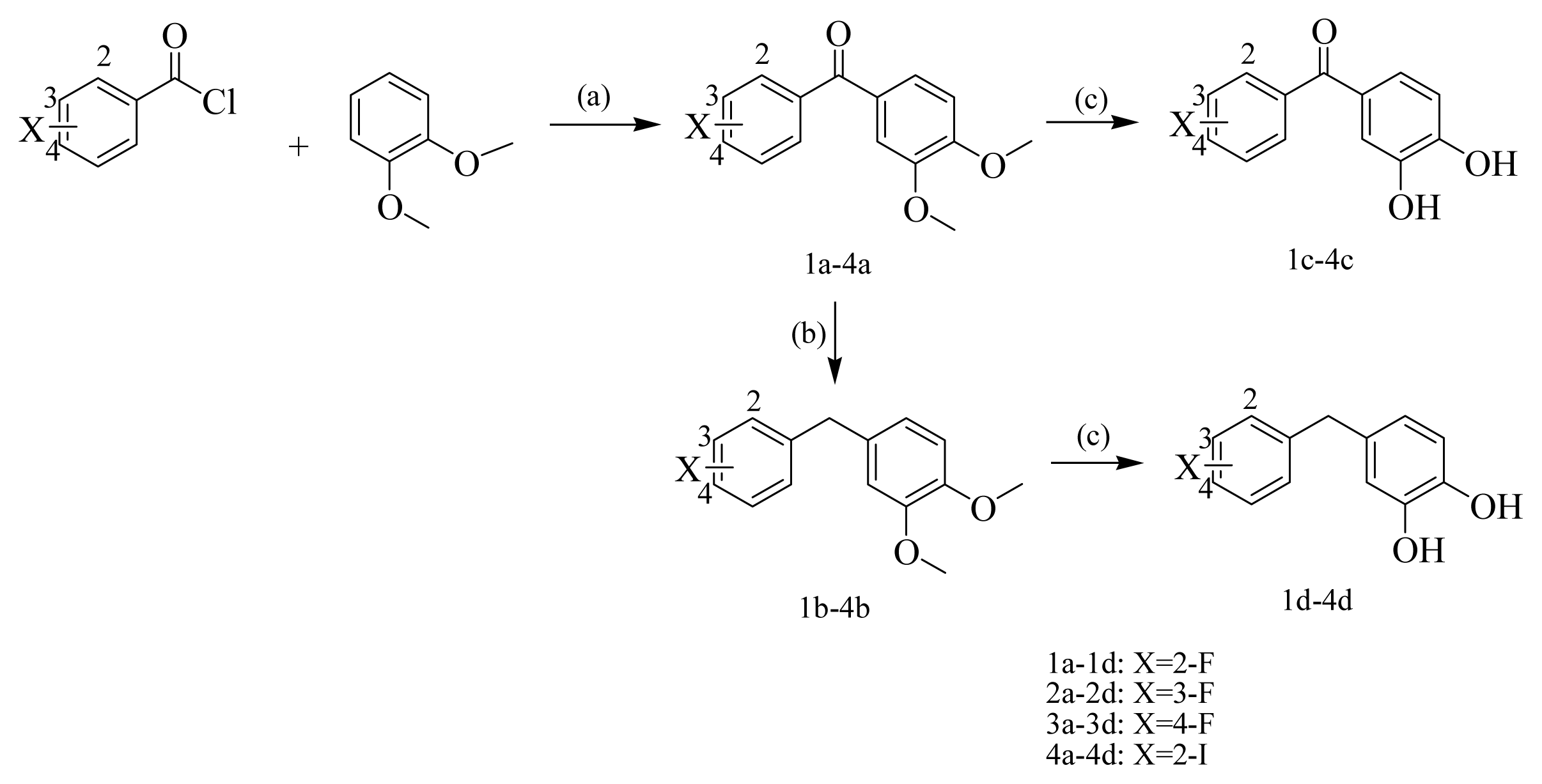
3.2. Typical Procedures for the Preparation of Halophenol Derivatives
3.2.1. Preparation of compounds 1a–4a
2-fluoro-3′,4′-dimethoxyl benzophenone (1a)
3-fluoro-3′,4′-dimethoxyl benzophenone (2a)
4-fluoro-3′,4′-dimethoxyl benzophenone (3a)
2-iodo-3′,4′-dimethoxyl benzophenone (4a)
3.2.2. The Preparation of Compounds 1b–4b
2-fluoro-3′,4′-dimethoxyl diphenylmethane (1b)
3-fluoro-3′,4′-dimethoxyl diphenylmethane (2b)
4-fluoro-3′,4′-dimethoxyl diphenylmethane (3b)
2-iodo-3′,4′-dimethoxyl diphenylmethane (4b)
3.2.3. The Preparation of Compounds 1c–4c
2-fluoro-3′,4′-dihydroxyl benzophenone (1c)
3-fluoro-3′,4′-dihydroxyl benzophenone (2c)
4-fluoro-3′,4′-dihydroxyl benzophenone (3c)
2-iodo-3′,4′-dihydroxyl benzophenone (4c)
3.2.4. The Preparation of Compounds 1d–4d
2-fluoro-3′,4′-dihydroxyl diphenylmethane (1d)
3-fluoro-3′,4′-dihydroxyl diphenylmethane (2d)
4-fluoro-3′,4′-dihydroxyl diphenylmethane (3d)
2-iodo-3′,4′-dihydroxyl diphenylmethane (4d)
3.3. Pharmacology
4. Conclusions
Acknowledgments
- Conflict of InterestThe authors have declared no conflict of interest.
References
- Bolla, M; Rostaing-Puissant, B; Bottari, SP; Chedin, M; Marron-Charriere, J; Colonna, M; Berland, E; Chambaz, E. Protein tyrosine kinase activity in 350 T1/T2, N0/N1 breast cancer. Preliminary results. Breast Cancer Res Treat 1996, 39, 327–334. [Google Scholar]
- Hirabayashi, A; Mukaiyama, H; Kobayashi, H; Shiohara, H; Nakayama, S; Ozawa, M; Tsuji, E; Miyazawa, K; Misawa, K; Ohnota, H; et al. Structure-activity relationship studies of imidazo[1,2-c]pyrimidine derivatives as potent and orally effective Syk family kinases inhibitors. Bioorg Med Chem 2008, 16, 9247–9260. [Google Scholar]
- Jung, KJ; Lee, EK; Yu, BP; Chung, HY. Significance of protein tyrosine kinase/protein tyrosine phosphatase balance in the regulation of NF-κB signaling in the inflammatory process and aging. Free Radic Biol Med 2009, 47, 983–991. [Google Scholar]
- Shi, L; Feng, XE; Cui, JR; Fang, LH; Du, GH; Li, QS. Synthesis and biological activity of flavanone derivatives. Bioorg Med Chem Lett 2010, 20, 5466–5468. [Google Scholar]
- Hori, H; Nagasawa, H; Uto, Y; Ohkura, K; Kirk, KL; Uehara, Y; Shimamura, M. Design of hypoxia-targeting protein tyrosine kinase inhibitor using an innovative pharmacophore 2-methylene-4-cyclopentene-1,3-dione. Biochim Biophys Acta 2004, 1697, 29–38. [Google Scholar]
- Liechti, C; Sequin, U; Bold, G; Furet, P; Meyer, T; Traxler, P. Salicylanilides as inhibitors of the protein tyrosine kinase epidermal growth factor receptor. Eur J Med Chem 2004, 39, 11–26. [Google Scholar]
- Li, HQ; Zhu, TT; Yan, T; Luo, Y; Zhu, HL. Design, synthesis and structure–activity relationships of antiproliferative 1,3-disubstituted urea derivatives. Eur J Med Chem 2009, 44, 453–459. [Google Scholar]
- Kilic, Z; Isgor, YG; Olgen, S. Synthesis and pp60c-Src tyrosine kinase inhibitory activities of novel indole-3-imine and amine derivatives substituted at N1 and C5. Arch Pharm Chem Life Sci 2009, 342, 333–343. [Google Scholar]
- Olgen, S; Isgor, YG; Coban, T. Synthesis and activity of novel 5-substituted pyrrolo[2,3-d]pyrimidine analogues as pp60c-Src tyrosine kinase inhibitors. Arch Pharm Chem Life Sci 2008, 341, 113–120. [Google Scholar]
- Mazitschek, R; Giannis, A. Inhibitors of angiogenesis and cancer-related receptor tyrosine kinases. Curr Opin Chem Biol 2004, 8, 432–441. [Google Scholar]
- Li, J; Guo, SJ; Su, H; Han, LJ; Shi, DY. Total synthesis of bis-(2,3-dibromo-4,5- dihydroxyphenyl)-methane as potent PTP1B inhibitor. Chin Chem Lett 2008, 19, 1290–1292. [Google Scholar]
- Li, K; Li, XM; Ji, NY; Gloer, JB; Wang, BG. Urceolatin, a structurally unique bromophenol from Polysiphonia urceolata. Bioorg Med Chem 2007, 15, 6627–6631. [Google Scholar]
- Duan, XJ; Li, XM; Wang, BG. Highly brominated mono- and bis-phenols from the marine red alga symphyocladia latiuscula with radical-scavenging activity. J Nat Prod 2007, 70, 1210–1213. [Google Scholar]
- Shi, DY; Li, J; Guo, SJ; Han, LJ. Antithrombotic effect of bromophenol, the alga-derived thrombin inhibitor. J Biotechnol 2008, 136, S579. [Google Scholar]
- Popplewell, WL; Northcote, PT. Colensolide A: A new nitrogenous bromophenol from the New Zealand marine red alga Osmundaria colensoi. Tetrahedron Lett 2009, 50, 6814–6817. [Google Scholar]
- Oh, KB; Lee, JH; Lee, JW; Yoon, KM; Chung, SC; Jeon, HB; Shin, J; Lee, HS. Synthesis and antimicrobial activities of halogenated bis(hydroxyphenyl)methanes. Bioorg Med Chem Lett 2009, 19, 945–948. [Google Scholar]
- Wiemer, DF; Idler, DD; Fenical, W. Vidalols A and B, new anti-inflammatory bromophenols from the Caribbean marine alga Vidalia obtusiloba. Experientia 1991, 47, 851–853. [Google Scholar]
- Wang, W; Okada, Y; Shi, HB; Wang, YQ; Okuyama, T. Structures and aldose reductase inhibitory effects of bromophenols from the red alga Symphyocladia latiuscula. J Nat Prod 2005, 68, 620–622. [Google Scholar]
- Xu, XL; Song, FH; Wang, SJ; Li, S; Fan, X; Zhao, JL; Yang, YC; Shang, SQ; Yang, L; Shi, JG. Dibenzyl bromophenols with diverse dimerization patterns from the brown alga Leathesia nana. J Nat Prod 2004, 67, 1661–1666. [Google Scholar]
- Kurata, K; Taniguchii, K; Takashima, K; Hayashi, I; Suzuki, M. Feeding-deterrent bromophenols from Odonthalia corymbifera. Phytochemistry 1997, 45, 485–487. [Google Scholar]
- Shi, DY; Li, J; Guo, SJ; Su, H; Fan, X. The antitumor effect of bromophenol derivatives in vitro and Leathesia nana extract in vivo. Chin J Oceanol Limnol 2009, 27, 277–282. [Google Scholar]
- Zhao, WY; Feng, XE; Ban, SR; Lin, WH; Li, QS. Synthesis and biological activity of halophenols as potent antioxidant and cytoprotective agents. Bioorg Med Chem Lett 2010, 20, 4132–4134. [Google Scholar]
- Jager, W; Winter, O; Halper, B; Salamon, A; Sartori, M; Gajdzik, L; Hamilton, G; Theyer, G; Graf, J; Thalhammer, T. Modulation of liver canalicular transport processes by the tyrosine- kinase inhibitor genistein: Implications of genistein metabolism in the rat. Hepatology 1997, 26, 1467–1476. [Google Scholar]
- Majewska, E; Paleolog, E; Baj, Z; Kralisz, U; Feldmann, M; Tchorzewski, H. Role of tyrosine kinase enzymes in TNF-a and IL-1 induced expression of ICAM-1 and VCAM-1 on human umbilical vein endothelial cells. Scand J Immunol 1997, 45, 385–392. [Google Scholar]
- Han, XJ; Civiello, RL; Mercer, SE; Macor, JE; Dubowchik, GM. Synthesis of aza and fluorine-substituted 3-(piperidin-4-yl)-4,5-dihydro-1H-benzo[d][1,3]diazepin-2(3H)-ones. Tetrahedron Lett 2009, 50, 386–388. [Google Scholar]
- Xue, J; Kumar, V; Khaja, SD; Chandrasekaran, EV; Locke, RD; Matta, KL. Syntheses of fluorine-containing mucin core 2/core 6 structures using novel fluorinated glucosaminyl donors. Tetrahedron 2009, 65, 8325–8335. [Google Scholar]
- Tsirkone, VG; Tsoukala, E; Lamprakis, C; Manta, S; Hayes, JM; Skamnaki, VT; Drakou, C; Zographos, SE; Komiotis, D; Leonidas, DD. 1-(3-Deoxy-3-fluoro-β-d-glucopyranosyl) pyrimidine derivatives as inhibitors of glycogen phosphorylase b: Kinetic, crystallographic and modelling studies. Bioorg Med Chem 2010, 18, 3413–3425. [Google Scholar]
- Fehér, C; Urbán, B; Urge, L; Darvas, F; Bakos, J; Skoda-Foldes, R. Facile synthesis of 6-iodo-2,2′-dipivaloyloxy-1,1′-binaphthyl, a key intermediate of high reactivity for selective palladium-catalyzed monofunctionalization of the 1,1′-binaphthalene core. Tetrahedron Lett 2010, 51, 3629–3632. [Google Scholar]
- Castro, MD; Marzabadi, CH. Preparation and reactions of iodo sugars. Tetrahedron 2010, 66, 3395–3404. [Google Scholar]
- Mphahlele, MJ; Lesenyeho, LG; Makelane, HR. Synthesis of 1H-pyrrolo[3,2-c]quinoline derivatives via palladium-catalyzed heteroannulation of 2-aryl-3-iodo-4-(phenylamino)quinolines and 4-(N,N-allylphenylamino)-2-aryl-3-iodoquinolines. Tetrahedron 2010, 66, 6040–6046. [Google Scholar]

{kind=link}
{kind=link}
 | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Compd. | Substituted group
| IC50a (μM) | |||||||||
| Linker | R1 | R2 | R3 | R4 | R5 | R6 | R7 | R8 | R9 | ||
| 1c * | C=O | F | H | H | H | H | H | H | OH | OH | >50 |
| 1d * | CH2 | F | H | H | H | H | H | H | OH | OH | >50 |
| 2c * | C=O | H | F | H | H | H | H | H | OH | OH | >50 |
| 2d * | CH2 | H | F | H | H | H | H | H | OH | OH | >50 |
| 3c | C=O | H | H | F | H | H | H | H | OH | OH | >50 |
| 3d * | CH2 | H | H | F | H | H | H | H | OH | OH | >50 |
| 4c * | C=O | I | H | H | H | H | H | H | OH | OH | >50 |
| 4d * | CH2 | I | H | H | H | H | H | H | OH | OH | >50 |
| 5c | C=O | Br | OH | OH | OH | Br | H | H | OH | OH | >50 |
| 5d | CH2 | Br | OH | OH | OH | Br | H | H | OH | OH | >50 |
| 6c | C=O | Cl | OH | OH | OH | Cl | H | H | OH | OH | 2.97 |
| 6d | CH2 | Cl | OH | OH | OH | Cl | H | H | OH | OH | 3.96 |
| 7c | C=O | H | Br | OH | H | H | Br | H | OH | OH | >50 |
| 7d | CH2 | H | Br | OH | H | H | Br | H | OH | OH | 6.34 |
| 8c | C=O | Cl | H | H | H | H | H | H | OH | OH | 17.7 |
| 8d | CH2 | Cl | H | H | H | H | H | H | OH | OH | 14.8 |
| 9c | C=O | H | Cl | H | H | H | H | H | OH | OH | 17.8 |
| 9d | CH2 | H | Cl | H | H | H | H | H | OH | OH | 12.9 |
| 10c | C=O | H | H | Cl | H | H | H | H | OH | OH | 41.6 |
| 10d | CH2 | H | H | Cl | H | H | H | H | OH | OH | 6.97 |
| 11c | C=O | H | H | H | H | H | Br | H | OH | OH | 16.0 |
| 11d | CH2 | H | H | H | H | H | Br | H | OH | OH | 6.26 |
| 12c | C=O | H | H | H | H | H | Br | Br | OH | OH | >50 |
| 12d | CH2 | H | H | H | H | H | Br | Br | OH | OH | >50 |
| 13c | C=O | OH | H | H | Br | H | Br | H | OH | OH | >50 |
| 13d | CH2 | OH | H | H | Br | H | Br | H | OH | OH | 5.05 |
| Control (Genistein) | 13.6 | ||||||||||
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Feng, X.E.; Zhao, W.Y.; Ban, S.R.; Zhao, C.X.; Li, Q.S.; Lin, W.H. Structure–Activity Relationship of Halophenols as a New Class of Protein Tyrosine Kinase Inhibitors. Int. J. Mol. Sci. 2011, 12, 6104-6115. https://doi.org/10.3390/ijms12096104
Feng XE, Zhao WY, Ban SR, Zhao CX, Li QS, Lin WH. Structure–Activity Relationship of Halophenols as a New Class of Protein Tyrosine Kinase Inhibitors. International Journal of Molecular Sciences. 2011; 12(9):6104-6115. https://doi.org/10.3390/ijms12096104
Chicago/Turabian StyleFeng, Xiu E, Wan Yi Zhao, Shu Rong Ban, Cheng Xiao Zhao, Qing Shan Li, and Wen Han Lin. 2011. "Structure–Activity Relationship of Halophenols as a New Class of Protein Tyrosine Kinase Inhibitors" International Journal of Molecular Sciences 12, no. 9: 6104-6115. https://doi.org/10.3390/ijms12096104