Mechanism of Sphingosine 1-Phosphate- and Lysophosphatidic Acid-Induced Up-Regulation of Adhesion Molecules and Eosinophil Chemoattractant in Nerve Cells

Abstract
:1. Introduction
2. Results and Discussion
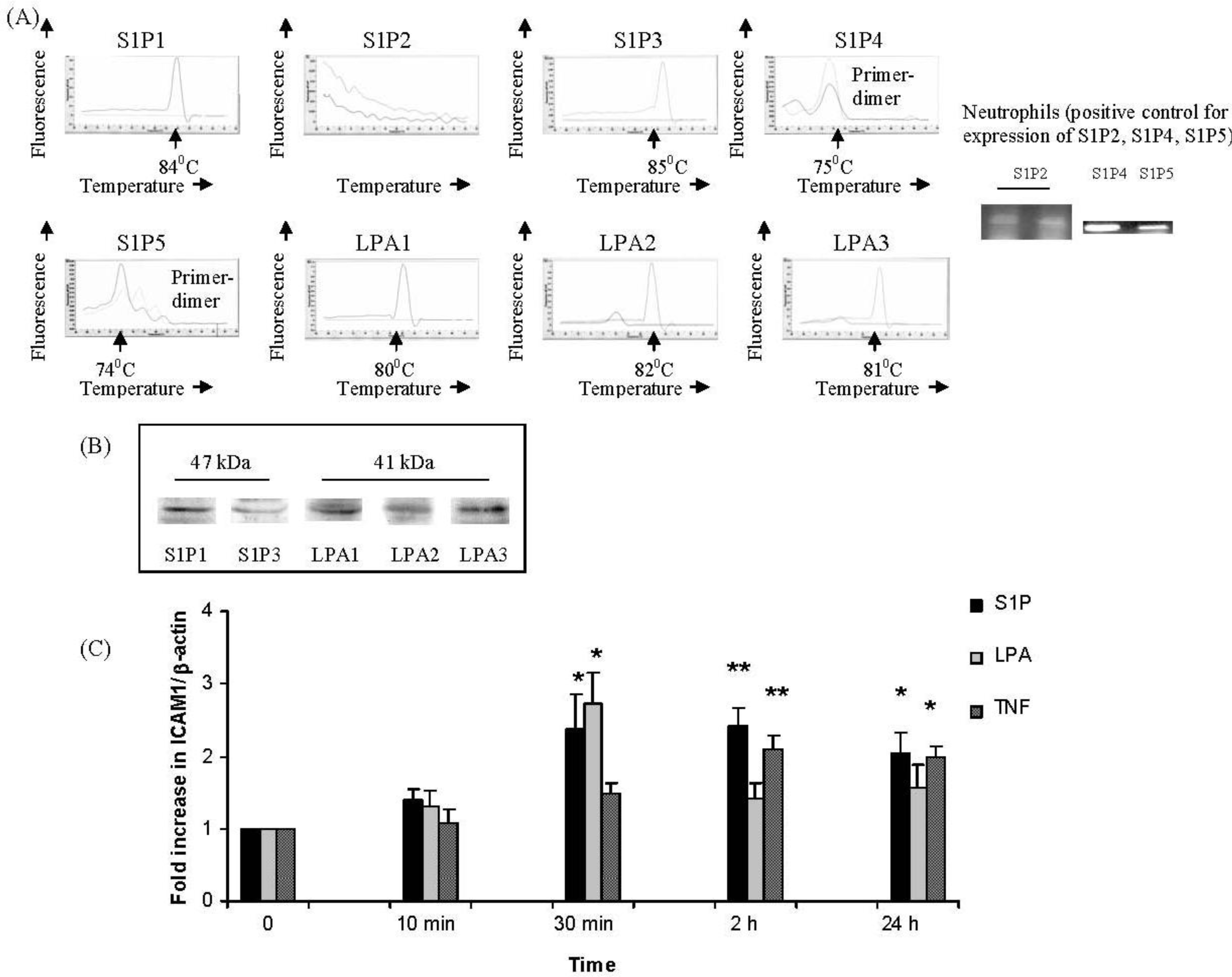
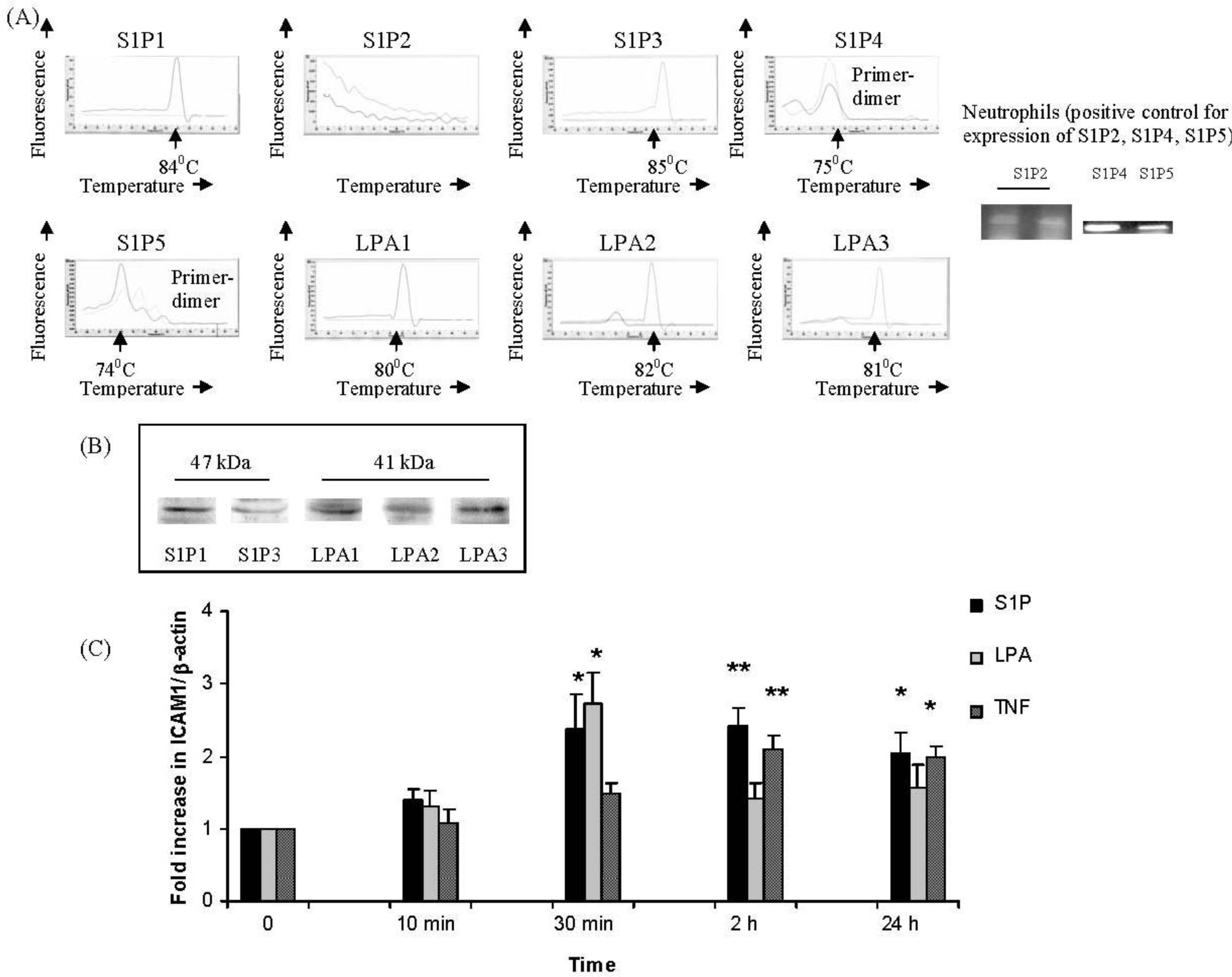
2.1. IMR32 Cells Express S1P1, S1P3 and LPA1–3; S1P and LPA Induce Up-Regulation of ICAM-1 Transcription
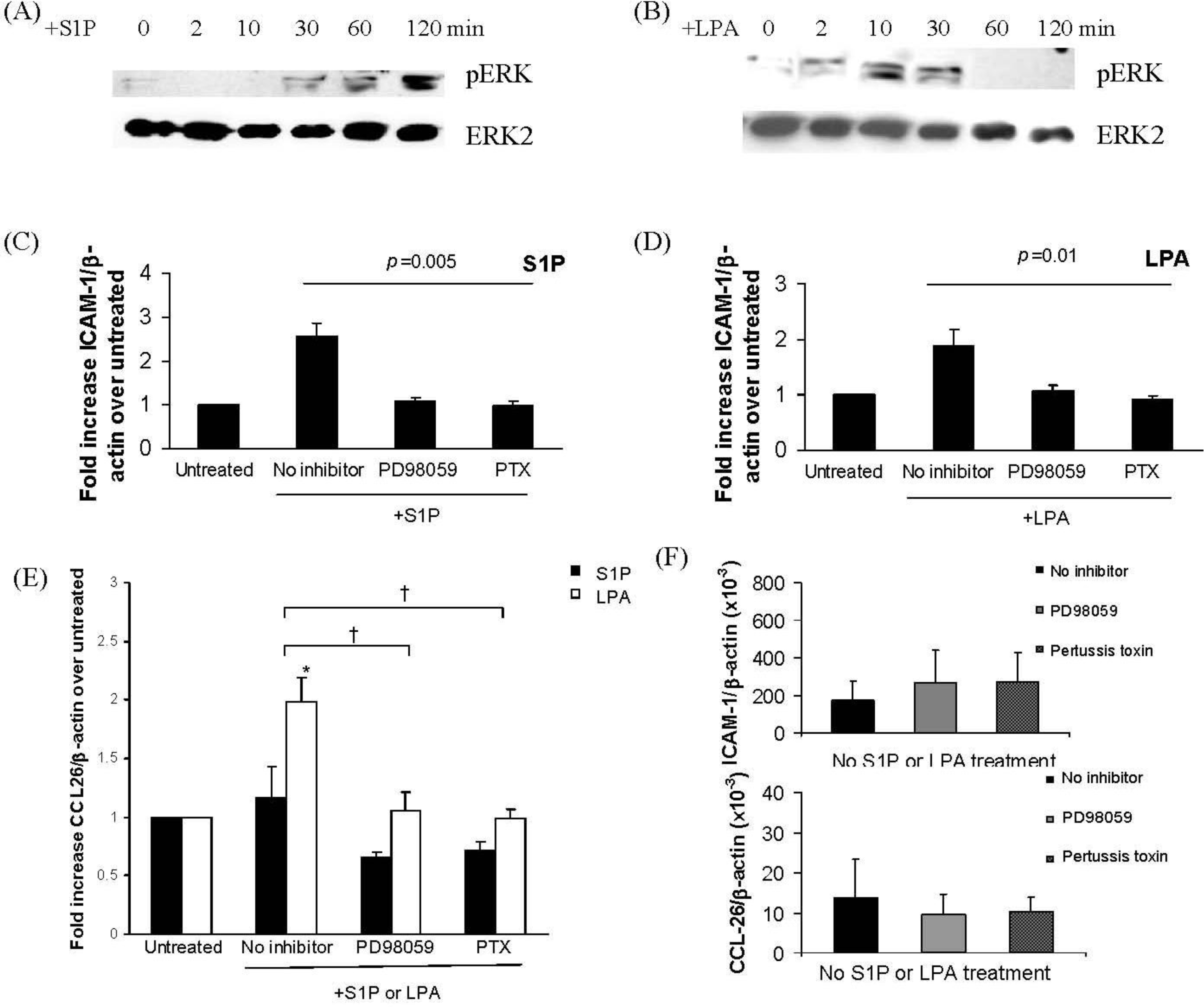
2.2. S1P and LPA Induce ERK Activation in IMR32 Cells with Different Time Courses with Consequenct Gi-Protein Coupled and ERK-Dependent Up-Regulation of ICAM-1 and/or CCL-26
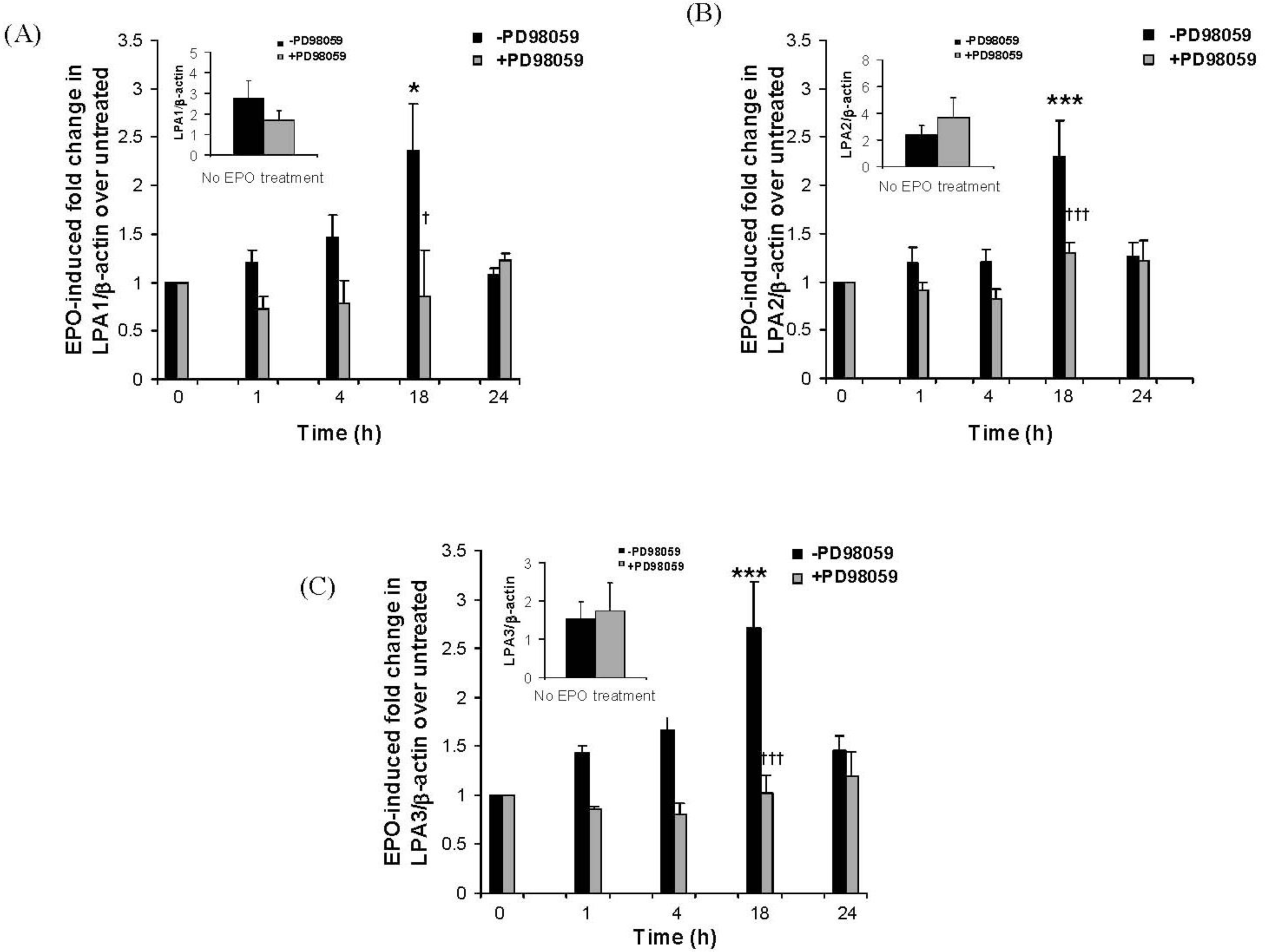
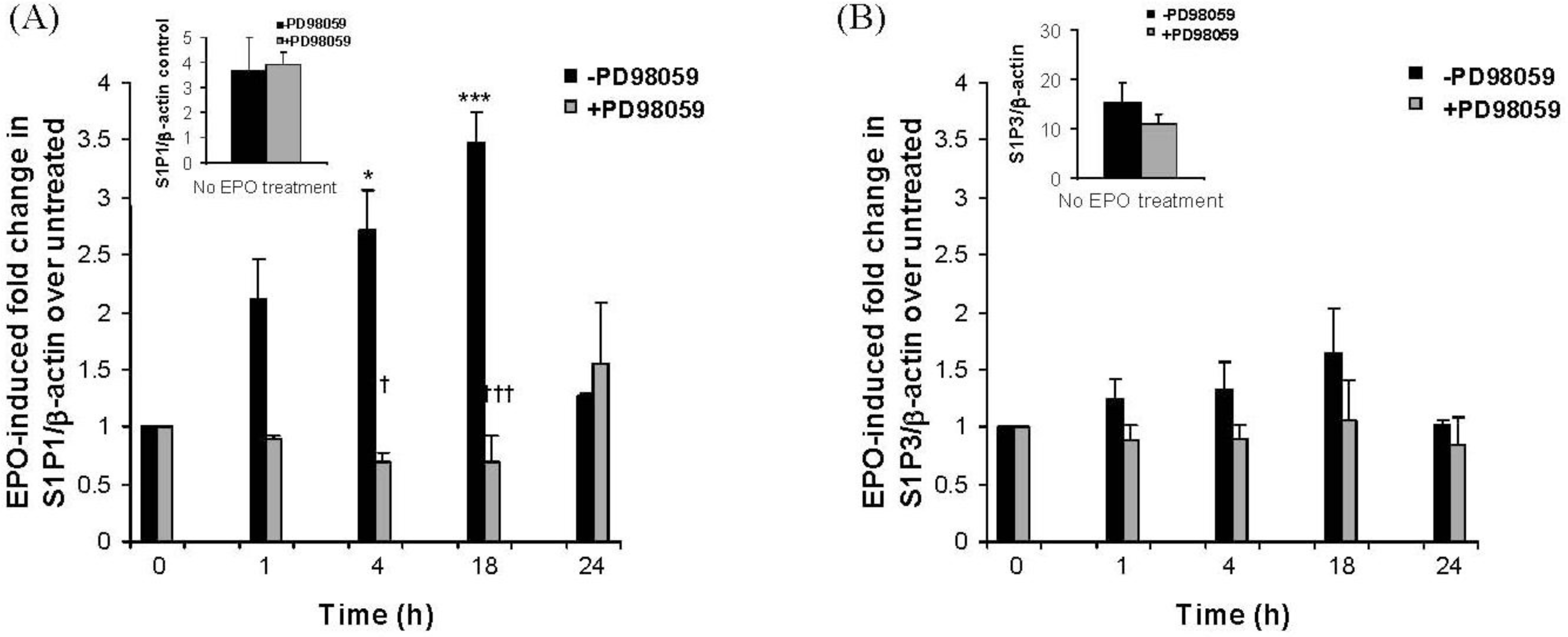
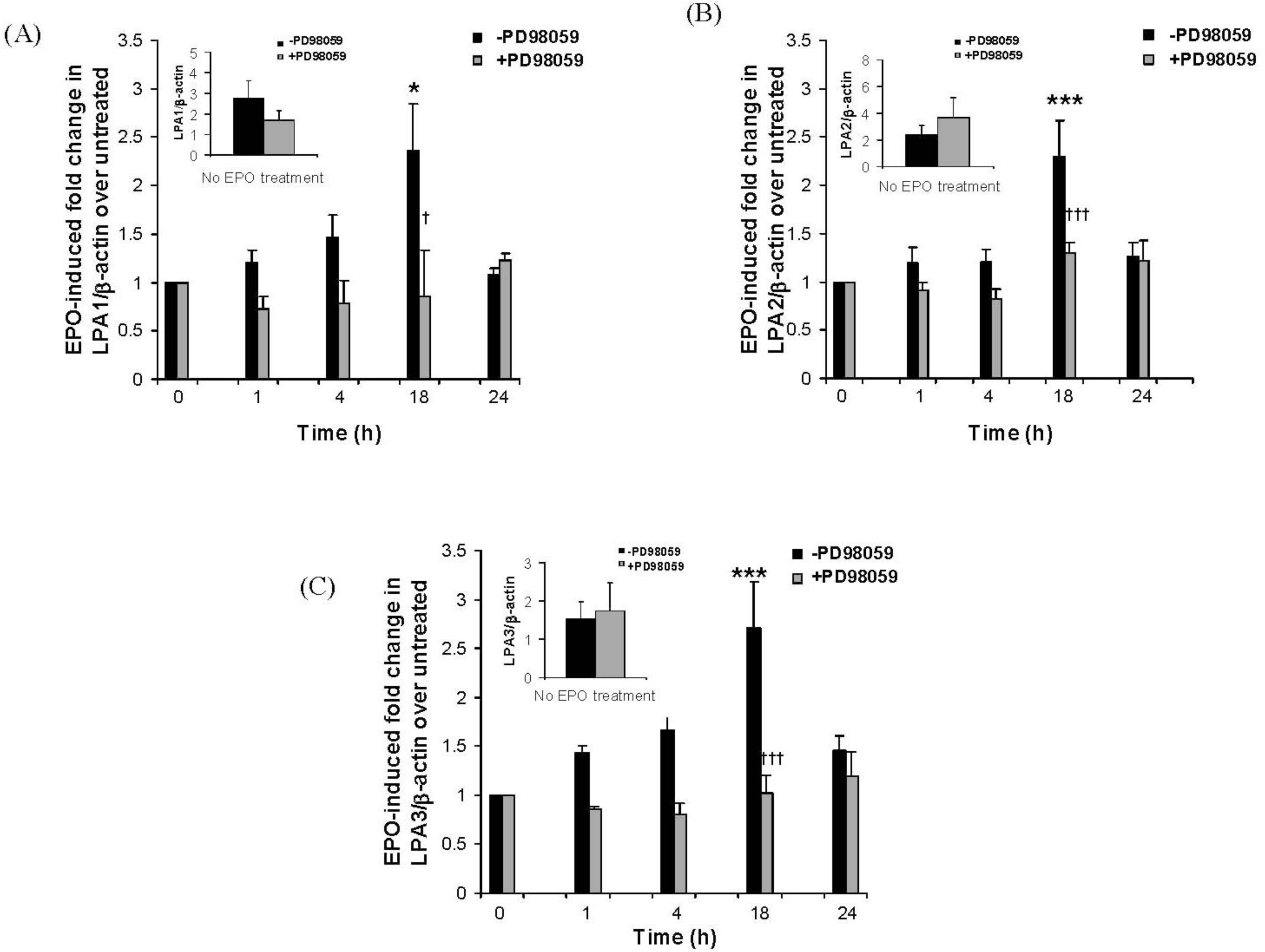
2.3. The Eosinophil Granule Protein EPO Induces Up-Regulation of S1P1, LPA1, LPA2 and LPA3 Receptors Expression
3. Experimental Section
4. Conclusion
References
- Fryer, AD; Jacoby, DB. Plasticity of cholinergic and tachykinergic nerves: The convergence of the twain. Am. J. Physiol. Lung Cell. Mol. Physiol 2002, 283, L907–L908. [Google Scholar]
- Rothenberg, ME; Hogan, SP. The eosinophil. Annu. Rev. Immunol 2006, 24, 147–174. [Google Scholar]
- Evans, CM; Fryer, AD; Jacoby, DB; Gleich, GJ; Costello, RW. Pretreatment with antibody to eosinophil major basic protein prevents hyperresponsiveness by protecting neuronal M2 muscarinic receptors in antigen-challenged guinea pigs. J. Clin. Invest 1997, 100, 2254–2262. [Google Scholar]
- Sheahan, P; Walsh, RM; Walsh, MA; Costello, RW. Induction of nasal hyper-responsiveness by allergen challenge in allergic rhinitis: the role of afferent and efferent nerves. Clin. Exp. Allergy 2005, 35, 45–51. [Google Scholar]
- Fryer, AD; Stein, LH; Nie, Z; Curtis, DE; Evans, CM; Hodgson, ST; Jose, PJ; Belmonte, KE; Fitch, E; Jacoby, DB. Neuronal eotaxin and the effects of CCR3 antagonist on airway hyperreactivity and M2 receptor dysfunction. J. Clin. Invest 2006, 116, 228–236. [Google Scholar]
- Kingham, PJ; McLean, WG; Sawatzky, DA; Walsh, MT; Costello, RW. Adhesion-dependent interactions between eosinophils and cholinergic nerves. Am. J. Physiol. Lung Cell. Mol. Physiol 2002, 282, L1229–L1238. [Google Scholar]
- Sawatzky, DA; Kingham, PJ; Court, E; Kumaravel, B; Fryer, AD; Jacoby, DB; McLean, WG; Costello, RW. Eosinophil adhesion to cholinergic nerves via ICAM-1 and VCAM-1 and associated eosinophil degranulation. Am. J. Physiol. Lung Cell. Mol. Physiol 2002, 282, L1279–L1288. [Google Scholar]
- Sawatzky, DA; Kingham, PJ; Durcan, N; McLean, WG; Costello, RW. Eosinophil-induced release of acetylcholine from differentiated cholinergic nerve cells. Am. J. Physiol. Lung Cell. Mol. Physiol 2003, 285, L1296–L1304. [Google Scholar]
- Kingham, PJ; McLean, WG; Walsh, MT; Fryer, AD; Gleich, GJ; Costello, RW. Effects of eosinophils on nerve cell morphology and development: The role of reactive oxygen species and p38 MAP kinase. Am. J. Physiol. Lung Cell. Mol. Physiol 2003, 285, L915–L924. [Google Scholar]
- Walsh, MT; Curran, DR; Kingham, PJ; Morgan, RK; Durcan, N; Gleich, GJ; McLean, WG; Costello, RW. Effect of eosinophil adhesion on intracellular signaling in cholinergic nerve cells. Am. J. Respir. Cell Mol. Biol 2004, 30, 333–341. [Google Scholar]
- Morgan, RK; Kingham, PJ; Walsh, MT; Curran, DC; Durcan, N; McLean, WG; Costello, RW. Eosinophil adhesion to cholinergic IMR-32 cells protects against induced neuronal apoptosis. J. Immunol 2004, 173, 5963–5970. [Google Scholar]
- Morgan, RK; Costello, RW; Durcan, N; Kingham, PJ; Gleich, GJ; McLean, WG; Walsh, MT. Diverse effects of eosinophil cationic granule proteins on IMR-32 nerve cell signaling and survival. Am. J. Respir. Cell Mol. Biol 2005, 33, 169–177. [Google Scholar]
- Curran, DR; Morgan, RK; Kingham, PJ; Durcan, N; McLean, WG; Walsh, MT; Costello, RW. Mechanism of eosinophil induced signaling in cholinergic IMR-32 cells. Am. J. Physiol. Lung Cell. Mol. Physiol 2005, 288, L326–L332. [Google Scholar]
- Meyer zu Heringdorf, D; Jakobs, KH. Lysophospholipid receptors: Signalling, pharmacology and regulation by lysophospholipid metabolism. Biochim. Biophys. Acta 2007, 1768, 923–940. [Google Scholar]
- Noguchi, K; Ishii, S; Shimizu, T. Identification of p2y9/GPR23 as a novel G protein-coupled receptor for lysophosphatidic acid, structurally distant from the Edg family. J. Biol. Chem 2003, 278, 25600–25606. [Google Scholar]
- Rosen, H; Goetzl, EJ. Sphingosine 1-phosphate and its receptors: An autocrine and paracrine network. Nat. Rev. Immunol 2005, 5, 560–570. [Google Scholar]
- Lee, CW; Rivera, R; Gardell, S; Dubin, AE; Chun, J. GPR92 as a new G12/13- and Gq-coupled lysophosphatidic acid receptor that increases cAMP, LPA5. J. Biol. Chem 2006, 281, 23589–23597. [Google Scholar]
- Buccoliero, R; Futerman, AH. The roles of ceramide and complex sphingolipids in neuronal cell function. Pharmacol. Res 2003, 47, 409–419. [Google Scholar]
- Colombaioni, L; Garcia-Gil, M. Sphingolipid metabolites in neural signalling and function. Brain Res. Rev 2004, 46, 328–355. [Google Scholar]
- Fukushima, N. LPA in neural cell development. J. Cell. Biochem 2004, 92, 993–1003. [Google Scholar]
- Fukushima, N; Shano, S; Moriyama, R; Chun, J. Lysophosphatidic acid stimulates neuronal differentiation of cortical neuroblasts through the LPA(1)-G(i/o) pathway. Neurochem. Int 2007, 50, 302–307. [Google Scholar]
- Oskeritzian, CA; Milstien, S; Spiegel, S. Sphingosine-1-phosphate in allergic responses, asthma and anaphylaxis. Pharmacol. Ther 2007, 115, 390–399. [Google Scholar]
- Zhao, Y; Natarajan, V. Lysophosphatidic acid signaling in airway epithelium: Role in airway inflammation and remodeling. Cell. Signal 2009, 21, 367–377. [Google Scholar]
- Ammit, AJ; Hastie, AT; Edsall, LC; Hoffman, RK; Amrani, Y; Krymskaya, VP; Kane, SA; Peters, SP; Penn, RB; Spiegel, S; et al. Sphingosine 1-phosphate modulates human airway smooth muscle cell functions that promote inflammation and airway remodeling in asthma. FASEB J 2001, 15, 1212–1214. [Google Scholar]
- Georas, SN; Berdyshev, E; Hubbard, W; Gorshkova, IA; Usatyuk, PV; Saatian, B; Myers, AC; Williams, MA; Xiao, HQ; Liu, M; et al. Lysophosphatidic acid is detectable in human bronchoalveolar lavage fluids at baseline and increased after segmental allergen challenge. Clin. Exp. Allergy 2007, 37, 311–322. [Google Scholar]
- Goetzl, EJ; Wang, W; McGiffert, C; Huang, MC; Graler, MH. Sphingosine 1-phosphate and its G protein-coupled receptors constitute a multifunctional immunoregulatory system. J. Cell. Biochem 2004, 92, 1104–1114. [Google Scholar]
- Mackle, T; Gendy, SS; Walsh, M; McConn-Walsh, R; Costello, RW; Walsh, MT. Role of sphingosine 1-phosphate receptor expression in eosinophils of patients with allergic rhinitis, and effect of topical nasal steroid treatment on this receptor expression. J. Laryngol. Otol 2008, 122, 1309–1317. [Google Scholar]
- Roviezzo, F; Del Galdo, F; Abbate, G; Bucci, M; D’Agostino, B; Antunes, E; De Dominicis, G; Parente, L; Rossi, F; Cirino, G; De Palma, R. Human eosinophil chemotaxis and selective in vivo recruitment by sphingosine 1-phosphate. Proc. Natl. Acad. Sci. USA 2004, 101, 11170–11175. [Google Scholar]
- Rahaman, M; Costello, RW; Belmonte, KE; Gendy, SS; Walsh, MT. Neutrophil sphingosine 1-phosphate and lysophosphatidic acid receptors in pneumonia. Am. J. Respir. Cell Mol. Biol 2006, 34, 233–241. [Google Scholar]
- Idzko, M; Laut, M; Panther, E; Sorichter, S; Durk, T; Fluhr, JW; Herouy, Y; Mockenhaupt, M; Myrtek, D; Elsner, P; Norgauer, J. Lysophosphatidic acid induces chemotaxis, oxygen radical production, CD11b up-regulation, Ca2+ mobilization, and actin reorganization in human eosinophils via pertussis toxin-sensitive G proteins. J. Immunol 2004, 172, 4480–4485. [Google Scholar]
- Hashimoto, T; Ohata, H; Honda, K. Lysophosphatidic acid (LPA) induces plasma exudation and histamine release in mice via LPA receptors. J. Pharmacol. Sci 2006, 100, 82–87. [Google Scholar]
- Lin, DA; Boyce, JA. IL-4 regulates MEK expression required for lysophosphatidic acid-mediated chemokine generation by human mast cells. J. Immunol 2005, 175, 5430–5438. [Google Scholar]
- Hashimoto, T; Ohata, H; Momose, K; Honda, K. Lysophosphatidic acid induces histamine release from mast cells and skin fragments. Pharmacology 2005, 75, 13–20. [Google Scholar]
- Milara, J; Mata, M; Mauricio, MD; Donet, E; Morcillo, EJ; Cortijo, J. Sphingosine-1-phosphate increases human alveolar epithelial IL-8 secretion, proliferation and neutrophil chemotaxis. Eur. J. Pharmacol 2009, 609, 132–139. [Google Scholar]
- Lee, H; Lin, CI; Liao, JJ; Lee, YW; Yang, HY; Lee, CY; Hsu, HY; Wu, HL. Lysophospholipids increase ICAM-1 expression in HUVEC through a Gi- and NF-kappaB-dependent mechanism. Am. J. Physiol. Cell Physiol 2004, 287, C1657–C1666. [Google Scholar]
- Lin, CI; Chen, CN; Lin, PW; Chang, KJ; Hsieh, FJ; Lee, H. Lysophosphatidic acid regulates inflammation-related genes in human endothelial cells through LPA1 and LPA3. Biochem. Biophys. Res. Commun 2007, 363, 1001–1008. [Google Scholar]
- Lin, CI; Chen, CN; Lin, PW; Lee, H. Sphingosine 1-phosphate regulates inflammation-related genes in human endothelial cells through S1P1 and S1P3. Biochem. Biophys. Res. Commun 2007, 355, 895–901. [Google Scholar]
- Durcan, N; Costello, RW; McLean, WG; Blusztajn, J; Madziar, B; Fenech, AG; Hall, IP; Gleich, GJ; McGarvey, L; Walsh, MT. Eosinophil-mediated cholinergic nerve remodeling. Am. J. Respir. Cell Mol. Biol 2006, 34, 775–786. [Google Scholar]
- Kingham, PJ; Costello, RW; McLean, WG. Eosinophil and airway nerve interactions. Pulm. Pharmacol. Ther 2003, 16, 9–13. [Google Scholar]
- Blanchard, C; Durual, S; Estienne, M; Emami, S; Vasseur, S; Cuber, JC. Eotaxin-3/CCL26 gene expression in intestinal epithelial cells is up-regulated by interleukin-4 and interleukin-13 via the signal transducer and activator of transcription 6. Int. J. Biochem. Cell Biol 2005, 37, 2559–2573. [Google Scholar]
- Costello, RW; Schofield, BH; Kephart, GM; Gleich, GJ; Jacoby, DB; Fryer, AD. Localization of eosinophils to airway nerves and effect on neuronal M2 muscarinic receptor function. Am. J. Physiol 1997, 273, L93–L103. [Google Scholar]
- Berkman, N; Ohnona, S; Chung, FK; Breuer, R. Eotaxin-3 but not eotaxin gene expression is upregulated in asthmatics 24 hours after allergen challenge. Am. J. Respir. Cell Mol. Biol 2001, 24, 682–687. [Google Scholar]
- Bradford, MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem 1976, 72, 248–254. [Google Scholar]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Primer Sequence | Amplicon Length (Base Pairs) |
|---|---|---|
| S1P1 | 5′-ATCGTCCTGAGCGTCTTCAT-3′ (forward) 5′-CCAGGAAGTACTCCGCTCTG-3′ (reverse) | 95 |
| S1P2 | 5′-CCAAGCATTATGTGCTGTGC-3′ (forward) 5′-CAGAAGGAGGATGCTGAAGG-3′ (reverse) | 186 |
| S1P3 | 5′-ACCAGTACGTGGGGAAGTTG-3′ (forward) 5′-GGCAATCAAAACCATCAGGT-3′ (reverse) | 105 |
| S1P4 | 5′-CCAAGCGCTACATCCTCTTC-3 (forward) 5′-CAGAGGTTGGAGCCAAAGAC-3′ (reverse) | 221 |
| S1P5 | 5′-ACAACTACACCGGCAAGCTC-3′ (forward) 5′-GCCCCGACAGTAGGATGTT-3′ (reverse) | 218 |
| LPA1 | 5′-ATTTCACAGCCCCAGTTCAC-3′ (forward) 5′-CACCAGCTTGCTGACTGTGT-3′ (reverse) | 106 |
| LPA2 | 5′-CTGCTCCTGGATGGTTTAGG-3′ (forward) 5′-CTCGGCAAGAGTACACAGCA-3′ (reverse) | 95 |
| LPA3 | 5′-TTGCCTCTGCAACATCTCTG-3′ (forward) 5′-ATGATGAGGAAGGCCATGAG-3 (reverse) | 82 |
| ICAM-1 | 5′-CAAGGCCTCAGTCAGTGTGA-3′ (forward) 5′-CCTCTGGCTTCGTCAGAATC-3′ (reverse) | 129 |
| CCL26 | 5′-CCTCCTGAGTCTCCACCTTG-3′ (forward) 5′-TGGGAGCAGCTGTTACTGGT-3′ (reverse) | 115 |
| -actin | 5′-GGACTTCGAGCAAGAGATGG-3′ (forward) 5′-AGGAAGGAAGGCTGGAAGAG-3′ (reverse) | 118 |
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Costello, R.W.; Maloney, M.; Atiyeh, M.; Gleich, G.; Walsh, M.-T. Mechanism of Sphingosine 1-Phosphate- and Lysophosphatidic Acid-Induced Up-Regulation of Adhesion Molecules and Eosinophil Chemoattractant in Nerve Cells. Int. J. Mol. Sci. 2011, 12, 3237-3249. https://doi.org/10.3390/ijms12053237
Costello RW, Maloney M, Atiyeh M, Gleich G, Walsh M-T. Mechanism of Sphingosine 1-Phosphate- and Lysophosphatidic Acid-Induced Up-Regulation of Adhesion Molecules and Eosinophil Chemoattractant in Nerve Cells. International Journal of Molecular Sciences. 2011; 12(5):3237-3249. https://doi.org/10.3390/ijms12053237
Chicago/Turabian StyleCostello, Richard W., Michael Maloney, Mazin Atiyeh, Gerald Gleich, and Marie-Therese Walsh. 2011. "Mechanism of Sphingosine 1-Phosphate- and Lysophosphatidic Acid-Induced Up-Regulation of Adhesion Molecules and Eosinophil Chemoattractant in Nerve Cells" International Journal of Molecular Sciences 12, no. 5: 3237-3249. https://doi.org/10.3390/ijms12053237