Global Transcriptomic Profiling Using Small Volumes of Whole Blood: A Cost-Effective Method for Translational Genomic Biomarker Identification in Small Animals

Abstract
:1. Introduction
2. Results and Discussion
2.1. Results
2.1.1. Quantity and Quality Assessment of Total RNA Isolated from Rat Whole Blood Using the PAXgene and QSI Methods
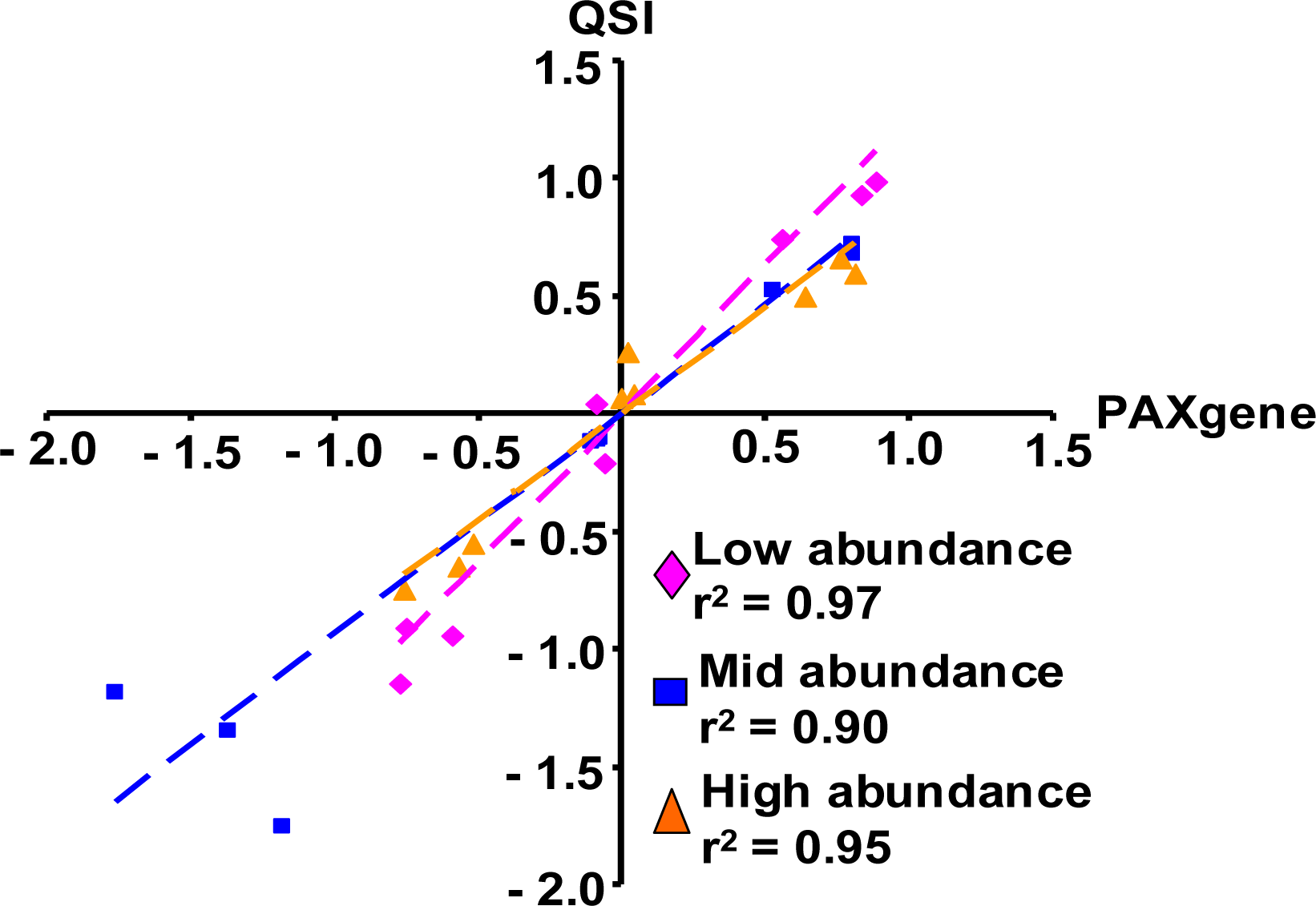
2.1.2. RNA Isolated from the PAXgene and QSI Methods Yield Comparable Affymetrix GeneChip® Array Performance
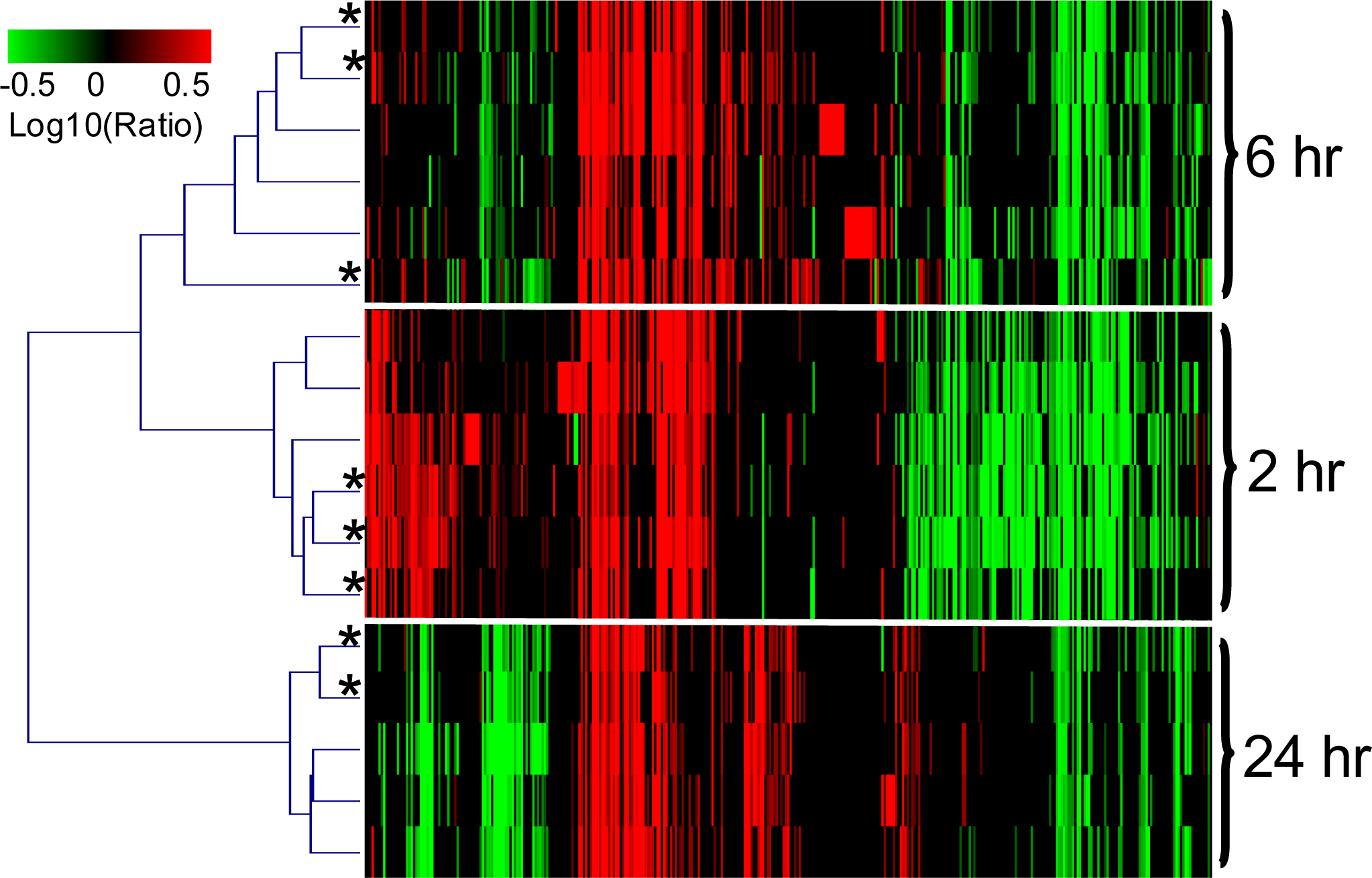
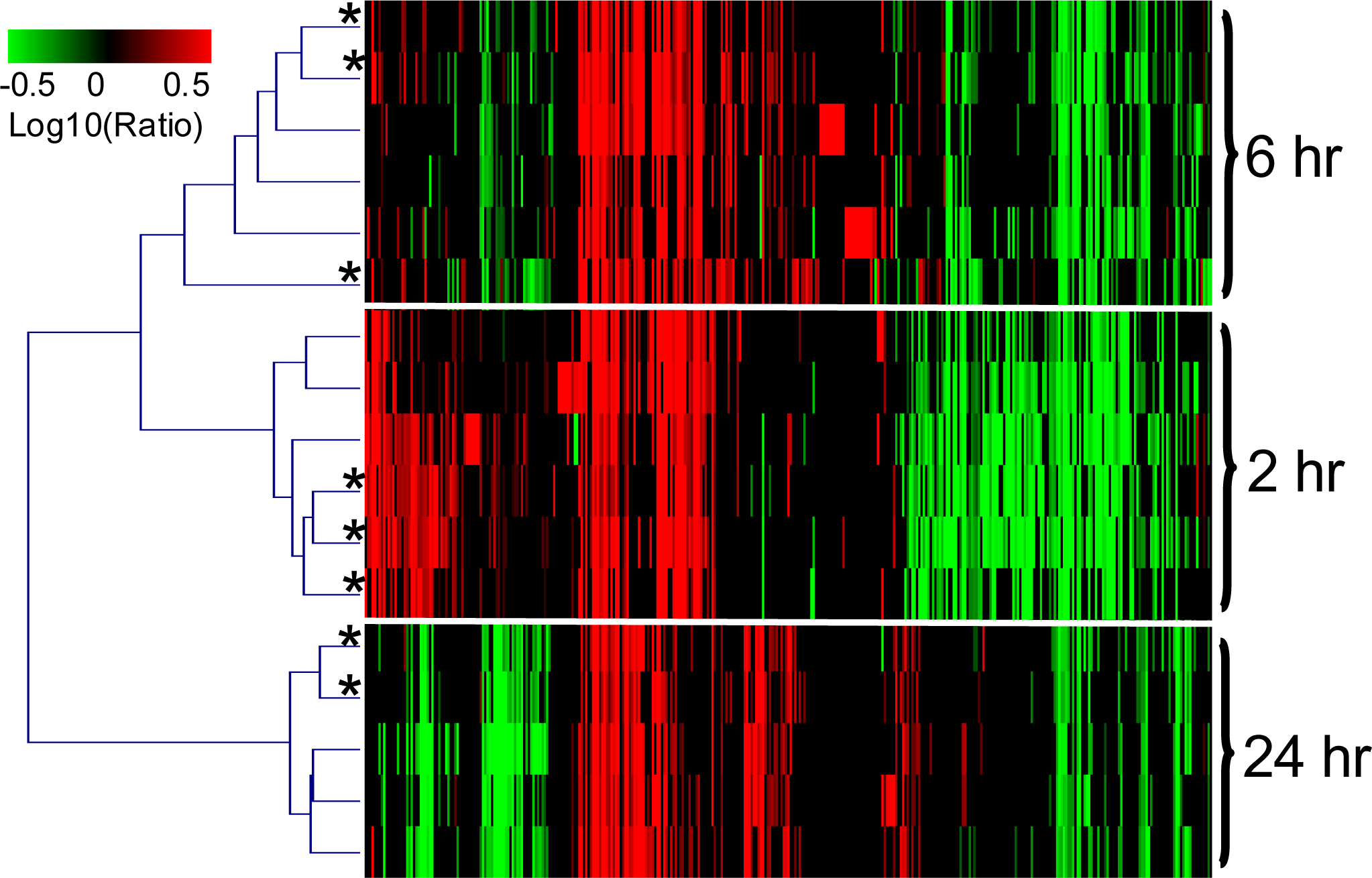
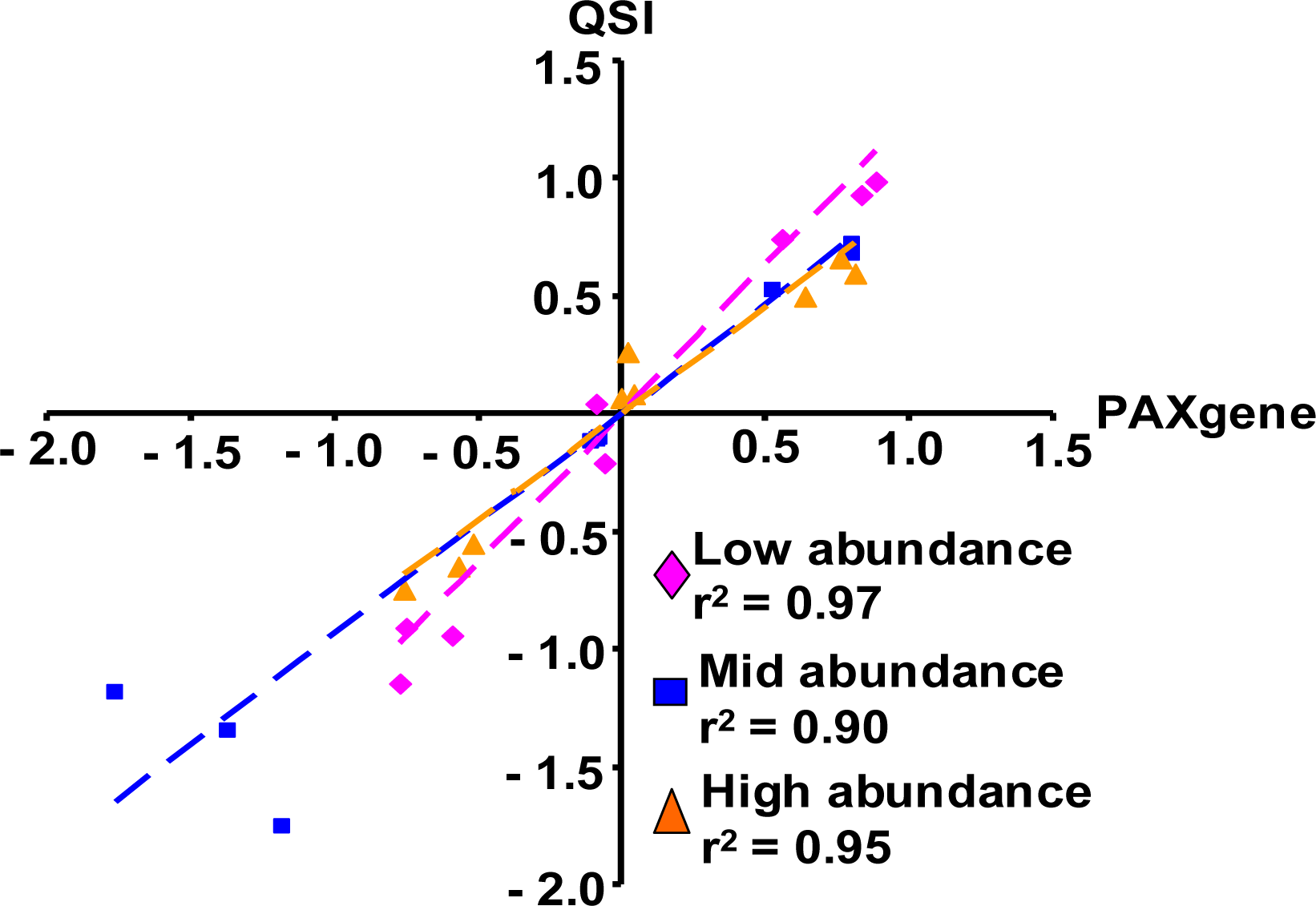
2.1.3. The PAXgene and QSI Methods Yield Comparable Blood Transcriptomic Profiles in Rats Exposed to LPS
2.1.4. Quantitative RT-PCR Validation of Microarray Results
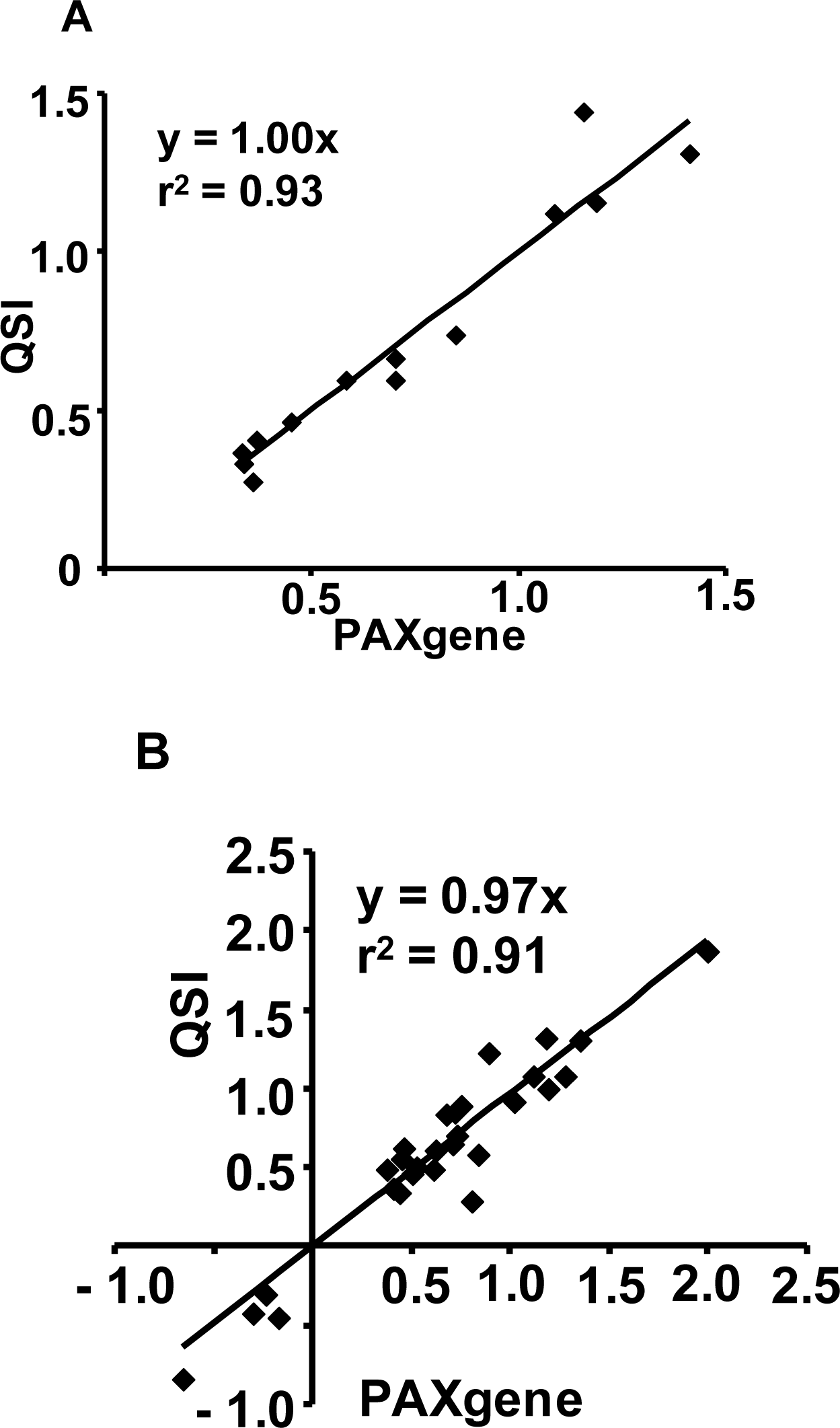
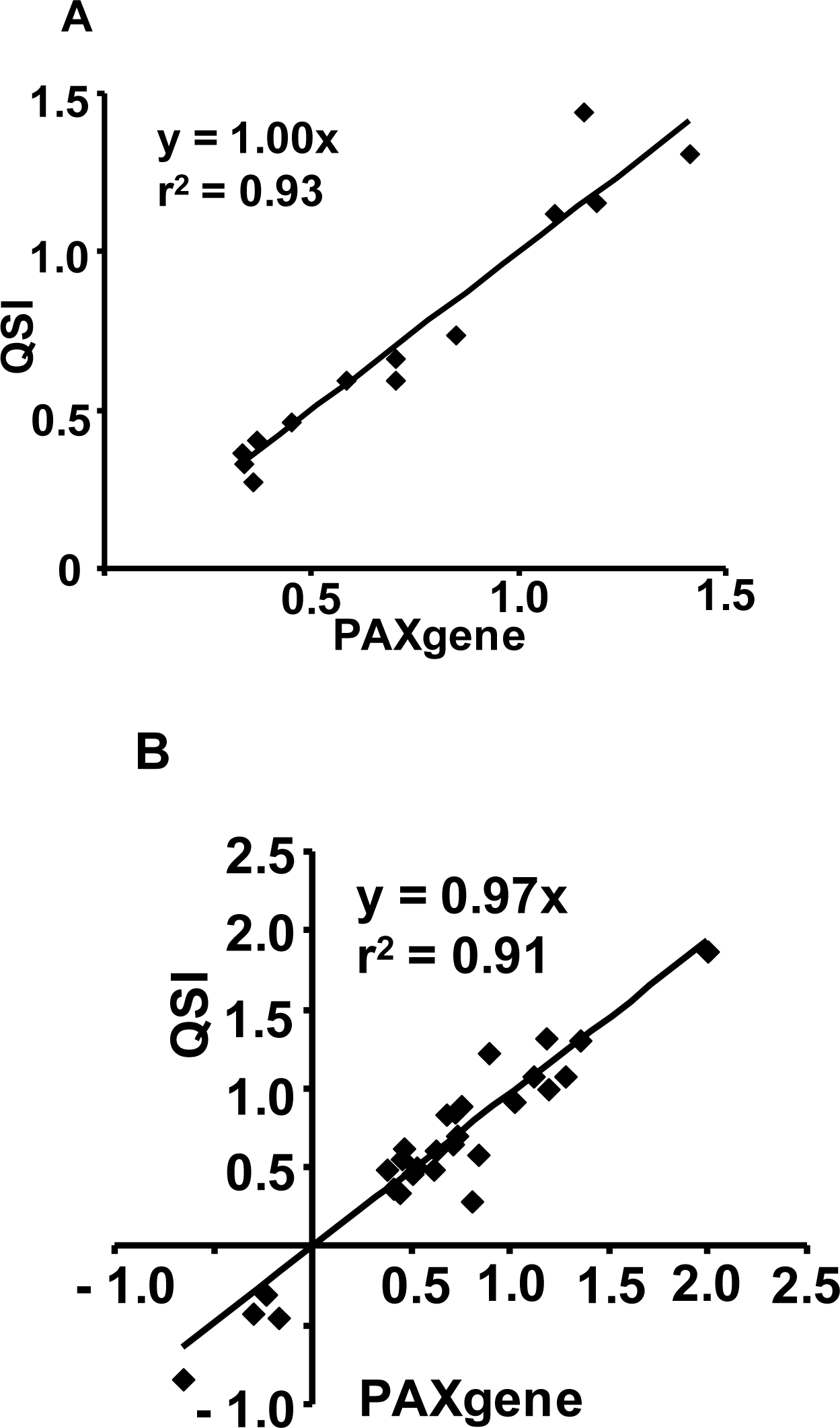
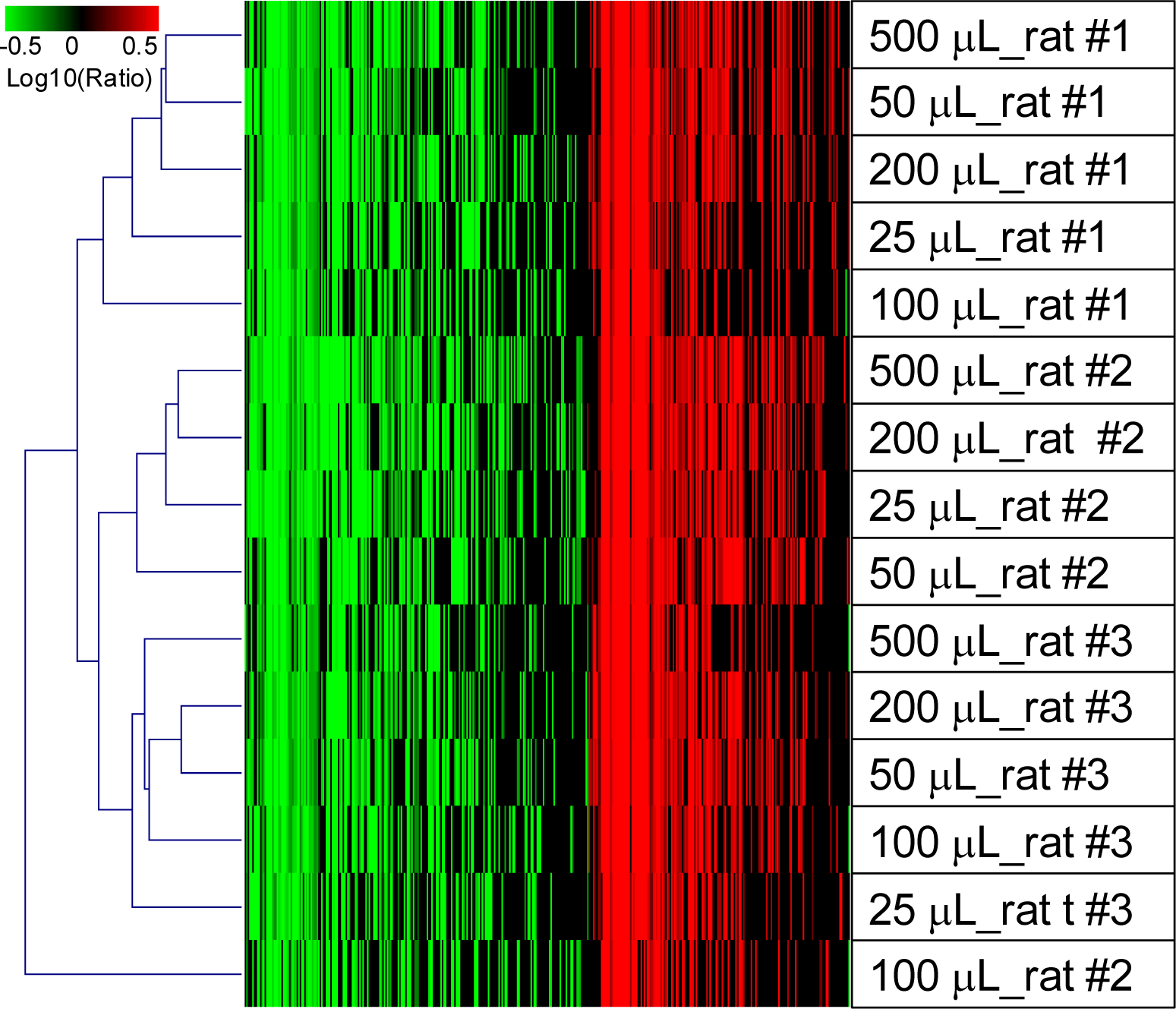
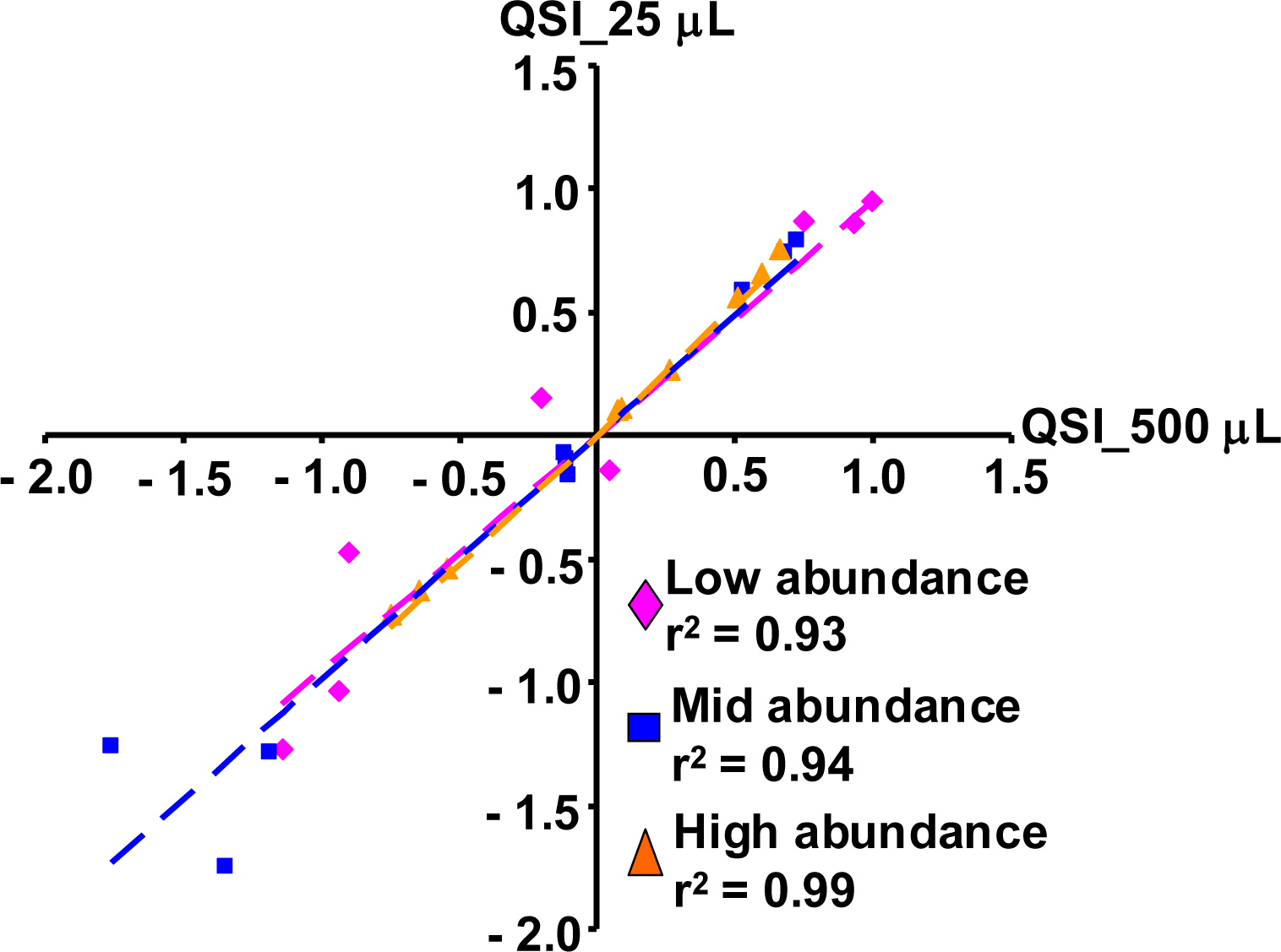
2.1.5. Evaluation of the QSI Method Using Small Volume of Whole Blood as Input
2.2. Discussion
3. Experimental Section
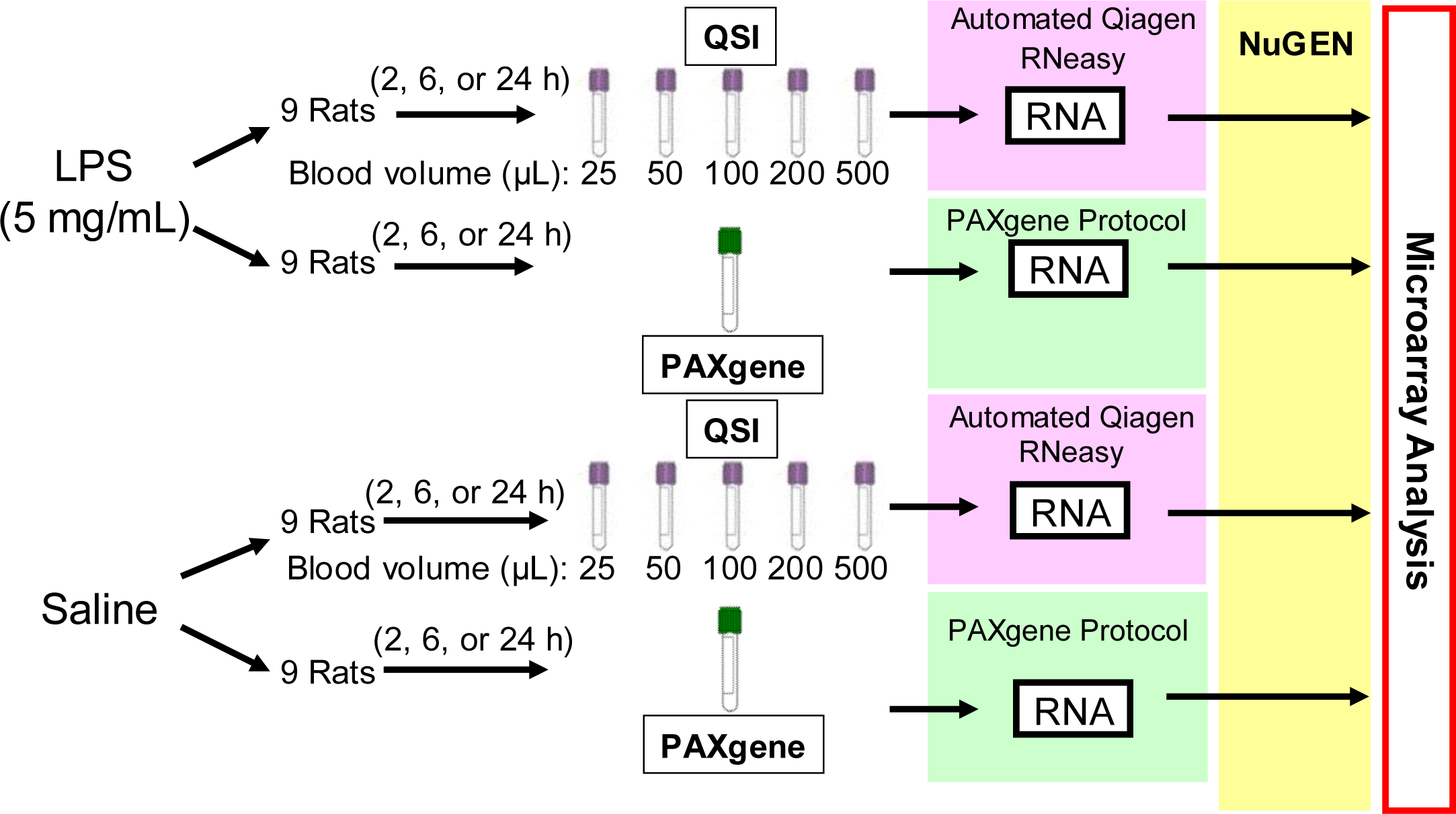
3.1. Animals, Treatment, and Sample Collection
3.2. RNA Isolation
3.3. Microarray Analysis
3.4. Real-Time Quantitative RT-PCR
3.5. Statistical Analysis
4. Conclusions
Supplementary Material
ijms-12-02502-s001.pdfAbbreviations:
| QSI | QIAzol-based RNA stabilization and isolation method |
| LPS | lipopolysaccharide |
| RIN | RNA Integrity Number |
| PBMC | peripheral blood mononuclear cells |
- Conflict of InterestThe authors declare no conflict of interest.
References
- Blomme, EA; Yang, Y; Waring, JF. Use of toxicogenomics to understand mechanisms of drug-induced hepatotoxicity during drug discovery and development. Toxicol. Lett 2009, 186, 22–31. [Google Scholar]
- Acharya, CR; Hsu, DS; Anders, CK; Anguiano, A; Salter, KH; Walters, KS; Redman, RC; Tuchman, SA; Moylan, CA; Mukherjee, S; et al. Gene expression signatures, clinicopathological features, and individualized therapy in breast cancer. JAMA 2008, 299, 1574–1587. [Google Scholar]
- Custodio, AB; Gonzalez-Larriba, JL; Bobokova, J; Calles, A; Alvarez, R; Cuadrado, E; Manzano, A; Diaz-Rubio, E. Prognostic and predictive markers of benefit from adjuvant chemotherapy in early-stage non-small cell lung cancer. J. Thorac. Oncol 2009, 4, 891–910. [Google Scholar]
- Fielden, MR; Eynon, BP; Natsoulis, G; Jarnagin, K; Banas, D; Kolaja, KL. A gene expression signature that predicts the future onset of drug-induced renal tubular toxicity. Toxicol. Pathol 2005, 33, 675–683. [Google Scholar]
- Umbright, C; Sellamuthu, R; Li, S; Kashon, M; Luster, M; Joseph, P. Blood gene expression markers to detect and distinguish target organ toxicity. Mol. Cell. Biochem 2010, 335, 223–234. [Google Scholar]
- Bushel, PR; Heinloth, AN; Li, J; Huang, L; Chou, JW; Boorman, GA; Malarkey, DE; Houle, CD; Ward, SM; Wilson, RE; Fannin, RD; Russo, MW; Watkins, PB; Tennant, RW; Paules, RS. Blood gene expression signatures predict exposure levels. Proc. Natl. Acad. Sci. USA 2007, 104, 18211–18216. [Google Scholar]
- Staratschek-Jox, A; Classen, S; Gaarz, A; Debey-Pascher, S; Schultze, JL. Blood-based transcriptomics: Leukemias and beyond. Expert Rev. Mol. Diagn 2009, 9, 271–280. [Google Scholar]
- Liew, CC; Ma, J; Tang, HC; Zheng, R; Dempsey, AA. The peripheral blood transcriptome dynamically reflects system wide biology: A potential diagnostic tool. J. Lab. Clin. Med 2006, 147, 126–132. [Google Scholar]
- Pahl, A. Gene expression profiling using RNA extracted from whole blood: Technologies and clinical applications. Expert Rev. Mol. Diagn 2005, 5, 43–52. [Google Scholar]
- Kim, SJ; Dix, DJ; Thompson, KE; Murrell, RN; Schmid, JE; Gallagher, JE; Rockett, JC. Effects of storage, RNA extraction, genechip type, and donor sex on gene expression profiling of human whole blood. Clin. Chem 2007, 53, 1038–1045. [Google Scholar]
- Debey, S; Schoenbeck, U; Hellmich, M; Gathof, BS; Pillai, R; Zander, T; Schultze, JL. Comparison of different isolation techniques prior gene expression profiling of blood derived cells: Impact on physiological responses, on overall expression and the role of different cell types. Pharmacogenomics J 2004, 4, 193–207. [Google Scholar]
- Rainen, L; Oelmueller, U; Jurgensen, S; Wyrich, R; Ballas, C; Schram, J; Herdman, C; Bankaitis-Davis, D; Nicholls, N; Trollinger, D; Tryon, V. Stabilization of mRNA expression in whole blood samples. Clin. Chem 2002, 48, 1883–1890. [Google Scholar]
- Asare, AL; Kolchinsky, SA; Gao, Z; Wang, R; Raddassi, K; Bourcier, K; Seyfert-Margolis, V. Differential gene expression profiles are dependent upon method of peripheral blood collection and RNA isolation. BMC Genomics 2008, 9, 474. [Google Scholar]
- Chai, V; Vassilakos, A; Lee, Y; Wright, JA; Young, AH. Optimization of the PAXgene blood RNA extraction system for gene expression analysis of clinical samples. J. Clin. Lab. Anal 2005, 19, 182–188. [Google Scholar]
- Wright, C; Bergstrom, D; Dai, H; Marton, M; Morris, M; Tokiwa, G; Wang, Y; Fare, T. Characterization of globin RNA interference in gene expression profiling of whole-blood samples. Clin. Chem 2008, 54, 396–405. [Google Scholar]
- Field, LA; Jordan, RM; Hadix, JA; Dunn, MA; Shriver, CD; Ellsworth, RE; Ellsworth, DL. Functional identity of genes detectable in expression profiling assays following globin mRNA reduction of peripheral blood samples. Clin. Biochem 2007, 40, 499–502. [Google Scholar]
- Vartanian, K; Slottke, R; Johnstone, T; Casale, A; Planck, SR; Choi, D; Smith, JR; Rosenbaum, JT; Harrington, CA. Gene expression profiling of whole blood: comparison of target preparation methods for accurate and reproducible microarray analysis. BMC Genomics 2009, 10. [Google Scholar] [CrossRef]
- Li, L; Ying, L; Naesens, M; Xiao, W; Sigdel, T; Hsieh, S; Martin, J; Chen, R; Liu, K; Mindrinos, M; Davis, R; Sarwal, M. Interference of globin genes with biomarker discovery for allograft rejection in peripheral blood samples. Physiol. Genomics 2008, 32, 190–197. [Google Scholar]
- Liu, J; Walter, E; Stenger, D; Thach, D. Effects of globin mRNA reduction methods on gene expression profiles from whole blood. J. Mol. Diagn 2006, 8, 551–558. [Google Scholar]
- Kennedy, L; Vass, JK; Haggart, DR; Moore, S; Burczynski, ME; Crowther, D; Miele, G. Hematopoietic Lineage Transcriptome Stability and Representation in PAXgene Collected Peripheral Blood Utilising SPIA Single-Stranded cDNA Probes for Microarray. Biomark. Insights 2008, 3, 403–417. [Google Scholar]
- Krawiec, JA; Chen, H; Alom-Ruiz, S; Jaye, M. Modified PAXgene method allows for isolation of high-integrity total RNA from microlitre volumes of mouse whole blood. Lab. Anim 2009, 43, 394–398. [Google Scholar]
- Robison, EH; Mondala, TS; Williams, AR; Head, SR; Salomon, DR; Kurian, SM. Whole genome transcript profiling from fingerstick blood samples: a comparison and feasibility study. BMC Genomics 2009, 10, 617. [Google Scholar]
- Fan, H; Hegde, PS. The transcriptome in blood: challenges and solutions for robust expression profiling. Curr. Mol. Med 2005, 5, 3–10. [Google Scholar]
- Eklund, AC; Turner, LR; Chen, P; Jensen, RV; deFeo, G; Kopf-Sill, AR; Szallasi, Z. Replacing cRNA targets with cDNA reduces microarray cross-hybridization. Nat. Biotechnol 2006, 24, 1071–1073. [Google Scholar]
- Dallas, PB; Gottardo, NG; Firth, MJ; Beesley, AH; Hoffmann, K; Terry, PA; Freitas, JR; Boag, JM; Cummings, AJ; Kees, UR. Gene expression levels assessed by oligonucleotide microarray analysis and quantitative real-time RT-PCR—how well do they correlate. BMC Genomics 2005, 6. [Google Scholar] [CrossRef] [Green Version]
- Rajeevan, MS; Vernon, SD; Taysavang, N; Unger, ER. Validation of array-based gene expression profiles by real-time (kinetic) RT-PCR. J. Mol. Diagn 2001, 3, 26–31. [Google Scholar]
- Viale, A; Li, J; Tiesman, J; Hester, S; Massimi, A; Griffin, C; Grills, G; Khitrov, G; Lilley, K; Knudtson, K; Ward, B; Kornacker, K; Chu, CY; Auer, H; Brooks, AI. Big results from small samples: evaluation of amplification protocols for gene expression profiling. J. Biomol. Tech 2007, 18, 150–161. [Google Scholar]
- Beekman, JM; Reischl, J; Henderson, D; Bauer, D; Ternes, R; Pena, C; Lathia, C; Heubach, JF. Recovery of microarray-quality RNA from frozen EDTA blood samples. J. Pharmacol. Toxicol. Methods 2009, 59, 44–49. [Google Scholar]
- Clement-Ziza, M; Gentien, D; Lyonnet, S; Thiery, JP; Besmond, C; Decraene, C. Evaluation of methods for amplification of picogram amounts of total RNA for whole genome expression profiling. BMC Genomics 2009, 10. [Google Scholar] [CrossRef]
- Ovation Whole Blood Solution Performance. Available online: http://www.nugeninc.com/tasks/sites/nugen/assets/File/technical_documents/techdoc_wb_sol_rep_01.pdf 2010 (access on 7 April 2011).
- Dauphinee, SM; Karsan, A. Lipopolysaccharide signaling in endothelial cells. Lab. Invest 2006, 86, 9–22. [Google Scholar]
- Talwar, S; Munson, PJ; Barb, J; Fiuza, C; Cintron, AP; Logun, C; Tropea, M; Khan, S; Reda, D; Shelhamer, JH; Danner, RL; Suffredini, AF. Gene expression profiles of peripheral blood leukocytes after endotoxin challenge in humans. Physiol. Genomics 2006, 25, 203–215. [Google Scholar]
- Guiding principles in the use of animals in toxicology. Toxicol. Appl. Pharmacol. 2002, 178, 4.
- Weng, L; Dai, H; Zhan, Y; He, Y; Stepaniants, SB; Bassett, DE. Rosetta error model for gene expression analysis. Bioinformatics 2006, 22, 1111–1121. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Method | RNA Yield (ng/μL whole blood) | RIN Score |
|---|---|---|
| PAXgene | 44.9 ± 5.6 | 9.0 ± 0.2 |
| QSI_500 | 21.0 ± 1.3 | 8.8 ± 0.4 |
| QSI_200 | 33.6 ± 5.4 | 9.5 ± 0.2 |
| QSI_100 | 79.4 ± 7.0 | 8.5 ± 0.5 |
| QSI_50 | 69.0 ± 6.3 | 8.8 ± 0.2 |
| QSI_25 | 65.4 ± 6.0 | 8.8 ± 0.2 |
| Method | % Present | GAPDH 3′/5′ Ratio | Actin 3′/5′/Ratio |
|---|---|---|---|
| PAXgene | 47.5 ± 2.1 | 2.7 ± 0.03 | 8.9 ± 0.3 |
| QSI_500 | 37.9 ± 3.6* | 2.5 ± 0.11 | 8.9 ± 1.2 |
| QSI_200 | 37.2 ± 2.3* | 2.7 ± 0.08 | 8.4 ± 0.3 |
| QSI_100 | 33.7 ± 5.1* | 2.9 ± 0.15 | 10.1 ± 1.1 |
| QSI_50 | 37.0 ± 3.1* | 2.8 ± 0.12 | 9.8 ± 1.1 |
| QSI_25 | 36.5 ± 4.4* | 2.7 ± 0.14 | 8.5 ± 0.6 |
| Pathway | RNA Isolation Methods | Total # of Genes in Pathway | # of Regulated Genes in Pathway* | % Regulated |
|---|---|---|---|---|
| Interferon Signaling | PAXgene | 30 | 13 | 43 |
| QSI_500 | 12 | 40 | ||
| QSI_25 | 13 | 43 | ||
| IL-10 Signaling | PAXgene | 70 | 23 | 33 |
| QSI_500 | 26 | 37 | ||
| QSI_25 | 23 | 33 | ||
| Activation of IRF by Cytosolic Pattern Recognition Receptors | PAXgene | 74 | 23 | 31 |
| QSI_500 | 22 | 30 | ||
| QSI_25 | 25 | 34 | ||
| Death Receptor Signaling | PAXgene | 64 | 19 | 30 |
| QSI_500 | 18 | 28 | ||
| QSI_25 | 20 | 31 | ||
| Role of PKR in interferon Induction and Antiviral Response | PAXgene | 46 | 13 | 28 |
| QSI_500 | 14 | 30 | ||
| QSI_25 | 16 | 35 |
| Gene Symbol | Abundance Level | Regulated by LPS according to Microarray | Fold Change over Vehicle | |
|---|---|---|---|---|
| Microarray | RT-PCR | |||
| Tcf7 | High | Down | −4.25 ± 0.75 | −7.74 ± 1.59 |
| Nrp1 | Mid | Down | −19.22 ± 4.13 | −4.65 ± 0.27 |
| Map2k6 | Low | Down | −5.16 ± 0.64 | −16.92 ± 4.63 |
| Alas2 | High | No Change | 0.37 ± 0.69 | 0.55 ± 0.63 |
| Canx | Mid | No Change | −1.22 ± 0.02 | −1.10 ± 0.04 |
| Flad1 | Low | No Change | −1.23 ± 0.04 | −2.77 ± 0.31 |
| S100a9 | High | Up | 5.52 ± 0.62 | 16.70 ± 3.80 |
| Il1b | Mid | Up | 5.35 ± 0.99 | 9.19 ± 1.61 |
| Tnf | Low | Up | 6.04 ± 1.23 | 3.94 ± 0.76 |
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Fricano, M.M.; Ditewig, A.C.; Jung, P.M.; Liguori, M.J.; Blomme, E.A.G.; Yang, Y. Global Transcriptomic Profiling Using Small Volumes of Whole Blood: A Cost-Effective Method for Translational Genomic Biomarker Identification in Small Animals. Int. J. Mol. Sci. 2011, 12, 2502-2517. https://doi.org/10.3390/ijms12042502
Fricano MM, Ditewig AC, Jung PM, Liguori MJ, Blomme EAG, Yang Y. Global Transcriptomic Profiling Using Small Volumes of Whole Blood: A Cost-Effective Method for Translational Genomic Biomarker Identification in Small Animals. International Journal of Molecular Sciences. 2011; 12(4):2502-2517. https://doi.org/10.3390/ijms12042502
Chicago/Turabian StyleFricano, Meagan M., Amy C. Ditewig, Paul M. Jung, Michael J. Liguori, Eric A. G. Blomme, and Yi Yang. 2011. "Global Transcriptomic Profiling Using Small Volumes of Whole Blood: A Cost-Effective Method for Translational Genomic Biomarker Identification in Small Animals" International Journal of Molecular Sciences 12, no. 4: 2502-2517. https://doi.org/10.3390/ijms12042502
APA StyleFricano, M. M., Ditewig, A. C., Jung, P. M., Liguori, M. J., Blomme, E. A. G., & Yang, Y. (2011). Global Transcriptomic Profiling Using Small Volumes of Whole Blood: A Cost-Effective Method for Translational Genomic Biomarker Identification in Small Animals. International Journal of Molecular Sciences, 12(4), 2502-2517. https://doi.org/10.3390/ijms12042502